
Abstract
We are utilizing recent advances in ultrafast laser technology and recent discoveries in optimal shaping of laser pulses to significantly enhance the stand-off detection of explosives via control of molecular processes at the quantum level. Optimal dynamic detection of explosives is a method whereby the selectivity and sensitivity of any of a number of nonlinear spectroscopic methods are enhanced using optimal shaping of ultrafast laser pulses. We have recently investigated the Gerchberg–Saxton algorithm as a method to very quickly estimate the optimal spectral phase for a given analyte from its spontaneous Raman spectrum and the ultrafast laser pulse spectrum. Results for obtaining selective coherent anti-Stokes Raman spectra (CARS) for an analyte in a mixture, while suppressing the CARS signals from the other mixture components, are compared for the Gerchberg–Saxton method versus previously obtained results from closed-loop machine-learning optimization using evolutionary strategies.

Photo of an acousto-optic modulator based pulse shaper, with red lines denoting the laser beam path. Entering at the bottom left is the transform limited pulse (spectrogram inset shows wavelength versus time in a false color plot), and exiting at the bottom right is the the arbitrarily shaped pulse
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Explosives are the key component for most violent threats in the world today. They are used in improvised explosive devices, suicide bombs, and package bombs, and are also used as the means for dispersal of other weapons of mass effect, such as chemical, biological, or radiological weapons. A necessary step towards neutralizing such threats is the ability to detect and identify the explosives. Ideally, the detection would be as far “left of boom” as possible, i.e., at the bomb maker or materials supplier. For reasons of personnel safety, it is also highly desirable to detect and identify the explosives from large stand-off distances. There have been a number of reviews of the technology being developed to provide this capability [1, 2]. Progress to date for the development of stand-off detection technologies has been slow because: (1) only trace levels of observable material are available due to the low vapor pressures of many explosives and/or the small quantities of surface adsorbed explosive residues, (2) excitation of the sample must occur at a distance with subsequent return of the resultant signals back to the detector, (3) operation must occur in the presence of atmospheric turbulence and particulate matter, (4) there are stringent demands for high sensitivity and high signal-to-noise and (5) high selectivity is needed to detect a given target material in the presence of possibly very similar but benign background chemicals. Nevertheless, there are a large number of analytical methods that have been used to try to solve these problems, although there is to date still no complete solution [1, 2]. In particular, the number of these methods that can be used at stand-off distances is small, with most based on laser excitation such as stand-off Raman spectroscopy, stand-off coherent anti-Stokes Raman spectroscopy (CARS), and laser-induced breakdown spectroscopy (LIBS). Several groups are pushing the limits of the standard implementations of these stand-off spectroscopies via incremental improvements [3]. We are investigating a more revolutionary approach using the framework of optimal dynamic or quantum control. A wide variety of nonlinear spectroscopic methods are amenable to optimization using optimal dynamic detection of explosives (ODD-Ex) tools, as discussed in depth below.
ODD-Ex methodology
ODD-Ex operates by adapting the interrogating laser to the quantum molecular dynamics of the target analyte (explosive) [4, 5]. For a given nonlinear laser/molecule interaction leading to a signal, such as emission or ionization, there exists a laser pulse (i.e., a time-dependent optical field) that will optimize that signal [6]. Such an optimal laser pulse is not typically what is emitted by a commercial laser system, but rather the laser pulse must be adapted to suit the specific molecule and the spectroscopic signature of interest. There is a strong fundamental theoretical foundation underlying the existence of the optimal control field, as well as the features of the control search landscape, which suggests that the optimal solution can be readily discovered [6, 7]. Because the laser must control the behavior of the target molecules on their natural vibrational, rotational, and electronic timescales, a short (e.g., femtosecond) laser is needed. In the future, the ultimate solution would be a laser system capable of emitting on demand any of the hundreds to thousands of different types of pulses needed to interrogate and detect the hundreds to thousands of desired target explosive molecules. Such a commercial laser system is not available today. However, there exists widely available technology to externally shape raw femtosecond laser pulses into the desired complex time-dependent optical fields. This technology is now sufficiently mature to attack applied problems such as explosive detection.
The technology consists of a programmable phase and amplitude mask located at the Fourier plane of a zero dispersion stretcher [8, 9], as shown schematically in Fig. 1. A zero dispersion stretcher is in essence a subtractive double spectrometer; the programmable mask is simply placed at the central focal plane. As the various colors contained in the raw laser pulse are spatially separated at the Fourier plane, each spectral component passes through separate pixels of the mask, either a spatial light modulator (SLM) or an acousto-optic modulator (AOM). The modulator adjusts the phase and amplitude of each spectral component under computer control. When the pulse is spectrally reconstructed through the second spectrometer, the addition of spectral phase, and/or modulation of amplitude, causes the initially short, simple pulse to be more complicated in the time and frequency domains. This process provides a versatile, fast, computer-controlled, time-dependent optical field that can be shaped to control the interaction of the field with molecules, allowing manipulation of the molecular dynamics via the time-dependent molecular Hamiltonian.

Schematic representation of a pulse shaper, consisting of a stretcher (left hand side), a modulator, either SLM or AOM at the Fourier plane, and a compressor (right hand side) that reassembles the spectral components. M mirror; CM cylindrical mirror; G grating
This ability to shape femtosecond optical pulses is combined in ODD-Ex with advanced machine-learning algorithms to quickly determine the pulse shape that optimizes a particular desired molecular outcome. For detection, this outcome is a unique signature for a specific molecule. The concept of using an experimental feedback loop to determine optimal pulse shapes was first demonstrated by Judson and Rabitz [10] and has become the common practice in a wide range of applications [11]. For the application to the detection of explosives, first the optimal pulse’s shape for a given nonlinear spectroscopy and a given molecule is determined and placed into a library. Then, for detection use in the field, this library of optimal pulse shapes would be scanned at up to kilohertz rates to rapidly identify potential threat materials. The optimal pulse shapes are tested for robustness to interferences, backgrounds, and matrix materials, as well as the effects of turbulence and atmospheric conditions. It is the ability of ODD-Ex to produce a signal from a desired target analyte while suppressing signals from interfering materials [12] that sets it apart from other laser-spectroscopic methods. Another advantage is the capacity to exploit time-dependent dynamics for discrimination between very similar molecules [13].
ODD-CARS
We are concentrating at present on applying the ODD-Ex paradigm to CARS; we abbreviate this implementation as ODD-CARS. We have chosen CARS because it is a nonlinear form of Raman spectroscopy, one of the workhorse methods for stand-off detection of explosives. ODD-Ex methodology can be seen then to supplement an already existing stand-off spectroscopic method to make it even better. CARS provides for highly directional signal beams of high intensity, and the signals are anti-Stokes to the probe laser, both of which dramatically reduce the fluorescence interference that usually plagues ordinary Raman methods. Other authors have shown that femtosecond pulse shaping CARS implementations show significant promise for stand-off detection of explosives [14, 15]. Previously, we showed that ODD-Ex provides a framework to find pulse shapes that optimize CARS signals for a given analyte in a mixture, while suppressing signals from interfering materials by orders of magnitude [12]. This ability provides the capacity for achieving significant improvements in selectivity. We discuss these previous results in this paper alongside the potential time savings afforded by use of the Gerchberg–Saxton algorithm to estimate the optimal phase, versus the machine-learning algorithm optimization process.
A significant time is usually needed to determine the optimal pulse shape for ODD-CARS of a given analyte molecule using standard machine-learning ODD methodology, even incorporating the latest evolutionary strategy algorithms [16]. The experiment to determine the optimal shape that preserved the nitromethane CARS peaks while suppressing CARS features from the other materials in a mixture of nitromethane, toluene, and acetone, as shown in Fig. 2, necessitated nearly 12 h using a genetic algorithm (GA). While this approach is sufficient, we have been exploring the ability to more rapidly obtain the pulse shape optimized for a given analyte CARS spectrum using the Gerchberg–Saxton algorithm to produce a starting spectral phase.

The CARS spectrum of a mixture containing nitromethane and interfering species is given by the red dashed line. Shaping the pulse as shown in the right panel leads to the CARS spectrum given by the blue line. The nitromethane peaks marked by arrows remain, while the largest interfering peaks are significantly reduced. This is due to selective excitation of the nitromethane (from Ref. [12]; used with permission)
Gerchberg–Saxton algorithm
We use the Gerchberg–Saxton (GS) pulse shaping methodology described by Rundquist et al. [17], which is based on the well-known Gerchberg–Saxton phase retrieval algorithm [18]. The methodology requires knowing the target spectrum and the laser spectrum. For the implementation described here, we assumed that the two-photon excitation spectrum contains equivalent vibrational spectroscopic information as the CARS spectrum [19, 20], and that the analyte Raman spectrum can be used as the target in the GS methodology. The distinct advantages of the GS methodology are its independence of target complexity, its lack of cost functions and weight factors, and no parameters to optimize.
The GS methodology starts by taking the fast Fourier transform (FFT) of the experimentally measured pulse (ultrafast laser) spectrum, assuming random phase, into the time domain. The laser spectrum is simply measured using an integrating sphere and a spectrometer, and we assume that all spectral components are contained within the transform-limited pulse. The resulting amplitude is then replaced by the amplitude of the target (the target is set to be the FFT of the spontaneous Raman spectrum of the analyte), while retaining the phase. An inverse FFT (IFFT) is then performed to transform back to the spectral domain, where the amplitude is replaced with the input pulse (laser) spectrum and the phase is again retained without change. The process is then iterated many times until there are negligible residual changes in the retained phase. On the last iteration, the retained phase is then taken to be the desired phase mask optimized for the analyte molecule. The process is denoted schematically in Fig. 3. The main convergence was noted to occur in the first few iterations, with the remaining iterations producing minor incremental improvements of the result.

Flowchart representation of the Gerchberg–Saxton algorithm implementation
GS simulation
We first demonstrated the GS methodology for ODD-CARS using computer simulations. The simulations incorporated experimental Raman spectra, obtained using an Ahura First Defender portable Raman system (Thermo Scientific). The raw Raman spectra were background subtracted using a polynomial fit, as it was observed that non-zero regions between peaks resulted in incomplete suppression of non-analyte peaks in the GS simulated CARS spectrum. For the simulations, the Raman spectrum of neat nitromethane (Fig. 4 left) was used as the GS target of the desired analyte. The GS algorithm was tasked with extracting the nitromethane CARS spectrum out of the CARS spectrum of a 2:2:1 (volume) mixture of toluene, acetone, and nitromethane (Fig. 4 right). With the nitromethane Raman spectrum as the GS target spectrum in the GS algorithm, and the filamentation broadened ultrafast laser spectrum (Fig. 5 left top) as the GS laser, the final spectral phase from the GS process is shown in Fig. 5 left bottom. The simulated two-photon excitation spectrum of the 2:2:1 toluene/acetone/nitromethane mixture (the two-photon excitation spectrum contains the essential information needed for multiple vibrational excitation selectivity [19, 20]) using the nitromethane-specific GS spectral phase is shown in Fig. 5 right (red), which is overlaid on the nitromethane Raman spectrum (black). Note the nearly complete elimination of toluene and acetone spectral features, leaving only nitromethane peaks. Also note that the two-photon excitation relative spectral intensities are different from the expected nitromethane CARS peak intensities.

Left spontaneous Raman spectrum of nitromethane (background subtracted). Right spontaneous Raman spectrum of a mixture of 2:2:1 (volume) of toluene/acetone/nitromethane (background subtracted)

Left, top ultrafast laser spectrum after filamentation broadening. Left, bottom Gerchberg–Saxton spectral phase using the nitromethane Raman as the target. Right the red curve is the two-photon excitation spectrum of the 2:2:1 mixture of toluene/acetone/nitromethane in a computer simulation using the Gerchberg–Saxton spectral phase for nitromethane (shown bottom left in this figure). The black curve is the nitromethane Raman spectrum
Experiment
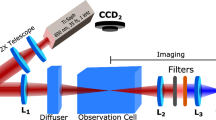
The experiments utilized 1 kHz repetition rate, 1 mJ energy, 25 fs pulses from a Coherent Legend Elite Ti/sapphire based ultrafast laser system, with central wavelength near 800 nm. These pulses were spectrally broadened via self-phase modulation in a filament produced by weakly focusing into a 1-m-long pipe filled with ∼40 kPa of argon gas [21, 22]. The broadened spectrum was passed through an all reflective pulse shaper, consisting of a 600 lines/mm grating, 500 mm focal length cylindrical mirror, turning mirror, 640 pixel dual mask spatial light modulator (CRi 640-D-VN SLM) and retroreflecting mirror which slightly displaces the output beam in the vertical direction to allow spatial separation of the incoming and outgoing beams. We utilized the multiphoton intrapulse interference phase scan (MIIPS) process [23] to produce close to transform-limited pulses after the filamentation in argon. The MIIPS phase was saved and added to the GS algorithm produced phase in all subsequent experiments. Finally, this pulse was passed through a long-wave pass filter (Omega Optical 740ALP) with 740 nm cut on edge to remove most of the interfering light in the anti-Stokes region.
Pulse shapes were measured using transient-grating frequency-resolved optical gating (TG-FROG) [24]. Both the TG-FROG and the CARS experiments were performed in folded box geometry [25]. For CARS experiments, a 2.5-nm full width at half maximum (FWHM) interference filter centered at 772.5 nm (Thin Film Imaging Technologies 785–2.5 narrowband filter tilted at ∼15° from normal incidence) was placed in the probe beam, stretching it temporally to ∼800 fs. The energies of the pump and Stokes pulses at the 1- or 5-mm path length liquid sample cells were attenuated to ∼2 μJ to avoid self-phase modulation in the sample. The probe energy of 0.5 μJ was limited by the interference filter. The beams had parallel linear polarization. The four wave mixing signal was isolated spatially using an iris and focused into a 0.3 m spectrometer (Princeton Instruments SP-320 with 600 l/mm grating) with ∼1 nm spectral resolution (∼100 μm slit width). The CARS signals were subsequently detected using a CCD camera (Princeton Instruments PIXIS 100) in single shot mode. Control, MIIPS, FROG, GS algorithm, and signal accumulation software was written in LabView (National Instruments). Final spectral analysis and figures were produced using Igor Pro software (Wavemetrics).
The ultrafast laser spectrum was measured using an Ocean Optics USB-2000 fiber coupled spectrometer, with mode homogenization obtained using an integrating sphere (Thorlabs IS200).
All chemicals were used as received: toluene—Acros spectrophotometric grade 99+%; acetone—Acros 99.6% for spectroscopy ACS reagent; nitromethane—Acros 99+% for analysis.
Results and discussion
The GS algorithm was used to derive a shaped pulse specific for nitromethane, as discussed above. The resulting spectral phase shown in Fig. 5 left was used to produce an experimental CARS spectrum in a 2:2:1 (by volume) mixture of toluene, acetone, and nitromethane. A CARS spectrum of that mixture using a transform-limited pulse (spectrally broadened and MIIPS compressed) was also obtained. The two spectra are overlaid in Fig. 6. This figure can be compared to the computer simulated spectra shown in Fig. 5 right, as well as to the GA optimized CARS spectrum in Fig. 2.

Experimental CARS spectra of a 2:2:1 mixture of toluene/acetone/nitromethane obtained using the filamentation broadened and MIIPS corrected transform-limited ultrafast laser pulse (black curve), and using the Gerchberg–Saxton spectral phase obtained using the nitromethane Raman spectrum as the GS target (red curve)
Two obvious differences can be seen. First, the peak CARS signal using the GS optimized spectral phase is significantly lower than using the transform-limited pulse (the spectra were normalized in Fig. 5 so that the largest nitromethane feature was the same height for both the GS and transform-limited spectra). In our earlier experiments using the GA optimization on two nitromethane peaks [12], the feedback included maximizing the intensity of those two peaks in a given ratio, while suppressing all other peaks (see Fig. 2). The GS algorithm has no such intensity criterion. Secondly, the toluene and acetone CARS peaks are not suppressed as much relative to the nitromethane peaks as in the GS computer simulation (compare Figs. 5 and 6). The theory of control landscapes states that the optimal solution for controllable systems is reachable given unlimited laser resources [6]. We are operating in a regime of restricted laser resources even using the filament-broadened laser, which we believe is the main cause of the observed incomplete suppression of interfering peaks.
Conclusions
We have shown that optimal dynamic detection methods can be used to produce selectivity in CARS spectra of an analyte in a mixture. Previously, we used evolutionary strategies to produce a spectral phase optimized for a desired target analyte molecule, e.g., nitromethane in a liquid mixture. That process, however, takes considerable time. We studied here the possibility of using the Gerchberg–Saxton algorithm to produce the selective spectral phase from the known analyte Raman spectrum and the ultrafast laser pulse spectrum. Computer simulations of the Gerchberg–Saxton methodology showed nearly complete suppression of interfering CARS features, while retaining the CARS spectrum of a desired analyte in a mixture (e.g., nitromethane in a mixture with toluene and acetone). The experimental demonstration of the Gerchberg–Saxton methodology also showed suppression of the interfering CARS features in the mixture similar in magnitude to that observed previously using a full experimental optimization via evolutionary strategies. However, the intensity of the CARS spectrum of the analyte was not retained. Further work is concentrating on using the GS algorithm to provide a starting phase selective for a given analyte, which could then be optimized using gradient searches to maximize the CARS intensity.
References
Moore DS (2004) Instrumentation for trace detection of high explosives. Rev Sci Instr 75:2499–2512
Moore DS (2007) Recent advances in trace explosives detection instrumentation. Sens Imaging 8:9–38
Moore DS, Goodpaster JV (2009) Explosives analysis. Anal Bioanal Chem 395:245–246
Moore DS, Rabitz H, McGrane SD, Greenfield M, Scharff RJ, Beltrani V, Roslund J (2009) Optimal dynamic detection of explosives. SPIE Proceed 7304:730413
Moore DS (2009) Optimal coherent control of sensitivity and selectivity in spectrochemical analysis. Anal Bioanal Chem 393:51–56
Chakrabarti R, Rabitz H (2007) Quantum control landscapes. Int Rev Phys Chem 26:671–735
Rabitz H (2004) Controlling quantum phenomena: why does it appear easy to achieve. J Modern Optics 51:2469–2475
Weiner AM (2000) Femtosecond pulse shaping using spatial light modulators. Rev Sci Instr 71:1929–1960
Dugan MA, Tull JX, Warren WS (1997) High-resolution acousto-optic shaping of unamplified and amplified femtosecond laser pulses. J Opt Soc Am B 14:2348–2358
Judson RS, Rabitz H (1992) Teaching lasers to control molecules. Phys Rev Lett 68:1500–1503
Nuernberger P, Vogt G, Brixner T, Gerber G (2007) Femtosecond quantum control of molecular dynamics in the condensed phase. Phys Chem Chem Phys 9:2470–2497
McGrane SD, Scharff RJ, Greenfield M, Moore DS (2009) Coherent control of multiple vibrational excitations for optimal detection. New J Physics 11:105047
Roth M, Guyon L, Roslund J, Boutou V, Courvoisier F, Wolf JP, Rabitz H (2009) Quantum control of tightly competitive product channels. Phys Rev Lett 102:253001
Katz O, Natan A, Silberberg Y, Rosenwaks S (2008) Standoff detection of trace amounts of solids by nonlinear Raman spectroscopy using shaped femtosecond pulses. Appl Phys Lett 92:171116
Li H, Harris A, Xu BW, Wrzesinski PJ, Lozovoy VV, Dantus M (2008) Coherent mode-selective Raman excitation towards standoff detection. Optics Express 16:5499–5504
Roslund J, Shir OM, Bäck T, Rabitz H (2009) Accelerated optimization and automated discovery with covariance matrix adaptation for experimental quantum control. Phys Rev A 80:043415
Rundquist A, Efimov A, Reitze DH (2002) Pulse shaping with the Gerchberg–Saxton algorithm. J Opt Soc Am B 19:2468–2478
Gerchberg RW, Saxton WO (1972) A practical algorithm for the determination of the phase from image and diffraction plane pictures. Optik 35:237
Urbanek DC, Berg MA (2007) Simultaneous time and frequency detection in femtosecond coherent Raman spectroscopy. I. Theory and model calculations. J Chem Phys 127:044306
Dhar L, Rogers JA, Nelson KA (1994) Time-resolved vibrational spectroscopy in the impulsive limit. Chem Rev 94:157–193
Gallmann L, Pfeifer T, Nagel PM, Abel MJ, Neumark DM, Leone SR (2007) Comparison of the filamentation and the hollow-core fiber characteristics for pulse compression into the few-cycle regime. Appl Phys B 86:561–566
Hauri D, Guandalini A, Eckle P, Kornelis W, Biegert J, Keller U (2005) Generation of intense few-cycle laser pulses through filamentation–parameter dependence. Optics Express 13:7541–7547
Lozovoy VV, Pastirk I, Dantus M (2004) Multiphoton intrapulse inteference 4. Characterization and compensation of the spectral phase of ultrashort laser pulses. Optics Lett 29:775
Trebino R, DeLong KW, Fittinghoff DN, Sweetser JN, Krumbügel MA, Richman BA, Kane DJ (1997) Measuring ultrashort laser pulses in the time–frequency domain using frequency-resolved optical gating. Rev Sci Instr 68:3277
Shirley JA, Hall RJ, Eckbreth AC (1980) Folded BOXCARS for rotational Raman studies. Optics Lett 5:380–382
Acknowledgments
The authors gratefully acknowledge the support of the US Department of Homeland Security. Los Alamos National Laboratory is an affirmative action equal opportunity employer operated by Los Alamos National Security, LLC for the National Nuclear Security Administration of the US Department of Energy under contract DE-AC52-06NA25396.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the 10th Anniversary Issue.
Rights and permissions
About this article
Cite this article
Moore, D.S., McGrane, S.D., Greenfield, M.T. et al. Use of the Gerchberg–Saxton algorithm in optimal coherent anti-Stokes Raman spectroscopy. Anal Bioanal Chem 402, 423–428 (2012). https://doi.org/10.1007/s00216-011-5348-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-5348-x