
Abstract
Glucantime, a pentavalent antimonial drug, is commonly used for the treatment of leishmaniasis but the presence of residual trivalent antimony, Sb(III), is thought to be responsible for toxic side-effects observed in patients. Numerous analytical studies have focused on determining Sb(III) concentrations in Glucantime but without reaching a consensus: results span over 3 orders of magnitude. In this study, we present a detailed new analytical approach showing that: (1) Sb(III) levels are much higher than previously reported and represent more than 30 % of total Sb; (2) determination of Sb(III) concentrations in acidic conditions is hampered by fast oxidation rates. This latter point explains the large variations in previously reported results of Sb(III) concentrations in Glucantime. Measurements were made here at a vibrated gold microwire electrode by stripping voltammetry enabling measurement of Sb(III) in acidic, neutral or alkaline conditions. The developed methods are sensitive (e.g., detection limits of 19 pM for 120 s deposition at pH 4.5), stable (<6 %, N = 100), precise (5 %, N = 5) and robust (same electrode used for weeks) at all pH values. In diluted solutions of Glucantime, Sb(III) levels were strongly dependent both on pH and ionic strength. At pH < 3, Sb(III) is oxidized with oxidation rates that increase as pH is decreased. At high pH, Sb(III) forms electro-inactive complexes. Highest Sb(III) levels were detected at pH ∼3 and at low ionic strength. The presence of several Sb(III) and Sb(V) species was demonstrated by different reduction waves obtained by stripping scanned voltammetry. As an implication of these unexpectedly high Sb(III) concentrations, an alternative model can be proposed for the mode of action of pentavalent antimonials against leishmaniasis, in which antimony complexes may act as molecular carrier of Sb(III) and release it specifically in the acidic intracellular compartment where the Leishmania parasites reside.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Leishmaniasis is a tropical disease affecting 12 million people worldwide in impoverished areas and causing the death of an estimated 50,000 people every year [1]. Leishmaniasis is caused by a protozoan parasite of the genus Leishmania, transmitted by the bite of a sandfly. Wild and domesticated animals and humans themselves can act as a reservoir of infection. Leishmania parasite is found as a motile promastigote in the sandfly, it transforms into an amastigote when engulfed by host macrophages, and resides in the acidic environment of secondary lysosomes [1]. In 2010, it was reported that 88 countries on 4 continents were confronted to the disease with an estimated 1.6 million new cases emerging every single year [1]. Of those, 500,000 are visceral, the most virulent form of the disease, usually fatal within 2 years if untreated. The cutaneous form is the most common one causing ulcers on the face, arms, and legs resulting in permanently disfiguring scars. The most disfiguring form is the mucotaneous that destroys the soft tissues of the nose, mouth, and throat. Since 1993, the number of cases is increasing, mostly due to movement of populations that spread the disease to non-immune collectivity. When occurring in urban areas, explosive epidemics may occur [1].
The most common treatment for leishmaniasis over the last 70 years is based on injectable pentavalent antimonials. Meglumine antimoniate (MA), commercialized as Glucantime, is the most used medication. There are newer and more effective treatments such as liposomal amphotericin B which has no side-effects and is very effective, but it is too expensive for developing countries [2]. Solutions of pentavalent antimonials are more affordable but the treatment is lengthy and present serious toxic side-effects [3]. The active ingredient in treating leishmaniasis is believed to be antimonite Sb(III) after in vivo reduction of antimoniate Sb(V) [4]. Toxic side-effects are believed to originate from Sb(III), either produced in vivo or present in the pentavalent antimony solution before being injected [5].
Numerous speciation studies have been carried out in an attempt to determine the amount of Sb(III) and Sb(V) species in MA solutions. These studies all agree in stating that both Sb(V) and Sb(III) are present and complexed, but they markedly differ in amounts of Sb(III) present in Glucantime. Concentrations as high as 15 mg mL−1 [6] and as low as 0.02 mg mL−1 [7] have been reported showing that Sb(III) determination in such matrix is far from trivial.
The structure and composition of MA is still unclear. Roberts et al. [8] showed that MA is a mixture of complexes formed between antimony Sb and N-methyl-d-glucamine (NMG), the major moiety being the cationic specie NMG–Sb+–NMG. Additional species containing up to four Sb atoms and five NMG and corresponding to the general form (NMG–Sb) n –NMG were also observed. Frezard et al. [9] later challenged these structures and suggested the presence of 1:1, 1:2, 2:2 and 2:3 Sb(V)–NMG complexes as zwitterionic species, Sb(V) carrying a negative charge and the amino group a positive charge. MA is thus a complex mixture of oligomers that exist in equilibrium in solution. After 10 times dilution of Glucantime solution in water, the osmolality increased by 45 % after 8 days suggesting hydrolysis to 1:1 Sb(V)–NMG complex [8, 9]. Bonding between Sb(V) and NMG ligands is achieved through hydroxyl groups. The resulting complexes thus behave like an acetal [10], i.e., they are easily cleaved by dilute acids (due to protonation of the hydroxyl groups) while they are resistant to hydrolysis in alkaline solutions.
Various analytical methods have been used for the speciation of Sb in MA solutions. They include hyphenated techniques such as high pressure liquid chromatography–inductively coupled plasma–mass spectrometry (HPLC-ICP-MS) [11], hydride generation atomic absorption spectroscopy [12, 13], spectrophotometry [14]. In most cases, the method is first developed in standard solutions (usually acidic due to better sensitivity) and applied as such in MA solutions.
Electrochemical stripping techniques are commonly used for Sb determination in aqueous media at various electrodes such as the mercury drop [15–20], mercury films [21–23], solid gold [24, 25], gold films [26–28], gold [29], or silver nanoparticles [30]. Sb(III) is the most electro-active form and speciation can be achieved by first measuring Sb(III) followed by total Sb after chemical reduction of Sb(V) with, e.g., potassium iodide [28] or sulfur dioxide [31, 32]. Detection of Sb(III) is almost always reported in acidic conditions although it can also be detected at higher pH [20, 33]. Sb(V) is electro-inactive unless very strong acidic and chloride solutions (up to 6 M HCl) are used (e.g., [24]) or unless a very low deposition potential (−1.8 V) is employed with mild acidic conditions (pH 1) [25].
Only few methods were developed for Sb(III) determination in Glucantime [6, 11, 18, 19, 22, 30, 34] and most of them used acidic conditions. Recently, we reported a sensitive method for the detection of Sb(III) in neutral pH seawater [33] using stripping potentiometry or stripping voltammetry at a gold wire electrode. We report here in our work on the speciation of Sb in solutions of Glucantime. It consists of: (1) the development of Sb(III) detection in solutions of varying pH (acidic to basic); (2) applications to Sb(III) and total Sb determinations in Glucantime; and (3) insights into the chemical speciation of Sb(III) using scanned stripping voltammetry (SSV) (also called pseudopolarography [35]). This technique is based on successive stripping analysis at varying deposition potentials E dep. By plotting the intensity of the peak against E dep, the presence of complexes having similar stability constants is indicated by the presence of one wave. Distinct waves indicate the presence of different complexes having different stability constants. This technique has been used in natural waters for the speciation of, e.g., Cu [36], Pb [37], or Cd [38]. To the best of our knowledge, it had never been used for Sb(III) in a pharmaceutical context.
Experimental
Apparatus
Experiments were performed using a μAutolabIII potentiostat (Ecochemie, now Metrohm) controlled by the GPES software (version 4.9). The working electrode (WE) was a gold wire (Goodfellow, UK) of 10 μm diameter (99.99 %, temper hard). The reference electrode (RE) was a Ag/AgCl/KCl(3 M) with a double bridge filled with 0.1 M NaNO3. The auxiliary electrode (AE) was an iridium wire (Goodfellow) of 150 μm diameters and an approximate length of 4–5 mm. Both WE and AE were homemade as previously described [39]. A small vibrating device (diameter of 4 mm, weight of 20 g) consisting in a off weight rotor (1.5 V, 150 Hz frequency) is attached to the WE to vibrate the electrode during the deposition step of a stripping experiment, in place of the usual magnetic stirrer to enhance mass transport [33]. A magnetic stirrer, controlled manually, was also used to ensure rapid mixing of the solution after sample or standard additions. Typically, at the end of the day, the electrode was cleaned in 0.5 M H2SO4 by hydrogen generation (−3 V for 30 s) and its surface monitored through the reduction charge of the oxide monolayer by cyclic voltammetry [40]. Electrodes were stored in Milli-Q water and were stable over periods of weeks.
Chemicals
Sb(III) and Sb(V) atomic absorption standard solutions (1 g L−1 or 8.21 mM) were from Aldrich and Sigma respectively. Clean HCl (11.6 M) was produced in-house by double distillation on a quartz condenser. HClO4 (60 %), sulfuric acid (98 %), orthoboric acid H3BO3, and sodium hydroxide NaOH were of AnalaR grade from BDH, UK. HNO3 (70 %), sodium acetate CH3COONa and potassium chloride KCl were of reagent grade from Fischer Scientific, UK. Potassium phosphate KH2PO4 was from Riedel-de-Haёn, Germany. pH of acetate buffer solutions were adjusted with HClO4. NaOH was used to adjust the phosphate buffer pH.
Glucantime® ampoules were from Sanofi-Aventis Farmacêutica Ltda (SP, Brazil) and are denoted MA1, MA2 (validity 10/2010, lot 5E5314), MA3 (04/2014, lot L 902520), and MA4 (07/2010, L 503644). MA1 was used to develop the analytical method (its validity date and lot number were unfortunately not available). MA2, MA3, and MA4 were opened just before Sb(III) determination. MA1 solutions were diluted 100- and 10,000-fold in either acid (HClO4, HCl) neutral (acetate, phosphate buffers) or basic solutions (NaOH) in polyethylene containers (50 ml) wrapped in aluminum foils to avoid potential photo-oxidation. For Sb(III) determination, these solutions were prepared immediately before addition of Glucantime in the voltammetric cell. MA1 solution (undiluted) was placed in a 30-ml polypropylene sterile tube, wrapped with aluminum paper to avoid potential photo-oxidation and was stored in the fridge at all time. No changes in Sb(III) speciation were noticed even after 9 months of storage, highlighting the long stability of these solutions.
Sb(III) and SbT determination
All measurements were done in a quartz cell. For Sb(III) determination, all solutions were deaerated with N2. Square wave anodic stripping voltammetry (SWASV) was mostly used but anodic stripping chronopotentiometry (ASC) was also suitable. Typical SW parameters were 50 Hz frequency, 4 mV step and 25 mV amplitude. A typical standard addition procedure was run as follows: five SWASV scans (deposition at −0.6 V for 10 s, desorption at −1.2 V for 1 s, 1 s equilibrium, stripping from −0.5 to 0.2 V) were first made in the background electrolyte to stabilize the electrode. The fifth scan was used as the background scan. Glucantime was then added to the cell (typical dilution factor of 107) and five new SWASV scans were recorded. A minimum of two standard additions were made with five SWASV scans obtained after each addition. The background scan was subtracted from all scans. Only the last three SWASV scans (after each addition) were used for quantification. The procedure was fully automated through the Project option of the GPES software. In solutions of pH > 3, a short conditioning step (−2.5 V for 1 to 5 s and +0.7 V for 2 s) was applied before each analysis which maintained high sensitivity.
SSV was achieved without or with background subtraction. In the latter case, a background scan was run at each deposition potential. Each analysis consisted in the following procedure:
-
1.
Conditioning: −2.5 V (1 s), +0.7 V (1 s), 1 s eq, stripping from 0.6 to 0.7 V
-
2.
SWASV Analytical scan: E dep (5–30 s), −1.2 V (1 s), 1 s eq, stripping from e.g. −0.3 to 0.2 (dependent on the pH)
-
3.
SWASV background scan: 0.7 V (2 s), −1.2 V (1 s), 1 s eq, stripping from e.g. −0.3 to 0.2 (dependent on the pH)
The half-wave potentials (E 1/2) of reduction waves obtained in SSV curves were determined using the Lingane modified equation ([35]):
where E d is the deposition potential, i p is the peak current and i p max is the maximum peak current. If there are several waves, i p max is the maximum peak current of the considered wave. When \( {i_{\mathrm{p}}}={{{i_{\mathrm{p}}^{\max }}} \left/ {2} \right.} \), then E d = E 1/2.
For determination of total Sb, the diluted Glucantime solution (F = 107) was acidified to pH 1 with HCl and UV-digested for 45 min. Differential pulse anodic stripping voltammetry was used (100 ms, 4 ms, 4 mV, 25 mV) using a deposition potential of −1.8 V for 10s, similar to previously reported [25]. No background subtraction was used.
Results
Detection of Sb(III) and Sb(V)
This section presents the establishment of optimum experimental conditions for detection of Sb(III) and Sb(V) at the gold wire electrode in synthetic solutions of various pH, giving analytical parameters such as detection limits, linear range, stability, and presenting the influence of deposition time and deposition potentials.
-
Sb(III)
Sb(III) can be determined by ASV or ASC in alkaline, neutral and acidic conditions. Figure 1 presents typical subtracted SWASV voltammograms of 60 nM Sb(III) at pH 12 (10 mM NaOH), pH 7 (10 mM phosphate buffer), pH 4.75 (10 mM acetate), and pH 2 (10 mM HClO4). The reversibility (fast electron transfer) was confirmed by the presence of a peak in both forward and backward scans of a SW procedure. SWASV and ASC peak potentials were shifted towards negative potentials at a rate of −49.2 and −49.9 mV/pH, respectively. This is ∼20 % lower than the expected value of 59 mV/pH predicted from Eq. 1:
Fig. 1 
Background subtracted scans of 60 nM Sb(III) at pH 12 (10 mM NaOH), pH 7 (10 mM KH2PO4), pH 4.75 (10 mM CH3COONa), and pH 2 (10 mM HClO4). SWASV parameters: −0.6 V (5 s), −1.2 V (1 s), 1 s eq, 50 Hz, 6 mV, 25 mV
$$ \mathrm{S}\mathrm{b}{{\left( {\mathrm{OH}} \right)}_3}+3{{\mathrm{e}}^{-}}+3{{\mathrm{H}}^{+}}\to \mathrm{S}{{\mathrm{b}}^0}+3{H_2}O $$(2)Similarly to detection of Sb(III) in seawater[33], the Sb(III) signal was much improved when a desorption potential (E des) procedure was implemented. It consists in the application of a low negative potential (−1.2 V) for a short time (1 s) just before the equilibrium and the stripping step. SSV curves obtained with and without E des in 10 mM CH3COONa (pH 4.91), 10 mM KH2PO4 (pH 6.11), 10 mM NaOH or 10 mM H2SO4 are shown in Fig. 2. In sulfuric acid (Fig. 2a), in acetate (Fig. 2b), or phosphate (Fig. 2c) buffer, the SSV displayed the expected sharp wave of a 3 electrons transfer only when E des was used. Without the application of E des, the peak was wider and not well-shaped when E dep ≳ −0.6 V. Similar behavior was obtained in UV-digested solutions indicating that anion adsorption (and not organic adsorption) is likely to be the cause of such effect. However, in HCl, the wave was sharp even without a desorption potential (Fig. 2a). The decrease observed in HCl at low deposition potentials (less than −1 V; Fig. 2a) is due to Sb(III) oxidation by chlorine that is generated at the auxiliary electrode during the deposition step, as previously reported in details for As(III) [39]. In HCl solutions, we only used a relatively high deposition potential to avoid such instability problems. At pH 12 (Fig 2d), no difference was observed with or without a desorption potential, showing that hydroxide do not interfere with the Sb stripping process. Unless otherwise stated, a desorption potential of −1.2 V was always used.
Fig. 2 
Influence of the application of the desorption potential (E des) on the Sb(III) peak derivative in 10 mM H2SO4 (triangle) or 10 mM HCl (circle) (A), 10 mM CH3COONa (B), 10 mM KH2PO4 (C) and 10 mM NaOH (D). Respective pH values: 2, 4.75, 6.1, and 12. A, B, C: 30 nM Sb(III), D: 60 nM Sb(III); E dep for 30s, E des = −1.2 V for 1 s
At pH 12, two peaks were obtained at −475 and −350 mV, the latter being ~30 % smaller than the former (Fig. 1). They correspond to the oxidation of Sb0 to SbIII (3 electrons transfer) and to the oxidation of SbIII to SbV (2 electrons transfer) [41]. The respective intensity of the peaks suggests that, at this pH, all Sb(III) formed during reoxidation of Sb0 remains adsorbed at the gold surface before being oxidized further to Sb(V). The two peaks are only present in neutral and alkaline conditions. At pH ≤ 6, only one peak was obtained indicating the non-adsorption of Sb(III) in this combined range of pH and potential. This is similar to the behavior of As(III) on the gold electrode [42] in non-chloride solutions.
Stability of the Sb(III) signal at different pH was assessed by running 50 successive SWASV or ASC measurements (E dep = −0.6 V for 5 s, E des = −1.2 V for 1 s). Maximum standard deviations of 6 % were obtained indicating the good stability of Sb(III) in these solutions. No systematic decrease was occurring for any of them. Highest sensitivities were obtained in acetate buffer (10 mM) at pH 4.75.
Sb(III) linear range was at least up to 100 nM for 30 s deposition. In a solution containing 20 nM Sb(III), the peak height varied linearly with deposition time up to 2 min. Applying a desorption potential every 30 s during deposition (30 s at E dep, 1 s at −1.2 V, 30 s at E dep, 1 s at −1.2 V, etc. as recently suggested [33]) linear variation was extended up to 5 min indicating that anion build-up at the electrode surface interferes with the stripping step.
Detection limits (LoDs) are low. In deaerated acetate buffer, LoDs of 19 and 130 pM were achieved using deposition time of 120 and 15 s, respectively (Table 1). LoDs were calculated as three times the standard deviation of ten consecutive analyses of low levels of Sb(III); 0.25 and 2 nM Sb(III) were used for t dep = 120 s and 15 s, respectively. The deposition potential was −0.25 V and 1 s desorption time (−1.2 V) was used. The LoD of 19 pM for Sb(III) is the second lowest ever reported, based on a recently reported review [43]. The most sensitive technique is obtained by cathodic stripping voltammetry at an in situ-plated bismuth film electrode [44] (16 pM for 30 s deposition time).
Table 1 Detection limits of Sb(III) E dep = −0.25 V, E des = −1.2 V (1 s) -
Sb(V)
Sb(V) is usually considered electro-inactive unless relatively strong acidic conditions are used. Using a 5 μm vibrated gold microwire electrode, we previously reported detection limits of 5 pM (300 s deposition) in 0.2 M HCl [25]. This method was used for determination of total Sb in acidified mineral, groundwater and seawater. In addition to the acidic conditions (0.1 M H2SO4 + 50 mM HCl), the detection of 100 nM Sb(V) was here also tested in neutral (10 mM phosphate and 10 mM acetate) and alkaline (10 mM sodium hydroxide) using deposition potentials down to −2.5 V. Figure 3 displays the SSV of Sb(V) at different pH values (12, 6, 4.7, and 0.6) together with voltammograms obtained in phosphate buffer (pH 6). A well-shaped peak (W 1/2 ∼ 50 mV) was obtained at all pH values, although much smaller at pH 12. This is in contrast with results obtained at the mercury electrode where only strong acidic conditions and high chloride concentrations can promote the reduction of Sb(V) [45]. However, in these acidic conditions, only a high-deposition potential was used to avoid problems related to hydrogen generation. In neutral or alkaline conditions at the gold electrode, a very low deposition potential was required but the resulting peaks were well-shaped. Such low E dep are not usually used due to interference from hydrogen generation. One can only assume that this is the reason why the electrochemical reduction of Sb(V) in alkaline, or neutral, conditions had never been reported before. The vibrated gold microwire electrode is largely insensitive to hydrogen generation [25] and is thus ideally suited for severe cathodic deposition conditions. To the best of our knowledge, this is the first report of Sb(V) detection by ASV in non-acidic conditions.
Fig. 3 
A Influence of deposition potentials on 100 nM Sb(V) in: (1) acid (0.1 M H2SO4 + 50 mM HCl); (2) 10 mM acetate buffer pH 4.9; (3) 10 mM phosphate buffer pH 6 and (4) 10 mM NaOH. B Typical SWASV voltammograms obtained at pH 6 (corresponding to curve 3 in A). SWASV parameters: 0.7 V (5 s), E dep (30s), 1 s eq, 50 Hz, 6 mV, 25 mV. No background subtraction. Solutions were purged
Similar to Sb(III), Sb(V) peak potentials varied linearly with pH at a rate of −47.3 mV.pH−1 with values similar to those displayed in Fig. 1. This is expected since the stripping process is the same (i.e. Sb0 to SbIII).



Detection of Sb(III) in Glucantime
Both Sb(V) and Sb(III) were detected in Glucantime in agreement with previous studies. However, we found that pH, ionic strength, and dilution factor strongly influenced the signal of Sb(III) in Glucantime.
-
pH effect
Two types of experiments were conducted at various pH: (1) stability on a 20-min time scale; (2) stability over several days.
-
1.
Figure 4 presents the time evolution of the Sb(III) peak derivative at different pH for the first 20 min after addition of MA1 in the voltammetric cell (overall dilution factor F dil = 1.67 · 106). For clarity purposes, the peak intensities were normalized to the first measurement (obtained ∼30 s after addition of MA1). It is clearly seen that at pH values below 6, the signal decreases with time. At pH < 4.75, all curves follow an apparent first-order kinetic at short time. Rate constants (given by the slope of the regression lines) increased with acidity (e.g. −1.03, −0.47, −0.26, and −0.1 min−1 in 50, 20, 10, and 5 mM H2SO4, respectively). In acetate buffer, the response is much more stable but still found to decrease while no significant decrease was observed in phosphate buffer, pH 6.
Fig. 4 
Time evolution of the Sb(III) peak after addition of Glucantime (dilution factor of 1.67 106) in solution of different pH. Curves 1, 2, 3, and 4: 50, 20, 10, and 5 mM H2SO4, respectively; 5, 6 and 7: 10 mM CH3COONa at pH 2.8, 4.1 and 4.75 respectively; 8: 10 mM KH2PO4, pH 6. SWASV parameters: 50 Hz, 6 mV, 25 mV, E dep = −1.2 V for 6 s (curves 1–4) and E dep = −0.6 V for 6 s (curves 5–8)
-
2.
MA1 solutions were diluted (F dil = 104) in solutions of various pH and Sb(III) concentrations measured at different intervals over 3 days. Best stability was obtained in NaOH (10 mM) with a small 10 % decrease after three days. In phosphate buffer, pH 7, no significant decrease was observed after 24 h. In acetate buffer (pH 4.75), the same concentrations were obtained 30 and 60 min after dilution of MA1 (9.8 ± 0.7 and 9.5 ± 0.8 mg/ml, respectively). However, a 50 and 85 % decrease were observed after 48 and 72 h, respectively. Glucantime solutions were thereafter always diluted in 10 mM NaOH, kept at room temperature in dark conditions, and prepared on the day.
It is thus apparent that the pH of Glucantime is of prime importance when determining Sb(III). Loss of Sb(III) signal was not due to adsorption on the cell as similar loss rates were obtained for a PTFE or a quartz cell. Adsorption of interfering species at the surface of the gold electrode was also discarded as same rate loss was observed irrespective of the deposition time (5, 10, or 30 s) or irrespective of the dilution factor (F = 1.6 106, 107 or 3.3 107). The loss of signal at low pH could be due to oxidation processes or formation of electro-inactive Sb(III) complexes (at the deposition potential used above i.e. −0.6 V). To gain insights into which of these two processes is most likely, Glucantime solutions were first diluted (F = 104) in solutions of various pH and left to equilibrate for a few hours. Sb(III) content was then determined in 10 mM acetate buffer (pH 4.75) after further dilution (F = 107) (Fig. 5). When Glucantime was first diluted in solution of pH > 4.75, Sb(III) concentration was found stable at ∼11 mg/ml. However, when diluted at pH 2, the concentration was four times lower while, at pH 1 and pH 0, the Sb(III) levels were below the detection limit. Although the pH during the voltammetric determination was the same for all solutions (4.75), a loss of Sb(III) signal is only observed for Glucantime solutions that experienced acidic conditions suggesting that this is a non-reversible process. The loss process is thus likely to be due to oxidation and not to the formation of electro-inactive Sb(III) complex. Additions of hydrazine sulfate (anti-oxidizing agent) up to 300 μM were however inefficient in preventing this oxidation. The mechanism of this oxidation is still unclear.
Fig. 5 
Influence of the pH of the solution that was used to store Glucantime (diluted 10,000-fold) on Sb(III) levels. MA1 sample was diluted in 1 M, 100 mM, 10 mM HClO4, 10 mM acetate buffer (pH 4.75), 10 mM phosphate buffer (pH 6.0), 10 mM borate buffer (pH 9) and in 10 mM NaOH. Sb(III) was determined after further dilution of these solutions (1,000 times) in 10 mM acetate buffer (pH 4.75)
-
1.
-
Ionic strength effect
The ionic strength of the solution placed in the voltammetric cell was found to significantly affect the amount of Sb(III) detected in MA solutions. This effect was first suspected after looking at the influence of HCl concentrations (Fig. 6a) on SWASV scans of Glucantime (F = 106). In this experiment, successive SWASV measurements were run every 11 s. After the fourth measurement (blank), Glucantime was added to the purged HCl solutions. After the 30th measurement, 100 nM Sb(III) standard was then added and from the increase in signal, used to calculate Sb(III) concentrations throughout the experiment. Sb(III) concentrations were higher and more stable at low HCl concentrations. In 0.1 M HCl, the signal decreased rapidly (as expected from above) while stable signals were obtained in 1 mM or 0.5 mM HCl. It is interesting to note that, after addition of 100 nM Sb(III) in Glucantime, the signal was stable, even at high HCl concentrations. Oxidation seems to only affect Sb(III) originally present in Glucantime and does not affect the added Sb(III), suggesting that the oxidant is therefore not in excess.
Fig. 6 
A Sb(III) concentrations in Glucantime vs time (see text) in HCl solutions of various concentrations. Glucantime diluted (F = 106). Numbers indicate the concentration of HCl in millimolar. Measurements run every 11 s. B Effect of addition of 4 mM NaCl on Sb(III) levels
The effect of ionic strength on Sb(III) concentrations in Glucantime was also tested in 1 mM HCl with addition of various amount of electrolyte. After adding 4 mM NaCl in 1 mM HCl, the concentration of Sb(III) almost halved (e.g., Fig. 6b). In the presence of 4 mM NaCl, 4 mM NaNO3 or 4 mM Ca(NO3)2, Sb(III) concentrations were found 42, 49, and 72 % lower than in 1 mM HCl (all determined by standard additions). This effect cannot be attributed to migration current since less than 5 % differences in Sb(III) peak heights were observed in 1 mM HCl containing either 1 or 10 mM NaCl. Increasing the ionic strength may thus favor the formation of a Sb(III) complex that is non-reducible at the deposition potential used here (E dep = −0.6 V).
-
Dilution effect
Dilution factor was found to play a role on Sb(III) concentrations only when Glucantime was diluted in acidic conditions. For instance, in 2 M HCl, Sb(III) was found at 2.36 ± 0.12 and 0.61 mg mL−1 for F = 102 and 104 respectively (after 2 weeks storage at room temperature). In 10 mM HCl, Sb(III) = 3.23 ± 0.12 and less than 1 mg mL−1 for F = 102 and 104, respectively. In contrast, in neutral phosphate solution (pH 6), Sb(III) concentrations were independent of the dilution factor (4.67 ± 0.07 and 4.56 ± 0.20 mg mL−1 for F = 104 and 105).



Sb speciation in Glucantime
Both Sb(III) and Sb(V) are present in Glucantime. Using SSV, we gained information not only on the oxidation state of Sb but also on the presence of various Sb(III) complexes. SSV curves were obtained in Glucantime solutions in conditions where Sb(III) was sufficiently stable, i.e., at pH ≥ 4.5 in acetate or phosphate buffer and at lower pH (1 mM HCl) with/without additions of electrolyte. SSV curves in Glucantime were obtained before/after additions of Sb(III) standards and with/without desorption potentials. Titration curves were also attempted to obtain complexation parameters.
-
SSV in acetate buffer (pH 4.7)
SSV curves of diluted (F = 107) UV and non-UV-digested Glucantime are shown in Fig. 7a and compared to the response of 10 nM Sb(III). The background electrolyte was 10 mM acetate buffer. Deposition potentials were varied from +1 down to −2.5 or −3 V. In acetate buffer, no peak was obtained at deposition potentials above 0.25 V. A sharp wave was observed from 0.2 V down to −0.15 V with a half-wave potential of −56 mV, followed by a plateau characteristic of diffusion limited conditions, similar to those presented in Fig. 2. The SSV curve of non-UV-digested Glucantime was very much different and can be divided in three distinct regions. Firstly, a peak was obtained at positive potentials, up to +0.9 V. This indicates the presence of Sb(III) complex(es), probably negatively charged, that adsorb onto the gold surface in this potential range. These adsorbed species are reduced during the equilibrium time (−0.3 V) and reoxidized during the stripping step. Secondly, for deposition potentials between −0.1 and −0.6 V, a broad wave was obtained with E 1/2 ∼ −0.26 V, i.e., ∼200 mV lower than E 1/2 obtained in acetate buffer. This wave indicates the presence of a stable Sb(III) complex (at this E dep and pH, the reduction of Sb(V) is highly unlikely) and the large shift (204 mV) suggests that this complex is irreversible, i.e., no dissociation of the complex occurs prior to Sb(III) reduction [46]. This is also supported by values of peak potentials that were the same in acetate and in MA, showing that complexes do not play a role during the stripping process, due to slow complexation kinetics [36, 47]. Thirdly, at lower E dep, the signal strongly increased at deposition potentials below −1.6 V up to approximately −2 V, generating a large wave with a half-wave potential of approximately −1.80 V. Very similar overall behavior was also obtained in phosphate buffer (not shown). To identify the origin of this large wave, SSV was achieved in UV-digested Glucantime (Fig. 7a). No signal was observed at high-deposition potentials (greater than −1.6 V) showing that UV digestion was efficient to oxidize all Sb(III) into Sb(V). A single wave was obtained from −1.6 down to −2.95 V with a half-wave value at −2.15 V, i.e., 250 mV more cathodic than the one observed in non-UV-digested Glucantime suggesting that these two waves might be different. However, comparing Glucantime and Glucantime + 50 nM Sb(V), the wave was located at the same potential (Fig. 7b), indicating that the increase in signal obtained at E dep < −1.6 V is due to Sb(V) reduction and not to the reduction of an irreversible Sb(III) complex.
Fig. 7 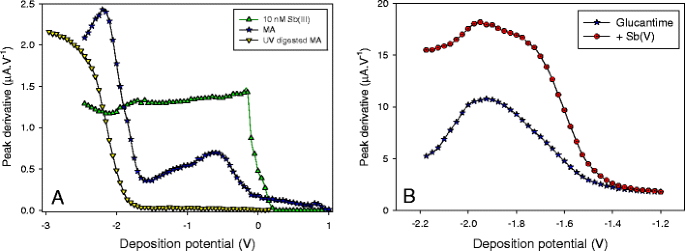
Speciation of Sb(III) in Glucantime—A 10 mM acetate buffer, pH 4.75; addition of 10 nM Sb(III), or Glucantime (F = 107) or UV-digested Glucantime (F = 107); B Glucantime (F = 3.33 106) in 10 mM phosphate buffer with/without addition of Sb(V) (addition corresponds to ∼80 mg mL−1). All values are background subtracted values
The anodic shift of the half-wave potential obtained in Glucantime after UV digestion is unexpected since, if Sb(V) was present as a complex in MA, only a negative shift could have been expected. There are two possible reasons for such effect. Firstly, the maximum peak current obtained at −2.2 V in non-UV-digested solution may be underestimated (due possibly to interferences present in MA) and should occur at lower potential values. If this is the case, E 1/2 would be overestimated. The second hypothesis is that one of the constituent of Glucantime promotes the reduction of Sb(V), shifting its reduction potential to higher values. This constituent would be organic as it is destroyed by UV digestion.
-
Influence of ionic strength on Sb(III) speciation
To get better insights into the ionic strength-induced changes in Sb(III) species, SSV of diluted Glucantime (F = 107) were run in 1 mM HCl at various concentrations of NaCl (0, 4, and 9 mM) and normalized to the sensitivity obtained in the corresponding background solution (i.e., 1 mM HCl + 0, 4, or 9 mM NaCl + 50 nM Sb(III)) (Fig. 8a). In the absence of NaCl, a wave between ∼0 and −0.35 V and a second wave between −0.35 and −1 V were observed. Upon addition of NaCl, the second wave strongly decreased until disappearing while the first wave remained unchanged. This suggests that addition of NaCl promotes the formation of a non-reducible complex at deposition potentials below −0.4 V.
Fig. 8 
SSV curves of Glucantime (F = 107) A in various NaCl concentrations (0, 4, and 9 mM) in 1 mM HCl (normalized to response obtained in 50 nM Sb(III)). B In 1 mM HCl + 1 mM NaCl before and after addition of 20 nM Sb(III), with (triangle) and without (stars) application of a desorption potential at −0.8 V
-
SSV at pH 3
SSV curves were also obtained in Glucantime (diluted 107 times in 1 mM HCl + 1 mM NaCl) before and after addition of Sb(III). Experiments were run with and without a desorption potential at E des = −0.8 V. At E dep > ∼0 V, the signal was much higher with applying E des than without. This means that, similar to acetate buffer (Fig. 7a), some Sb(III) species adsorb on the gold at these positive E dep values and only get reduced when the desorption potential is applied. At potentials < 0 V, the signal increased until reaching a plateau at E dep ∼ −0.5 and −0.65 V (with/without desorption potential, respectively). In both cases, a stronger increase was observed between 0 and −0.1 V. The position of this sharper increase suggests the presence of free, un-complexed, or reactive Sb(III) since it occurred at the same potential as the Sb(III) wave observed in standard solutions (Fig. 2). This wave was then followed by a larger wave which might represents more stable Sb(III) species.
When 20 nM Sb(III) were added to the solution, a sharp wave located at approximately −0.05 V was obtained. No other changes in the SSV curve were apparent showing that added Sb(III) remained as an un-complexed specie, either because the ligand was already saturated or the complexation kinetics are too slow for significant complexation to occur within the time frame of the experiment.
-
Titration curves
Titration experiments (F = 107) of Glucantime with addition of Sb(III) standard did not result in the expected curvature response characteristic of complexation (e.g., [48]). Instead, a linear relationship was always observed, suggesting once again that added Sb(III) remained un-complexed or complexation was too slow to take place within the experimental time (up to 15 min). Titration experiments were achieved in 10 mM acetate buffer (pH 4.9) using E dep = −0.1 V, potential at which the potential complexation of Sb(III) would be most likely seen (based on the SSV curve displayed in Fig. 7a). A similar linear relationship was obtained at E dep = −0.6 V in solution of pH > 4.
To summarize this section, the presence of at least three different Sb(III) species was shown: one that adsorbs onto the gold surface at positive potential, one that gets reduced at lower potentials (approximately −0.26 V in acetate) and the last one present as free or highly reactive Sb(III). While the first two were present in all tested solutions, the free reactive specie was only observed at pH 3. It is possible that a fourth class of Sb(III) complex exists but its presence may be hidden by the reduction of Sb(V) species. Our data also suggest that Sb(V) reduction may be facilitated by an organic constituent present in Glucantime.


Sb(III) and Sb(V) levels in Glucantime
-
Sb(III) determination
The concentration of Sb(III) was determined by standard additions either in acetate, phosphate, hydroxide or chlorhydric acid. The experiment time was kept to a minimum with short SWASV scans (5–10 s deposition at −0.6 V, 1 s desorption at −0.8 or −1.2 V) and short conditioning step in between (1 s at −2.5 V, 2-5 s at 0.8 V) to avoid instability effects due to oxidation processes. A standard addition procedure was achieved in less than 5 min. Figure 9 displays the concentration of Sb(III) obtained in 10 mM/1 mM solutions of varying pH. Rapid loss of the Sb(III) signal prevented analysis at pH < 2.5. Highest concentrations were found in 1 mM HCl (lowest ionic strength were not attempted as the Sb(III) signal became wider) or in 1 mM acetate buffer adjusted to pH 3.4 with HClO4 (Table 2). Concentrations were also determined in 1 mM HCl + 1 mM NaCl to avoid problems related to migration current that could occur at low ionic strength (Table 2). In 1 mM HCl, Sb(III) concentrations in MA1 were reproducible and found at 26.3 ± 1.54 mg mL−1 (N = 5; RSD = 5.8 %) at different days and using different electrodes. Importantly, measurements performed with various samples of Glucantime, although differing widely in validity date and lot number, showed very similar values of Sb(III) (Table 2). In addition, Sb(III) concentrations were greater at higher dilution factor. For instance, Sb(III) were 19.34 ± 0.80 and 25.07 ± 1.07 mg mL−1 at F = 106 and 107, respectively. Concentrations detected at pH > 3.5 (Fig. 9) decreased significantly showing that an increase in pH strongly modifies the chemical distribution of Sb(III) with formation of electro-inactive stable complexes.
Fig. 9 
Sb(III) concentrations determined from standard addition experiments as a function of pH in background electrolyte of 10 mM (triangle) or 1 mM (circle) buffer/acid concentrations
Table 2 Concentrations of Sb(III) detected in Glucantime (in milligram per milliliter) -
Sb total determination
Total Sb was determined only in MA1 and was found at 83.5 ± 3.5 mg mL−1, in general agreement with concentrations reported in other studies (Table 2).

Discussion
This study demonstrates that the determination of Sb(III) in Glucantime is prone to severe problems of instability in acidic conditions, most probably due to oxidation processes. Although it had not been explicitly shown before, this instability is expected from literature results. Table 3 gives an exhaustive list of analytical methods and associated conditions that were developed for the speciation of Sb in Glucantime ampoules. The reported Sb(III) concentrations varies over approximately 3–4 orders of magnitude. As a general trend, the concentration of Sb(III) are inversely related to acidity. For instance, [Sb(III)] was found at 1.6 10−3 mM in 1.5 M HCl while reported up to 84 mM in phosphate buffer, pH 6.8 [14]. Renedo et al. [22] developed an analytical method for the specific determination of Sb(III) in acidic conditions (1.5 M HCl) but, surprisingly, no values was given for Sb(III) in Glucantime. Only one exception is to be noticed: Franco et al. [6] reported a high Sb(III) concentration in 2 M HCl. However, they might have overlooked the potential reduction of Sb(V) in strong acidic conditions such as 2 M HCl, as recently shown by HPLC-ICP-MS ([11]). In this latter work, authors reported determination of Sb(III) in 0.5 M HCl. They noticed a rapid loss of signal if the solution were oxygenated in contrast to purged solution which were more stable [11]. However, even in purged solution, the determination had to be achieved rapidly to avoid loss of signal [49]. Flores et al. [12] reported instability of the Sb(III) signal with time after addition of citric acid (pH 1.86). They observed a 50 % decrease of the absorbance signal after only 2 min and analyzed Sb(III) using the shortest contact time of 5 s between the Glucantime solution and the citric acid. They also noticed that the loss of signal was only obtained after addition of the Glucantime solution, but not after addition of the Sb(III) standard. This is in agreement with our observations: the signal of Sb(III) originally present in MA is unstable in acidic conditions, but not the signal of added Sb(III) (see Fig. 6a—0.1 M HCl). If the signal loss is due to oxidation, one should assume that the oxidants are not present in excess.
Our observations of the pH effect on Sb(III) determination is consistent with previous reports [10] suggesting that Sb is complexed through hydroxyl groups that are easily cleaved in acidic conditions while stable to hydrolysis in alkaline solutions (called acetal compounds). Although no study has focused on the characterization of Sb(III) complex in meglumine antimoniate solutions, it is likely that Sb(III) complexation occurs through hydroxyl groups in a similar way as Sb(V). According to our data, Sb(III) complexes may be in part non-reducible within the potential range tested here, especially in non-acidic conditions. Upon acidification, Sb(III) would first be released due to protonation of hydroxyl groups and then would be lost, probably through oxidation. Using HPLC-ICP-MS, it was however suggested that only an acid concentration of 5 M HCl was sufficient to completely release Sb(III) [11].
At pH of 4.5 and 6, Sb(III) is present in Glucantime under various forms: complexes that adsorb onto the gold surface at positive potential, complexes that get reduced at lower potentials (Fig. 7a and 8) and possibly other more stable Sb(III) complexes that cannot be reduced or whose reduction is “hidden” by Sb(V) reduction (Fig. 7a). At pH 3, some free Sb(III) or reactive Sb(III) complexes can also be present (Fig. 8b). When Sb(III) is added during the standard addition procedure, it remains as a “free” specie (Fig. 8b) suggesting either that the ligand is saturated (and anything added remains as free) or that the association rate constant is very low (longer than the time scale of the experiment). Quantification of Sb(III) is thus problematic since: (1) not all species have similar diffusion coefficient; (2) it is possible that not all Sb(III) species are electro-active. Concentrations determined here by standard additions are thus only strictly correct if (1) all Sb(III) species have the same diffusion coefficient as the added Sb(III) and (2) if all Sb(III) species are electro-active at the deposition potential used here. If one of these two conditions is not fulfilled, the concentrations displayed in Fig. 9 and Table 2 are underestimated.
In any case, our results suggest that concentrations of Sb(III) in Glucantime are higher than previously reported. Concentrations determined in 1 mM HCl are approaching 30 mg/mL−1, i.e., more than 30 % of total Sb. The pharmacological relevance of the present results should be considered in light of the prodrug model for the mode of action of Glucantime against Leishmania parasite [2]. According to this model, the antileishmanial activity of pentavalent antimonials depends on the release of Sb(III) as the final active form of the metal. It is usually proposed that Sb(III) is produced through in vivo reduction of Sb(V). The high level of Sb(III) in Glucantime, as reported in the present study, leads us to propose an alternative model, in which antimony complexes may act as molecular carrier of Sb(III) and release it specifically in the acidic intracellular compartment where Leishmania parasite resides [46]. Even though the toxicity of pentavalent antimonials is attributed also to Sb(III) [2, 5], the pH dependence of Sb(III) release from MA solutions as supported by the present data, may account for the relative specificity of MA drugs against the parasite.
To the best of our knowledge, the electroactivity of Sb(V) at neutral pH by anodic stripping voltammetry had never been reported before. It can only be assumed that the unique ability of the vibrated wire electrode to withstand low deposition potentials without surface blockage is of prime importance in achieving Sb(V) detection in neutral conditions. Although it was not used in this work, it is likely that the detection of Sb(V) in such conditions is interesting: no need for strong acidic (corroding) conditions nor for chemical reducing agents. Finally, the low detection limits achieved at this sensor allowed high dilution factor to be used (up to 108), simultaneously reducing the influence of interfering compounds that may adsorb on the electrode.
Conclusion
The commonly used procedure to develop an analytical method in standard solutions and applying it “as it is” in a different matrix is not suitable for MA solutions. This work shows that: (1) acidic conditions promote the oxidation of Sb(III), explaining the large variations in Sb(III) concentrations previously reported in Glucantime; (2) Sb(III) levels are much higher than previously reported; (3) Sb(III) is present in MA under various forms which are pH dependent (higher pH promotes higher complex stability); (4) reliable determination of Sb(III) concentrations is hampered because of the different chemical forms between Sb(III) present in Glucantime and Sb(III) added. Sb(III) concentrations can only be estimated assuming that: (a) complexes have a similar diffusion coefficient as added Sb(III); (b) strong Sb(III) complexes are not present (i.e., all Sb(III) species are reduced at the chosen deposition potential). The oxidation process responsible for the loss of Sb(III) in acidic conditions is unclear and so is the effect of ionic strength. To clarify these different points, access to a synthetic Sb(III)–NMG complexes solution, as recently reported [11], would certainly help. The determination of Sb(III) in Glucantime is certainly a challenging task not yet solved.
References
WHO report (2010) Neglected tropical diseases: working to overcome the global impact of neglected tropical diseases,. WHO Press. http://www.who.int/neglected_diseases/2010report/en/
Frezard F, Demicheli C, Ribeiro RR (2009) Pentavalent Antimonials: New Perspectives for Old Drugs. Molecules 14(7):2317–2336. doi:10.3390/molecules14072317
Marsden PD (1985) Pentavalent antimonials: Old drug for new diseases. Rev Soc Bras Med Trop 18:187–198
Brochu C, Wang JY, Roy G, Messier N, Wang XY, Saravia NG, Ouellette M (2003) Antimony uptake systems in the protozoan parasite Leishmania and accumulation differences in antimony-resistant parasites. Antimicrob Agents Chemother 47(10):3073–3079. doi:10.1128/aac.47.10.3073-3079.2003
Dzamitika SA, Falcao CAB, de Oliveira FB, Marbeuf C, Garnier-Suillerot A, Demicheli C, Rossi-Bergmann B, Frezard F (2006) Role of residual Sb(III) in meglumine antimoniate cytotoxicity and MRP1-mediated resistance. Chemico-Biol Interactions 160(3):217–224. doi:10.1016/j.cbi.2006.01.008
Franco MA, Barbosa AC, Rath S, Dorea JG (1995) ANTIMONY OXIDATION-STATES IN ANTILEISHMANIAL DRUGS. AmJTrop Med Hyg 52(5):435–437
Lukaszczyk L, Zyrnicki W (2010) Speciation analysis of Sb(III) and Sb(V) in antileishmaniotic drug using Dowex 1 x 4 resin from hydrochloric acid solution. J Pharm Biomed Anal 52(5):747–751. doi:10.1016/j.jpba.2010.02.009
Roberts WL, McMurray WJ, Rainey PM (1998) Characterization of the antimonial antileishmanial agent meglumine antimoniate (Glucantime). Antimicrob Agents Chemother 42(5):1076–1082
Frezard F, Martins PS, Barbosa MCM, Pimenta AMC, Ferreira WA, de Melo JE, Mangrum JB, Demicheli C (2008) New insights into the chemical structure and composition of the pentavalent antimonial drugs, meglumine antimoniate and sodium stibogluconate. J Inorg Biochem 102(4):656–665. doi:10.1016/j.jinorgbio.2007.10.010
Carrio J, de Colmenares M, Riera C, Gallego M, Arboix M, Portus M (2000) Leishmania infantum: Stage-specific activity of pentavalent antimony related with the assay conditions. Exp Parasitol 95(3):209–214. doi:10.1006/expr.2000.4537
Seby F, Gleyse C, Grosso O, Plau B, Donard OFX (In press) Speciation of antimony in injectable drugs used for leishmaniasis treatment (Glucantime®) by HPLC-ICP-MS and DPP. Analytical and Bioanalytical Chemistry. doi:10.1007/s00216-012-6427-3
Flores EMD, dos Santos EP, Barin JS, Zanella R, Dressler VL, Bittencourt CF (2002) Determination of antimony(III) and total antimony by hydride generation atomic absorption spectrometry in samples of injectable drugs used for leishmaniasis treatment. J Anal At Spectrom 17(8):819–823. doi:10.1039/b203166c
Flores EMD, Paula FR, da Silva FEB, de Moraes DP, Paniz JNG, dos Santos EP, Dressler VL, Bittencourt CF (2003) Selective determination of Sb(III) in drugs by flow injection hydride generation AAS. At Spectrosc 24(1):15–21
Rath S, Jardim WF, Dorea JG (1997) A simple spectrophotometric procedure for the determination of antimony (III) and (V) in antileishmanial drugs. Fresen J Anal Chem 358(4):548–550. doi:10.1007/s002160050466
Quentel F, Filella M (2002) Determination of inorganic antimony species in seawater by differential pulse anodic stripping voltammetry: stability of the trivalent state. Anal Chim Acta 452(2):237–244
Postupolski A, Golimowski J (1991) Trace Determination of Antimony and Bismuth in Snow and Water Samples by Stripping Voltammetry. Electroanalysis 3(8):793–797
Waller PA, Pickering WF (1995) Determination of Antimony(III) and Antimony(V) by Differential-Pulse Anodic-Stripping Voltammetry. Talanta 42(2):197–204
Gomez Gonzalez MJ, Dominguez-Renedo O, Arcos-Martinez MJ (2007) Speciation of antimony by adsorptive stripping voltammetry using pyrogallol. Talanta 71(2):691–698. doi:10.1016/j.talanta.2006.05.025
Gomez Gonzalez MJ, Dominguez-Renedo O, Arcos-Martinez MJ (2006) Speciation of antimony by adsorptive stripping voltammetry using pyrogallol red. Electroanalysis 18(12):1159–1166. doi:10.1002/elan.200603515
Gedaminskiene V, Krapukaityte A, Armalis S (2005) AdSV determination of Sb(III) at a hanging mercury drop electrode applying a gallocyanine ligand. Chem Anal 50(6):973–980
Adeloju SB, Young TM, Jagner D, Batley GE (1998) Anodic stripping potentiometric determination of antimony on a combined electrode. Analyst 123(9):1871–1874
Dominguez-Renedo O, Gomez Gonzalez MJ, Arcos-Martinez MJ (2009) Determination of Antimony (III) in Real Samples by Anodic Stripping Voltammetry Using a Mercury Film Screen-Printed Electrode. Sensors 9(1):219–231. doi:10.3390/s90100219
Tanguy V, Waeles M, Vandenhecke J, Riso RD (2010) Determination of ultra-trace Sb(III) in seawater by stripping chronopotentiometry (SCP) with a mercury film electrode in the presence of copper. Talanta 81(1–2):614–620. doi:10.1016/j.talanta.2009.12.050
Santos JR, Lima J, Quinaz MB, Rodriguez JA, Barrado E (2007) Construction and evaluation of a gold tubular electrode for flow analysis: Application to speciation of antimony in water samples. Electroanalysis 19(6):723–730
Salaün P, Gibbon-Walsh K, van den Berg CMG (2011) Beyond the Hydrogen Wave: New Frontier in the Detection of Trace Elements by Stripping Voltammetry. Anal Chem 83(10):3848–3856. doi:10.1021/ac200314q
Wang E, Sun W, Yang Y (1984) Potentiometric Stripping Analysis with a Thin-Film Gold Electrode for Determination of Copper, Bismuth, Antimony, and Lead. Anal Chem 56(11):1903–1906
Huang HL, Jagner D, Renman L (1987) Flow Constant-Current Stripping Analysis for Antimony(III) and Antimony(V) with Gold Fiber Working Electrodes - Application to Natural-Waters. Anal Chim Acta 202:123–129
Tanaka T, Ishiyama T, Okamoto K (2000) Determination of antimony in steel by differential pulse anodic stripping voltammetry at a rotating gold film electrode. Anal Sci 16(1):19–23
Dominguez-Renedo O, Arcos-Martinez MJ (2007) Anodic stripping voltammetry of antimony using gold nanoparticle-modified carbon screen-printed electrodes. Anal Chim Acta 589(2):255–260. doi:10.1016/j.aca.2007.02.069
Dominguez-Renedo O, Arcos-Martinez MJ (2007) A novel method for the anodic stripping voltammetry determination of Sb(III) using silver nanoparticle-modified screen-printed electrodes. Electrochem Commun 9(4):820–826. doi:10.1016/j.elecom.2006.11.016
Brihaye C, Gillain G, Duyckaerts G (1983) Determination of Traces of Metals by Anodic-Stripping Voltammetry at a Rotating Glassy-Carbon Ring-Disc Electrode .3. Evaluation of Linear Anodic-Stripping Voltammetry with Ring Collection for the Determination of Cadmium, Lead and Copper in Pure Water and High-Purity Sodium-Chloride, and of Cadmium, Lead, Copper, Antimony and Bismuth in Sea-Waterco. Anal Chim Acta 148:51–57, APR
Gillain G, Brihaye C (1985) A ROUTINE SPECIATION METHOD FOR A POLLUTION SURVEY OF COASTAL SEA-WATER. Oceanol Acta 8(2):231–235
Salaün P, Gibbon-Walsh K, Alves GMS, Soares HMVM, Van den Berg CMG (2012) Determination of arsenic and antimony in seawater by voltammetric and chronopotentiometric stripping using a vibrated gold microwire electrode. Anal Chim Acta 746:53–62. doi:10.1016/j.aca.2012.08.013
Santos VS, Santos WDR, Kubota LT, Tarley CRT (2009) Speciation of Sb(III) and Sb(V) in meglumine antimoniate pharmaceutical formulations by PSA using carbon nanotube electrode. J Pharm Biomed Anal 50(2):151–157. doi:10.1016/j.jpba.2009.04.008
Croot PL, Moffett JW, Luther GW (1999) Polarographic determination of half-wave potentials for copper-organic complexes in seawater. Mar Chem 67(3–4):219–232
Gibbon-Walsh K, Salaün P, van den Berg CMG (2012) Pseudopolarography of copper complexes in seawater using a vibrating gold microwire electrode. J Phys Chem A 116(25):6609–6620
Pizeta I, Billon G, Omanovic D, Cuculic V, Garnier C, Fischer JC (2005) Pseudopolarography of lead (II) in sediment and in interstitial water measured with a solid microelectrode. Anal Chim Acta 551(1–2):65–72
Tsang JJ, Rozan TF, Hsu-Kim H, Mullaugh KM, Luther GW (2006) Pseudopolarographic determination of Cd2+ complexation in freshwater. Environ Sci Technol 40(17):5388–5394. doi:10.1021/es0525509
Salaün P, Planer-Friedrich B, van den Berg CMG (2007) Inorganic arsenic speciation in water and seawater by anodic stripping voltammetry with a gold microelectrode. Anal Chim Acta 585(2):312–322. doi:10.1016/j.aca.2006.12.048
Salaün P, van den Berg CMG (2006) Voltammetric detection of mercury and copper in seawater using a gold microwire electrode. Anal Chem 78(14):5052–5060
Salaün P, Gibbon-Walsh KB, Alves GMS, Soares HMVM, van den Berg CMG (2012) Determination of arsenic and antimony in seawater by voltammetric and chronopotentiometric stripping using a vibrated gold microwire electrode. Anal Chim Acta 746:53–62. doi:10.1016/j.aca.2012.08.013
Gibbon-Walsh K, Salaün P, van den Berg CMG (2010) Arsenic speciation in natural waters by cathodic stripping voltammetry. Anal Chim Acta 662(1):1–8. doi:10.1016/j.aca.2009.12.038
Toghill KE, Lu M, Compton RG (2011) Electroanalytical Determination of Antimony. Int J Electrochem Sci 6(8):3057–3076
Zong P, Nagaosa Y (2009) Determination of antimony(III) and (V) in natural water by cathodic stripping voltammetry with in-situ plated bismuth film electrode. Microchimica Acta 166(1–2):139–144. doi:10.1007/s00604-009-0176-9
Lingane JJ, Nishida F (1947) POLAROGRAPHIC CHARACTERISTICS OF CHLORO COMPLEXES OF +5 ANTIMONY. J Am Chem Soc 69(3):530–533
Feldmann J, Salaün P, Lombi E (2009) Critical review perspective: elemental speciation analysis methods in environmental chemistry - moving towards methodological integration. Environ Chem 6(4):275–289. doi:10.1071/en09018
Omanovic D, Branica M (2004) Pseudopolarography of trace metals. Part II. The comparison of the reversible, quasireversible and irreversible electrode reactions. J Electroanal Chem 565(1):37–48. doi:10.1016/j.jelechem.2003.09.026
Coale KH, Bruland KW (1988) COPPER COMPLEXATION IN THE NORTHEAST PACIFIC. Limnol Oceanogr 33(5):1084–1101
Seby F (personnal communication)
Gomez Gonzalez MJ, Dominguez-Renedo O, Arcos-Martinez MJ (2005) Simultaneous determination of antimony(III) and antimony(V) by UV–vis spectroscopy and partial least squares method (PLS). Talanta 68(1):67–71. doi:10.1016/j.talanta.2005.04.059
Acknowledgments
P.S. benefited from an EPSRC fellowship (EP/E061303) and acknowledges the Life Science Interface (LSI) program for financial support. F.F. is a recipient of a research fellowship from the Brazilian agency CNPq.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Salaün, P., Frézard, F. Unexpectedly high levels of antimony (III) in the pentavalent antimonial drug Glucantime: insights from a new voltammetric approach. Anal Bioanal Chem 405, 5201–5214 (2013). https://doi.org/10.1007/s00216-013-6947-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-6947-5