
Abstract
Methoxetamine is a new ketamine derivative designer drug which has recently become available via the Internet marketed as “legal ketamine”. It is a new dissociative recreational drug, acting as an NMDA receptor antagonist and dopamine reuptake inhibitor. The objective of this study was to develop on-line automated sample preparation using a TurboFlow device coupled with liquid chromatography with ion-trap mass spectrometric detection for measurement of methoxetamine in human plasma. Samples (100 μL) were vortex mixed with internal standard solution (ketamine-d4 in acetonitrile). After centrifugation, 20 μL of the supernatant was injected on to a 50 mm × 0.5-mm C18XL Turboflow column. The retained analytes were then back-flushed on to a 50 mm × 3-mm (3 μm) Hypersil Gold analytical column for chromatographic separation, then eluted with a formate buffer–acetonitrile gradient. Methoxetamine and the IS were ionized by electrospray in positive mode. Parent [M + H]+ ions were m/z 248.1 for methoxetamine and m/z 242.0 for the IS. The most intense product ions from methoxetamine (m/z 203.0) and the IS (m/z 224.0) were used for quantification. The assay was accurate (96.8–108.8 % range) and precise (intra and inter-day coefficients of variation <8.8 %) over the range of 2.0 (lower limit of quantification) to 1000.0 ng mL−1 (upper limit of quantification). No matrix effect was observed. This method has been successfully applied to determination of plasma concentrations of methoxetamine in the first French hospitalization case report after acute intoxication; the plasma concentration was 136 ng mL−1.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The effect of the Internet on the emergence of new synthetic drugs seems to be increasingly problematic for the health-monitoring systems [1]. New synthetic drugs, known as “designer drugs”, “legal highs”, or “research chemicals” (RCs) are sometimes used in pharmacology research or appear directly on the Internet where they are sold at low cost [2]. There have been few studies on methoxetamine (MXE), which has recently become available via the Internet and sold to consumers as a “legal ketamine” in the form of a white powder.
Psychoactive products users across Europe and, especially, in the UK [3–5] seem to have increased interest in the drug which first appeared in 2010 [6]. Most of the time, consumers use either oral or insufflation routes of administration [1] but intramuscular, intravenous, and rectal administration have also been described [7]. The chemical structure of MXE is very close to that of ketamine: it is the 3-methoxy, N-ethyl analogue of ketamine. It acts both as an N-methyl-d-aspartate receptor antagonist and a dopamine reuptake inhibitor [1, 6]. It is more lipophilic than ketamine, there is a delay before the beginning of its effects, and its half-life is much longer than that of ketamine. Ketamine and analogues, for example MXE, are used as recreational drugs for their dissociative and psychedelic effects [7].
The clinical effects of methoxetamine are poorly known, and most studies describe symptoms from Web forum discussions in which consumers describe their experience. Amongst the symptoms, confusion, analgesia, numbness, anxiety, and sympathomimetic clinical features [8], for example tachycardia, hypertension, and mydriasis are the most common. They seem to be compatible with ketamine-induced adverse effects [1, 2]. One recent study with analytical confirmation [9] described reversible cerebellar toxicity that manifests as a lack of coordination after consumption of MXE.
As designer drugs appear increasingly faster on the Web, they are a true challenge for toxicologists, who must be aware of new chemicals and of ways of identifying them. Considering the lack of pharmacologic studies, some authors try to collect data from different sources, for example Web consumers, to understand their acute toxicity [10]. In 2012, methoxetamine was included in a chemical overview of “legal highs” of interest to the toxicologist [11]. Recent reports describe, mostly, clinical cases [2, 8, 9] of acute toxicity of methoxetamine with, sometimes, analytical confirmation [8, 9]. However, none of these studies concern identification and the quantification of the compound in human samples.
In these studies, MXE has been identified—but not quantified—in serum samples by use of a liquid chromatography–tandem mass spectrometry (LC–MS–MS) toxicological screening method [2] or by gas chromatography–mass spectrometry (GC–MS) after purchase of the drug from an Internet site [7]. Serum methoxetamine concentrations have recently been determined in the same laboratory by use of GC–MS [8, 9]. However, none of these studies include the description and validation of an efficient method for identification and quantification of the drug in human and powder samples.
The objective of this study was to develop and validate an automated method with on-line extraction using turbulent-flow coupled with liquid chromatography with mass spectrometric detection, suitable for determination of concentrations of methoxetamine in human plasma for analytical confirmation in cases of drug consumption. This method has been applied to plasma methoxetamine determination in one case report that is, as far as we are aware, the first French case report of hospitalization after methoxetamine acute intoxication.
Experimental
Chemicals and reagents
Methoxetamine hydrochloride (2-(3-methoxyphenyl)-2-(ethylamino)cyclohexanone, C15H21NO2; monoisotopic mass 247.3; Fig. 1) and the internal standard (IS), ketamine-d4 hydrochloride (C13H13 C12D4NO; monoisotopic mass: 278.2; Fig. 2) were purchased from LGC Standards (Molsheim, France). HPLC-grade acetonitrile, ammonium formate, and formic acid were supplied by Sigma–Aldrich (Paris, France). HPLC-grade methanol was from Prolabo (Paris, France). Ultra-pure water was obtained by reverse osmosis using a Direct-Q UV3 apparatus (Millipore, Molsheim, France). All other chemicals were of analytical grade.

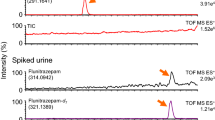
Chromatograms obtained from drug-free plasma spiked with methoxetamine at a final concentration of 5.0 ng mL−1. From top to bottom: chromatograms of the parent ion m/z = 242.0 and its product ion m/z = 224.0 of ketamine-d4, and of parent ion m/z = 248.1 and its product ion m/z = 203.0 of methoxetamine

Chromatograms obtained from the plasma from the case report subject. Methoxetamine concentration was 136.0 ng mL−1. From top to bottom: chromatograms of the parent ion m/z = 242.0 and its product ion m/z = 224.0 of ketamine-d4, and of parent ion m/z = 248.1 and its product ion m/z = 203.0 of methoxetamine
Working solutions, calibration standard, and quality controls
Stock solutions of methoxetamine (1 g L−1) and the IS (100 mg L−1) were prepared in methanol. Working solutions of methoxetamine for use as calibration standards (CS) were prepared at three concentrations (1.0, 0.1, and 0.01 mg L−1) by dilution of the stock solution with methanol. Another 1 g L−1 stock solution was prepared for quality control (QC) at the same concentrations by dilution in methanol. A working solution of the IS (0.5 mg L−1) was obtained by dilution of the stock solution with acetonitrile.
Calibration curves were prepared by spiking blank plasma with appropriate volumes of the previously mentioned working solutions to produce CS containing 2.0, 5.0, 10.0, 20.0, 50.0, 100.0, 200.0, 500.0, and 1000.0 ng mL−1. QC samples were also prepared in blank plasma at concentrations of 3.0, 30.0, 300.0, and 750.0 ng mL−1. Blank human plasma samples collected on acid citric dextrose (ACD) were obtained from a local blood bank (EFS, Versailles, France). CS and QC solutions were stored at −20 °C.
Sample preparation
One-hundred microlitres of the IS working solution (0.5 mg L−1) was added to each of 1.5-mL Eppendorf tubes containing 100 μL plasma. After vortex mixing and centrifugation (14,000 × g, 10 min), 50 μL supernatant was transferred into injection vials for analysis. This simple sample preparation, before to injection on to the TurboFlow column enables preservation of loading columns and extends column life [12].
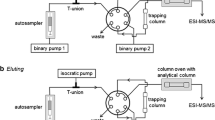
TurboFlow LC–MS–MS system and conditions
The TurboFlow device consists of a Cohesive Aria TLX-1 System (version 1.1.1, ThermoFisher Scientific) equipped with a multiple column module, a quaternary loading pump, a binary eluting pump, and a CTC PAL 2.2.0 autosampler. For loading pumps, system eluents used were (B) 5 mmol L−1 ammonium formate and (C) acetonitrile. For the eluting pump, system eluents used were (A) 2 mmol L−1 ammonium formate–formic acid buffer, pH 3.8 and (C) acetonitrile. The entire experiment was controlled by Aria operating software 1.1.1. Data were processed with ThermoFisher Scientific LCQuan 2.5.6 SP1 quantitative software using ThermoFisher Scientific Xcalibur software 2.0.7 SP1 data system software.
In the first step, 20 μL supernatant from sample preparation was injected on to a 50 mm × 0.5 mm C18XL TurboFlow column under turbulent flow (100 % B, 2.0 mL min−1, 30 s). In the second step, retained analytes were flushed from the TurboFlow column using elution solvent (A–C, 30:70 v/v) stored in a holding loop and focussed through a T-piece on to a Hypersil Gold C18 analytical column (3 μm particle size, 50 mm × 0.5 mm i.d.; ThermoFisher Scientific). In the third step, during gradient elution (A–C, 30:70 to 60:40 v/v) from the analytical column, the TurboFlow column was washed with eluent C (100 %). In the fourth step, the loop was refilled with the elution solvent (A–C, 30:70 v/v) while the chromatographic separation was proceeding on the analytical column. In the fifth step, the loading system was re-equilibrated with 100 % eluent B. Before re-equilibration of the analytical column (step 7), the composition of the mobile phase was inverted from that in the analytical column (step 6) to clean it from interferences before the next injection. The total analysis time was less than 10 min, including column re-equilibration. Eluent flow was diverted to waste for the first 2 min after each injection on to the TurboFlow column and MS–MS data were acquired from 2 min until 6.5 min. The gradient elution and valve-switching profile are summarized in Table 1.
Compounds were detected by use of an LCQ Deca XP Plus mass spectrometer (ThermoFisher Scientific) equipped with an electrospray ionization (ESI) source. Nitrogen (N2MID350 nitrogen generator; Parker Hannifin France, Contamine-sur-Avre, France) was used as sheath and auxiliary gas at a pressure of 30 and 5 arbitrary units, respectively. The ESI source was set in positive ionization mode, and an ion-spray potential of +4.0 kV was applied. The scan time was 0.03 ms-1. Capillary temperature was set to 250 °C under a potential of +15.0 V. The system was tuned by using a continuous 3 μL min−1 infusion of methoxetamine (1 mg L−1 in the mobile phase). Acquisition was performed in full-production ion (MS2) mode. Protonated molecular ions [M + H]+ of methoxetamine (m/z 248.1) and the IS (m/z 242.0) were trapped with a mass resolution of 1.0 atomic mass unit (amu) and fragmented with a collision energy of 25 % and 27 % respectively. Chromatographic data acquisition was performed using Xcalibur software (version 2.0.7, ThermoFisher Scientific). Post-analysis processing was carried out using LC Quan software (version 2.5.6, ThermoFisher Scientific).
Method validation procedure
The method was validated for selectivity, linearity, sensitivity, accuracy, precision, matrix effect, and recovery according to US Food and Drug Administration guidelines.
Selectivity, carry over
To investigate whether endogenous matrix constituents interfered with the assay, drug-free matrix blank samples, zero samples, and samples spiked at the LLOQ were analysed in accordance with the described procedure. Assay selectivity was defined by evidence of non-interference at retention times and ion channels identical with those for methoxetamine and the IS in the blank samples. A blank sample was also analysed immediately after the highest CS in each run to monitor carry over of methoxetamine and the IS. Serum and plasma methoxetamine concentrations were determined for fifteen patients randomly selected to evaluate the effect of blood collection tubes. Plasma was obtained with three anticoagulants: lithium heparin, ethylene diamine tetraacetate (EDTA), and sodium fluoride.
Linearity
Calibration curves included a blank sample, a zero sample, and nine CS over the concentration range 2 ng mL−1 (lower limit of quantification, LLOQ) to 1000 ng mL−1 (upper limit of quantification, ULOQ). Ten calibration curves were obtained over a period of twenty-one days for study of linearity. Quantification was achieved by plotting the peak area ratios of methoxetamine to the internal standard. Back-calculated concentrations of the CS had to be within 85–115 % of the nominal concentrations.
Lower limit of quantification
The LLOQ was defined as the lowest concentration for which accuracy was between 80 and 120 % and precision with a coefficient of variation (CV) of ±20 % or less was obtained over ten measurements.
Limit of detection
The LOD was defined as the lowest concentration for which the full MS–MS spectrum could be identified with a signal-to-noise ratio greater than 3.
Accuracy and precision
Accuracy (measured value/nominal value) and precision (coefficient of variation) were determined for the four QC levels. For the intraday assay, ten replicates of each QC level were processed on the same day. For the inter-day assay, each QC level was processed ten times three different days over a period of two weeks. The values obtained were analysed by analysis of variance (ANOVA), which separated the intra-day and inter-day standard deviation and consequently the corresponding coefficients of variation (CV). Accuracy within the range 85–115 % of the nominal values and precision with a CV of ±15 % were required, except for the LLOQ for which a range of 80–120 % and a CV of ±20 % were accepted for accuracy and precision, respectively.
Matrix effect and overall method recovery
To investigate analyte recovery, aqueous solutions of methoxetamine (5, 50, and 500 ng mL−1) were prepared with the IS. After the sample-preparation step, prepared solutions were:
-
1.
subjected to the complete TurboFlow procedure; and
-
2.
analysed with the TurboFlow system bypassed (i.e. injection directly on to the analytical column).
The mean peak areas for each analyte subjected to the complete TurboFlow procedure were compared with those obtained from injection on to the analytical column only, with the latter assumed to represent 100 % recovery. Overall method recovery of the IS had to be ±15 % of methoxetamine recovery.
Three other solutions at the same concentrations were prepared in pooled blank plasma. To evaluate the matrix effect, after the precipitation step, plasma and aqueous samples were both subjected to the complete TurboFlow procedure. The mean peak areas of each analyte from the pooled blank plasma solutions were compared with those observed with the aqueous solutions.
Case report
This analytical method was applied to a non-fatal case of intoxication with methoxetamine. As far as we are aware, this was the first case described in our country. The subject, a 24-year-old student with a history of depression and consumption of illegal drugs (ketamine, cocaine, and cannabis) was found by firemen on the pavement, in a town square, in the middle of the night. He was hospitalized in a general hospital and admitted to the intensive-care unit. Clinical examination revealed temporo-spatial disorientation associated with amnesia, nystagmus, and bilateral mydriasis. A stroke was first suspected, but brain scan was normal. As a small plastic bag containing white powder with a straw was found among his personal effects, intoxication was suspected. Urine and blood were then collected at the hospital admission time for toxicological laboratory tests.
Results and discussion
TurboFlow LC–MS–MS analysis
On-line extraction for toxicological analysis should be increasingly used, because this technology is very simple, fully automated, and less time-consuming than manual methods of sample preparation. Use of ion-trap detection is of interest when looking for new compounds, for example designer drugs, because their formulae are sometimes unknown and this technology saves the full spectrum with good sensitivity. Many TurboFlow applications have been described since the 1990s and can be applied in a routine toxicological laboratory [12]. Recently, two TurboFlow applications have been described for therapeutic drug monitoring of tyrosine kinase inhibitors [13] and azole antifungal drugs [14]. This kind of application should certainly increase in the future, because it furnishes rapid analytical results enabling adjustment of drug dosing.
Because stable-isotope-labelled methoxetamine was not available, ketamine-d4 was chosen because of its similar chemical structure. In previous analytically confirmed studies [8, 9], the authors used pyribenzamine as internal standard, which is less similar to methoxetamine than ketamine-d4, which nowadays seems the better candidate.
The mass spectra of methoxetamine and the IS contain the parent protonated [M + H]+ ions at m/z 248.1 for methoxetamine and at m/z 242.0 for the IS, and the MS–MS spectrum of methoxetamine shows that the most intense product ion is observed at m/z 203.0, which was used for quantification, and at m/z 224.0 for the IS.
Under the optimized conditions, the IS and methoxetamine were eluted with retention times of 5.2 and 5.5 min, respectively. The chromatogram obtained after extraction of drug-free plasma spiked with methoxetamine at a final concentration of 5.0 ng mL−1 is shown in Fig. 1.
Method validation
Selectivity and carry over
No interferences from the constituents of drug-free human serum or of plasma samples from hospital patients were observed at the retention times and ion channels of methoxetamine and the IS. When a blank sample was analysed immediately after the highest calibration standard, mean carry over was <0.2 % for methoxetamine and the IS (data not shown). No interference was caused by the different anticoagulants or by haemolysed samples.
Linearity and limit of quantification
Quantification was achieved by linear regression analysis, which was the best fitting model as determined by bias analysis. The nine-point calibration curve was highly linear in the concentration range 2–1000 ng mL−1 with 1/x weighting factor; correlation coefficients ranged from 0.9970 to 0.9998. Interday CV ranged from 1.4 to 15.5 % and bias ranged from 0.1 to 8.9 % for the back-calculated concentrations of the nine calibration standards. The LLOQ was 2 ng mL−1, the CV was 15.5 %, and accuracy was 100.9 % for a signal-to-noise ratio >10. The ULOQ was 1000 ng mL−1. Half and tenth dilutions of the QC samples were tested and back-calculated concentrations of diluted QC samples were accurate compared with the nominal concentrations.
Limit of detection
The LOD was 1.0 ng mL−1 with a signal-to-noise ratio greater than 3.
Precision and accuracy
Intraday and interday precision and accuracy for QC samples are summarized in Table 2. Intraday precision ranged from 4.0 to 7.5 % and accuracy ranged from 98.9 to 106.1 %. Interday precision ranged from 3.2 to 8.8 % and accuracy from 98.4 to 106.7 %. These data indicate that this enables accurate, precise, and reproducible quantification of methoxetamine throughout a wide dynamic range.
Matrix effect and overall method recovery
In plasma, matrix effect and overall recovery of the method were independent of methoxetamine concentration (Table 3). Matrix effect was in the range 3–10 % and overall TurboFlow recovery was in the range 86–91 %. Matrix effect for the IS was less than 4.0 % and overall recovery was in the range 89–92 %. These results are indicative of the absence of significant matrix effect. On-line extraction using the TurboFlow device preceded by the precipitation step usually reduces the matrix effect compared with acetonitrile precipitation alone, as previously described for a few studies [12].
Case report
Urine immunoassay was negative for cocaine but positive for cannabis. No blood or urine confirmation was carried out. Alcohol blood concentration was 1.2 g L−1 and acetaminophen was quantified in plasma at 1.0 mg L−1 (therapeutic range 20.0–30.0 mg L−1).
This validated TurboFlow LC–MS–MS method was successfully applied to our patient plasma sample and enabled quantification of methoxetamine at 136 ng mL−1. The chromatogram obtained after extraction of 100 μL plasma is shown in Fig. 2. The purity of MXE in the powder was 35 %, associated with 20 % acetaminophen. Wood et al. have described three analytically confirmed cases with serum concentrations in the range 90–200 ng mL−1 [8]. The serum concentrations were between 120 and 200 ng mL−1 after nasal insufflation of the drug and 90 ng mL−1 after dissolving approximately 200 mg of the powder in water and then drinking. Moreover, Shields et al. reported serum methoxetamine concentrations for three patients that ranged from 160 to 240 ng mL−1 after nasal insufflation of the drug [9]. Methoxetamine was quantified, in both studies, by use of single ion monitoring on a Shimadzu GC–MS system over a calibration range of 5 to 1000 ng mL−1 [8, 9]. Few data are available about blood concentrations of methoxetamine because reports of MXE poisoning have not been systematically analytically confirmed. Nevertheless, our result seems to be in accordance with the other two previous studies mentioned.
Reversible cerebellar toxicity has been described as one symptom that should alert clinicians to the possibility of methoxetamine exposure [9]. Although our patient was not suffering from cerebellar ataxia, observed signs and symptoms, for example confusion, mydriasis, and nystagmus were consistent with the adverse effects of ketamine derivatives and most studies of methoxetamine report this kind of symptom in their case reports [2. 8, 9].
Conclusion
Methoxetamine is just one among many other new chemicals that seems to be increasingly available on the Internet. It seems important that this kind of case, for example ours, is supported by a confirmatory toxicological analytical method. With the method developed and validated in this work, determination of blood concentrations of methoxetamine by use of an LC–MS–MS method preceded by an online extraction, is nowadays possible. The online extraction using the TurboFlow device enables very rapid detection and quantification of MXE in human plasma and can be applied every time MXE acute toxicity is suspected for rapid analytical confirmation.
References
Corazza O, Schifano F, Simonato P, Fergus S, Assi S, Stair J, Corkery J, Trincas G, Deluca P, Davey Z, Blaszko U, Demetrovics Z, Moskalewicz J, Enea A, di Melchiorre G, Mervo B, di Furia L, Farre M, Flesland L, Pasinetti M, Pezzolesi C, Pisarska A, Shapiro H, Siemann H, Skutle A, Sferrazza E, Torrens M, van der Kreeft P, Zummo D, Scherbaum N (2012) Phenomenon of new drugs on the Internet: the case of ketamine derivative methoxetamine. Hum Psychopharmacol 27(2):145–149
Hofer KE, Grager B, Muller DM, Rauber-Luthy C, Kupferschmidt H, Rentsch KM, Ceschi A (2012) Ketamine-like effects after recreational use of methoxetamine. Ann Emerg Med 60(1):97–99
Wood DM, Hunter L, Measham F, Dargan PI (2012) Limited use of novel psychoactive substances in South London nightclubs. QJM, Jun 19. [Epub ahead of print]
Schmidt MM, Sharma A, Schifano F, Feinmann C (2011) "Legal highs" on the net-Evaluation of UK-based Websites, products and product information. Forensic Sci Int 206(1–3):92–97
Westwell AD, Hutchings A, Caldicott DG (2012) The identification and chemical characterization of a new arylcyclohexylamine, methoxetamine, using a novel Emergency Department toxicosurveillance tool. Drug Test Anal [Epub ahead of print]
Rosenbaum CD, Carreiro SP, Babu KM (2012) Here today, gone tomorrow…and back again? A review of herbal marijuana alternatives (K2, Spice), synthetic cathinones (bath salts), kratom, Salvia divinorum, methoxetamine, and piperazines. J Med Toxicol 8(1):15–32
Ward J, Rhyee S, Plansky J, Boyer E (2011) Methoxetamine: a novel ketamine analog and growing health-care concern. Clin Toxicol (Phila) 49(9):874–875
Wood DM, Davies S, Puchnarewicz M, Johnston A, Dargan PI (2012) Acute toxicity associated with the recreational use of the ketamine derivative methoxetamine. Eur J Clin Pharmacol 68(5):853–856
Shields JE, Dargan PI, Wood DM, Puchnarewicz M, Davies S, Waring WS (2012) Methoxetamine associated reversible cerebellar toxicity: three cases with analytical confirmation. Clin Toxicol (Phila) 50(5):438–440
Wood DM, Dargan PI (2012) Novel Psychoactive Substances: How to Understand the Acute Toxicity Associated With the Use of These Substances. Ther Drug Monit
Gibbons S (2012) 'Legal highs'—novel and emerging psychoactive drugs: a chemical overview for the toxicologist. Clin Toxicol (Phila) 50(1):15–24
Couchman L (2012) Turbulent flow chromatography in bioanalysis: a review. Biomed Chromatogr 26(8):892–905
Couchman L, Birch M, Ireland R, Corrigan A, Wickramasinghe S, Josephs D, Spicer J, Flanagan RJ (2012) An automated method for the measurement of a range of tyrosine kinase inhibitors in human plasma or serum using turbulent flow liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 403(6):1685–1695
Couchman L, Buckner SL, Morgan PE, Ceesay MM, Pagliuca A, Flanagan RJ (2012) An automated method for the simultaneous measurement of azole antifungal drugs in human plasma or serum using turbulent flow liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 404(2):513–523
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abe, E., Ricard, F., Darrouzain, F. et al. An automated method for measurement of methoxetamine in human plasma by use of turbulent flow on-line extraction coupled with liquid chromatography and mass spectrometric detection. Anal Bioanal Chem 405, 239–245 (2013). https://doi.org/10.1007/s00216-012-6470-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-6470-0