
Abstract
A freeze-dried mussel tissue certified reference material (CRM-FDMT1) containing multiple groups of shellfish toxins has been prepared. Toxin groups present in the material include okadaic acid and the dinophysistoxins, azaspiracids, yessotoxins, pectenotoxins, spirolides and domoic acid. In this work, analytical methods have been examined for the characterisation of the candidate CRM. A comprehensive extraction procedure was developed, which gave good recovery (>98%) for all lipophilic toxins studied. A fast liquid chromatography–mass spectrometry (LC-MS) method was developed that separates the major toxins according to the MS ionisation mode of optimum sensitivity. Matrix effects associated with analysis of these extracts using the developed LC-MS method were assessed. Standard addition and matrix-matched calibration procedures were evaluated to compensate for matrix effects. The methods and approaches will be used for the precise characterisation of the homogeneity and stability of the various toxins in CRM-FDMT1 and for the accurate assignment of certified values. The developed methods also have excellent potential for application in routine regulatory monitoring of shellfish toxins.

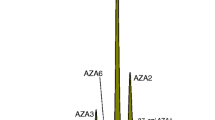
LC-MS/MS chromatogram of CRM-FDMT1: a mussel tissue CRM containing multiple groups of phycotoxins
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Laboratories involved in shellfish toxin regulation are moving away from bioassay procedures towards the use of chemical analytical methods. In the European Union liquid chromatography–mass spectrometry (LC-MS) has recently replaced the mouse bioassay (MBA) as the reference method for most classes of lipophilic shellfish toxins [1]. Reference materials (RMs) are essential for validation of alternative methods to the MBA. A project was undertaken to prepare and characterise a multi-toxin certified reference material (CRM) in response to the requirements of analysts and laboratories. The preparation of this freeze-dried mussel tissue (Mytilus edulis) material (CRM-FDMT1) containing six of the major groups of shellfish toxins was detailed in the previous article [2], which also described characterisation of the CRM’s physical properties. The next phase of work in this project, described in this paper, was the development of analytical methods for the characterisation of the lipophilic toxins present in the material for homogeneity, stability, and certification purposes. This includes extraction and instrumental methods for the precise and accurate determination of the various toxins present. This presented a significant challenge as CRM-FDMT1 contains many toxins from several toxin groups, including okadaic acid (OA) and the dinophysistoxins (DTXs), azaspiracids (AZAs), yessotoxins (YTXs), pectenotoxins (PTXs), spirolides (SPXs) and domoic acid (DA).
LC-MS is rapidly becoming the technique of choice for the simultaneous determination of the multiple groups of lipophilic shellfish toxins [3–7]. The method developed by Quilliam et al. [3], which uses an acidic mobile phase separation on a C8 column has been adapted by a number of researchers and is used widely for multi-toxin detection and monitoring programs. However, there are two limitations of this method: (a) the low pH mobile phase is not well suited for YTXs and (b) there is an overlap of retention times for toxins that are best analysed in opposing ionisation modes (i.e. OA, DTXs and YTXs in negative; AZAs and PTXs in positive). The latter issue has been mitigated through the use of newer generation mass spectrometers that are capable of rapid polarity switching [5]. While this method can provide good sensitivity and a rapid analysis time, not all MS instruments are capable of rapid polarity switching and quantitative performance may be decreased even on instruments with this capability. Improved chromatography for YTXs has been reported with the use of a neutral pH mobile phase based on ammonium acetate [6]. Poor peak shapes for YTX at acidic pH is due to an interaction between the sulphate anion of YTX and the protonated silanol groups of the stationary phase at the low pH. More recently, an alkaline pH mobile phase based on ammonium hydroxide has been used to improve the performance of YTX and the separation of toxins [7].
The aim of the current work was to develop an LC-MS method that: (a) provides separation of toxins into groups for optimal ionisation efficiency, without the need for an MS capable of rapid polarity switching; (b) has good chromatography for all the major toxins present in the CRM and (c) has a fast run time without the high back pressures associated with ultra performance chromatography and microparticulate columns. Thus, a variety of LC columns were tested with mobile phases of varying pH values to adjust the selectivity of separation.
A procedure for extraction of toxins from CRM-FDMT1 prior to LC-MS analysis should be effective for the various toxins groups. However, the varying chemical properties of the different toxins mean that this task is not necessarily straightforward. Although there have been a variety of procedures reported in the literature, most of these have been directed at specific toxin groups. Methanol (MeOH) has been shown to be more effective than acetone for extraction of the highly lipophilic AZAs from mussel tissues, although exhaustive recovery testing was not done in that work [8]. For the OA group of toxins, an exhaustive 3-step extraction procedure was applied in the certification of a CRM for OA and DTX1 in which 100% MeOH was used in the first step, followed by 80% MeOH for the second two steps [9]. A separate study on the extraction of OA group toxins from a variety of shellfish showed that use of 90% MeOH gave a higher yield than 80% MeOH in a single step procedure [10]. Unfortunately, no recovery information was supplied. Feasibility work on the production of freeze-dried RMs for shellfish toxins revealed that a 2-step procedure using 100% MeOH gave >95% recovery of AZA and OA group toxins [11]. Another study found that 90% MeOH was more effective than 80% MeOH for a wide variety of lipophilic toxins, though it was noted that repeated extraction of the sample was required for high recovery [4]. These points and the requirement for accuracy and precision in the characterisation of a CRM are considered in this study during selection of extraction methods for CRM-FDMT1.
Matrix effects are a major issue for quantitation in LC-MS analysis, and a number of studies have demonstrated the problems that arise in shellfish toxin determinations [4, 6, 12, 13]. Matrix effects result in the alteration of the instrumental signal due to suppression or enhancement of analyte ionisation in the MS ion source. Possible reasons for the effects in electrospray ionisation (ESI) have been discussed [14]. Matrix effects present a significant problem for characterising and assigning accurate concentration values to CRM-FDMT1. Various techniques have been applied to deal with the problem in the shellfish toxin field. Dilution of the tissue extracts has been shown to reduce levels of suppression [4] but this approach generally requires the use of a more sensitive MS system. Solid phase extraction (SPE) and liquid–liquid partitioning clean-up methods have been reported for shellfish samples [15, 16], but it is difficult to apply such techniques to the wide range of toxins present in CRM-FDMT1. Procedures using matrix-matched calibration (MMC) [17] and standard addition [12] have proven to be effective. Each of these techniques have limitations either in their ability to adequately recover and detect all toxins, compensate entirely for the matrix effect problem, or to be cost-effective in terms of labour and use of expensive materials and equipment.
This paper presents a multi-toxin extraction and LC-MS method for the characterisation of CRM-FDMT1. Work was focused only on the lipophilic toxins present in the CRM, as DA has very different properties and suitable methods are already available for this analyte. An assessment of matrix effects in the analysis of CRM-FDMT1 extracts is described and approaches taken to deal with the problem are discussed.
Materials and methods
Standards, chemicals and tissue materials
MeOH and acetonitrile (MeCN) were purchased from Caledon (Georgetown, ON, Canada). Ammonium acetate (NH4Ac), formic acid, ammonium formate and ammonium hydroxide were purchased from BDH laboratory reagents (Toronto, ON, Canada). Shellfish toxin calibrants (CRMs and non-certified RMs) were obtained from the NRC’s Certified Reference Material Program (CRMP; Halifax, NS, Canada), as were two new mussel tissue reference materials, CRM-FDMT1 and CRM-Zero-Mus. PTX1 standard was supplied by Prof. T. Suzuki, and PTX2sa was provided by Dr. Chris Miles. Distilled water was further purified using a Milli-Q purification system (Millipore Corp., Billerica, MA, USA).
Blue mussels (M. edulis) contaminated with OA, DTX2 and AZAs, provided by the Irish Marine Institute, were harvested from Bruckless on the Northwest Coast of Ireland.
Calibration and spiking stock solutions
A mixed standard solution containing OA, DTX1, DTX2, YTX, 45-OH-YTX, PTX2, PTX2sa, PTX11, AZA1, AZA2 and AZA3 was prepared gravimetrically in MeOH using commercially available CRMs and non-certified RMs from CRMP. This was used for selectivity testing with various LC conditions. A separate stock solution containing OA, YTX, AZA1 and PTX2 was used for preparing a dilution series for instrument calibration. Concentrations ranged from 92–130 ng/mL. A set of 13-desMe-SPX-C calibration solutions was prepared in MeOH with 0.1% formic acid (0.2 to 10 ng/mL). Finally, a stock spiking solution for standard addition containing OA, YTX, PTX2 and AZA1 was prepared gravimetrically in MeOH at levels of 150, 231, 84 and 407 ng/mL, respectively. This was used for standard addition and matrix match suitability testing.
LC-MS/MS analysis
Analyses were performed using an Agilent 1200 LC (Palo Alta, CA, USA) coupled to an AB-Sciex (Concord, ON, Canada) QTRAP 4000 MS equipped with a turbospray ionisation source. A binary mobile phase was used, with A = water and B = MeCN/H2O (95:5), each containing 5 mM NH4Ac (pH 6.8). A Phenomenex Synergi MaxRP C12 column (50 × 2 mm i.d., 3 μm) maintained at 20°C was eluted at 300 μL/min with a linear gradient of 25–100% B over 5 min, followed by a hold at 100% B for 3 min, a return to 25% B over 0.1 min and an equilibration at 500 μL/min for 2 min until the next run. The first 2.5 min of column effluent were diverted to waste, while an auxiliary flow of MeCN–H2O (50:50 v/v) was directed to the source. An injection volume of 5 µL was used for standards and sample extracts. The MaxRP column should be washed following the manufacturers instruction upon completion of sample sequences. Selective reaction monitoring (SRM) transitions were acquired in negative and/or positive ionisation modes (Table 1). Initial column testing was carried out also using acidic (2 mM ammonium formate, 50 mM formic acid) and basic (6.7 mM ammonium hydroxide) mobile phases.
Extraction of CRM-FDMT1 and samples for LC-MS analysis
CRM-FDMT1 was reconstituted prior to extraction by weighing 0.35 g portions of powder from thoroughly mixed bottles into 50 mL centrifuge tubes and adding 1.65 g portions of deionised water. The tubes were capped, vortexed for 30 s to combine the powder and the water and then sonicated for 60 s. This corresponds to a reconstituted water content of approximately 83.2% based on analyses of the CRM-FDMT1 powder [2].
Reconstituted samples (n = 3) were extracted using two variations of a three-step liquid–solid extraction (LSE) procedure. The first procedure used 100% MeOH for all three steps of the extraction, while the second procedure used 80%, 90% and 100% MeOH in steps 1, 2 and 3, respectively. For extraction, 7 mL of solvent was added to the reconstituted sample and vortex mixed for 3 min using a multiple tube vortex mixer (Model DVX-2500, VMR Int., PA, USA). The extract was centrifuged at 3,950×g for 10 min (Sorvall Legend RT+, Thermo Scientific, Osterode, Germany), and the resulting supernatant was decanted into a 25-mL volumetric flask. A fresh portion of solvent was added to the pellet and re-extracted for 3 min at 10,000 rpm using an Omni-prep multiple sample homogeniser (Omni Int., Kennesaw, GA, USA). The Omni-prep probes were cleaned by operating in centrifuge tubes containing 7 mL of fresh solvent. The wash solvent was retained for use in the third step of the respective extractions. After centrifugation and combining the extract from the second step with that of the first step, the solvent retained from washing the Omni-prep probe was added to the pellet and the sample was extracted a third time by vortexing as described for the first step. The supernatant was combined with that from the first two steps in the 25-mL flask and made up to volume with 100% MeOH. To test toxin recoveries from CRM-FDMT1, the remaining sample pellets were extracted with an additional 4.5 mL of 100% MeOH using the Omni-prep homogeniser, and after centrifugation, the supernatants were made up to 5 mL with MeOH. Aliquots of the various extracts and recovery testing samples were passed through 0.45 μm regenerated cellulose filters (Millipore Corp, Billerica, MA, USA) prior to analysis by LC-MS or further work. Triplicate samples were prepared for each method.
A rapid analysis approach was investigated for high throughput monitoring using the LC-MS method under higher flow rate conditions. To facilitate this, a simple and quick extraction was performed by adding 8 mL of MeOH to 2 g of shellfish tissue homogenate in a 50-mL centrifuge tube and homogenising using the Omni-prep as above. After centrifugation, the supernatant was decanted into a 10-mL flask and made up to volume with MeOH. An aliquot was filtered for analysis as above.
Matrix effect evaluation and compensation
For evaluation of matrix effects, serial two- and fourfold dilutions of a 100% MeOH extract of CRM-FDMT1 were prepared in triplicate using a Hamilton Microlab diluter (Hamilton Company, Reno, NV, USA) using dilution with pure MeOH. For standard addition experiments, extracts of CRM-FDMT1 and CRM-Zero-Mus (prepared using the three-step exhaustive extraction procedure with 100% MeOH) were spiked with varying volumes of the “stock spiking solution” as follows. A Hamilton Microlab diluter was first used to dispense 440 μL aliquots of the extracts in HPLC vials. Increasing volumes of the stock spiking solution were then added to the extract aliquots, and the final volumes of all samples were made to 550 μL with MeOH. This ensured a consistent matrix concentration at each spike level. An equivalent set of spikes was prepared using pure MeOH in place of sample extract. The concentration ranges in the spike solutions are shown in Table 3.
Results and discussion
LC-MS/MS method selection
Reversed phase LC separation with an acidic mobile phase is commonly used for the analysis of lipophilic shellfish toxins [3–5]. However, toxin groups such as OA/DTXs, YTXs and PTXs, which are preferably analysed in opposing ionisation modes, often co-elute. Grouping of toxins according to the ionisation mode of optimum sensitivity was a major goal in development of the LC-MS method for CRM-FDMT1 characterisation. A variety of LC columns packed with particle sizes of 3 μm or less were tested by running a short gradient from 25–100% organic, followed by a hold at 100% and then column re-equilibration. The separation selectivity was adjusted by varying the mobile phase pH: acidic (2 mM ammonium formate, 5 mM formic acid; pH 2.3) [3], neutral (5 mM NH4Ac; pH 6.8) [6] and, for columns that had higher pH working ranges, a basic mobile phase was tested (6.7 mM ammonium hydroxide; pH 11) [7]. Some retention data observed on four different columns is provided in the Electronic Supplementary Material. Figure 1 shows the influence of pH on retention times of various lipophilic toxins on a Gemini-NX C18 column. The recently reported multi-toxin method using a basic pH mobile phase achieved an efficient grouping of toxins and presents a promising alternative to the MBA for testing of shellfish samples because of its applicability for the entire range of regulated lipophilic shellfish toxins [7]. However, in testing this method for application to CRM-FDMT1 using a Gemini-NX C18 column, broader peaks with significant fronting were observed for AZAs when compared to analysis using acidic or neutral pH mobile phases (data not shown). While Gerssen et al [7] used a different LC column in their initial study, they also reported broader peaks and reduced resolution for AZAs. For this reason and because of the restricted choice of columns for basic mobile phase operations, this approach was not favoured for work on CRM-FDMT1 as AZAs are major toxin group present in the material. Stobo et al. previously reported the neutral pH for toxin analysis [6] but the column and mobile phase combination used in that instance did not achieve a complete separation of toxins according to ionisation mode as desired for CRM-FDMT1 characterisation. Following thorough testing, the LC-MS method selected for CRM-FDMT1 was based on the use of a neutral pH mobile phase (pH 6.8) in combination with a MaxRP C12 stationary phase.

Effect of mobile phase pH on lipophilic toxin separation affected by mobile phase pH. Gradient elution from 25% to 100% MeCN at 250 μL/min on a Gemini-NX C18 (3 μm, 50 × 2.1 mm) using acidic (pH 2.3), neutral (pH 6.8) and basic (pH 11) mobile phases
Figure 2 shows a chromatogram from the LC-MS analysis of a mixture of lipophilic toxin standards using the selected method. In a relatively fast analysis (7 min acquisition time and 10.5 min total run time including equilibration), the OA and YTX group toxins eluted separately from PTX2 and the AZA group thus allowing their determinations in a single run in negative and positive ionisation modes, respectively. Under these neutral pH conditions, the negatively ionised OA and YTX groups elute earlier than the PTX and AZA toxins, which are better detected in the positive ion mode. There was good resolution between OA and DTX2, which are monitored in the same SRM transition (R s = 2.25). Peak widths and asymmetries were acceptable for AZAs, while excellent peak shapes were observed for the YTXs and PTX2, thereby making the method suitable for use in characterisation of CRM-FDMT1.

Analysis of a standard mixture of lipophilic toxins by LC-MS using the MaxRP C12 column (2.5 μm, 50 × 2 mm) with neutral mobile phase (5 mM NH4Ac, pH 6.8). Isomeric toxins are indicated with an asterisk (*)
Only limited testing of various PTX analogues was performed due to their scarcity. It was determined, however, that PTX1, a toxin that is regulated, eluted near the beginning of positive ionisation mode region (data not shown). For application of this method to the monitoring of all regulated toxins, tight control of pH will be necessary to avoid PTX1 slipping into the negative ionisation region of toxins. As a safety precaution in the event of shifting retention times, an SRM transition for PTX1 can also be included in the negative mode region (873.5 > 137.1, DP −80 V, CE −75 eV).
SPXs, which are also present in CRM-FDMT1 [2], are still ionised at neutral pH, which results in some SPX analogues eluting in the negative ionisation window. However, for work on CRM-FDMT1, SPXs are preferably analysed in a separate run for two reasons: (a) there are a large number of spirolide analogues and these are more easily quantified in a second analysis and (b) improved stability has been reported for SPXs in acidic solution [18], so it is preferable to immediately take a portion of a fresh sample extract and acidify it. It is not suitable to acidify the entire sample extract because PTXs are unstable under such conditions (unpublished data).
CRM-FDMT1 reconstitution and extraction
Previous work on freeze-dried mussel tissue RMs for shellfish toxins has shown improved recovery and precision for lipophilic and hydrophilic toxins when the powder is reconstituted prior to extraction [11]. The effect of allowing the sample to sit at room temperature for a period of time following reconstitution was investigated as part of this study; however, extraction after 2 h did not result in further improvement of recoveries (data not shown).
Extraction of lipophilic toxins from shellfish matrices is carried out in most laboratories using an LSE approach in which solvent is added to a portion of the homogenised shellfish sample and extraction is performed by mixing or blending. The extract is typically centrifuged and filtered prior to analysis, but may undergo an additional clean-up (e.g. liquid–liquid partitioning). Frequently, the extraction procedure is carried out as a single step, which is generally sufficient for routine shellfish monitoring. However, such single step procedures do not provide complete recovery. The highest possible recovery of analyte is essential for CRM work in order to ensure accuracy of results. Therefore, repeated extraction of a sample with fresh portions of solvent is necessary to maximise recovery. The additional labour and time required for a procedure involving multiple extraction steps is acceptable. Two variations of an exhaustive and volumetrically controlled three-step extraction procedure were investigated for CRM-FDMT1. As 100% MeOH is commonly used as a generic lipophilic toxin extraction solvent [4, 11], and because recoveries of lipophilic toxins are typically higher with higher percentages of MeOH, the first LSE method tested used 100% MeOH as solvent for all three steps. Since lower percentages of MeOH have also been reported for extraction of some lipophilic toxins such as OA and YTX [4, 10, 19], a combination of 80%, 90% and 100% MeOH was also tested as the extraction solvent in the three respective steps of the LSE method.
The results of the extraction study are shown in Table 2. Triplicate samples were extracted for both methods tested, obtaining good precision for all analytes in the combined extracts (relative standard deviations all <5%). Recoveries were measured by doing a fourth extraction of the remaining sample pellet to determine residual toxin levels following the three step extraction methods.
With the exception of YTX recoveries were >99% for all analytes in both LSE procedures. The recovery was very good for YTX using 100% MeOH in each step of the extraction (>98%). An even higher recovery for YTX was obtained by using 80%, 90% and 100% MeOH in the respective steps of the mixed MeOH extraction procedure. The slower extraction observed for YTX fits with reports that this toxin is difficult to extract fully from shellfish tissues [4, 19]. Analysis of supernatants from the individual extraction steps showed a higher concentration in samples extracted with 100% MeOH compared to 80% MeOH in the first step of the procedure; however, it appears that further extractions of the sample result in comparable overall recoveries. For AZAs >99% recovery was obtained using 100% MeOH for the three analogues using both procedures. There is a suggestion of slightly lower recovery for AZA2 using the mixed extraction, presumably due to the increased lipophilicity of this analyte; however, the difference is not significant. Considering only the first step of the mixed extraction using 80% MeOH as solvent, the results clearly show that recoveries decreased as lipophilicity of the AZAs increased. So while recovery was approximately 83% for AZA3, it was lower for AZA2, at approximately 66%. This is an important consideration for single step extractions that might be used in routine monitoring. Interestingly, while overall recoveries are similar for the two methods, there are some small differences in concentrations measured. This may suggest that the percentage MeOH used for extraction can have an influence on matrix effects in LC-MS. However, further studies would be required to verify this.
Approximately 95% recovery for OA and AZA group toxins was previously reported using a two-step 100% MeOH procedure on a freeze-dried mussel matrix [11], so the improved recovery with the triplicate procedure reported here justifies the additional work involved. While both procedures tested gave similar results, 100% MeOH was selected as the extraction solvent to be used for further work on CRM-FDMT1. The excellent recoveries make the procedure suitable for assessing homogeneity and stability of the CRM.
Matrix effects in LC-MS analysis of CRM-FDMT1
Matrix effects are a well-known issue in the analysis of biological samples by ESI LC-MS resulting in under- or overestimation of analyte concentrations [14]. While the exact causes of matrix effects are not fully understood, a consensus is that co-extractives from the sample interfere with the ionisation process resulting in a suppressed or enhanced signal for the analyte in a matrix or extract when compared to the signal in a pure solvent. For the purpose of this study, matrix effects describe the difference in MS response for toxins in a shellfish tissue extract compared to methanolic solutions.
Many factors contribute to matrix effects in the analysis of shellfish, such as the species being analysed, and whether it has been processed (e.g. cooking). Additionally, aspects of the methodology can play a significant role in the level of matrix effects such as the type of instrument being used and the LC column selected for separation [13, 20]. The extraction procedure and clean-up steps used in preparation of samples can significantly alleviate or even confound the matrix effect issue so it is important that due consideration is paid to this aspect of the methodology.
As a first step, the effect of dilution of sample extracts was examined as a way to determine the extent of the problem with different toxins and as a possible means of reducing matrix effects. LC-MS analyses were conducted on neat sample extracts and on those diluted accurately with MeOH at two levels: twofold and fourfold. A plot of the response normalised according to dilution factor shows varying types and levels of matrix effects for the lipophilic toxins present in CRM-FDMT1 extracts (Fig. 3). Comparing the response in the neat and diluted extracts demonstrated that ion enhancement was occurring for the OA group toxins and that increasing the level of dilution reduced the problem. Minimal matrix effects were observed for YTX at these dilution levels; whereas for AZAs and PTX2, it appears that substantial ion suppression is taking place in concentrated extracts. For the dilutions tested, levels of matrix effects ranged from negligible to approximately 30% suppression or enhancement of signal depending on the analyte being considered. It is important to note that the limited dilution range tested makes it difficult to determine the level of dilution actually required to fully eliminate the problem. The dilution process could have been continued in order to observe when two or more consecutive dilutions gave equivalent results, which might be assumed to be the point at which the matrix effect has been eliminated. However, limits of detection (LOD) for some of the analytes became a more serious problem with extensive dilutions. Therefore, while some dilution is useful, full elimination of matrix effects may not be completely achieved unless a much more sensitive LC-MS system is used.

Dilution of 100% MeOH extract of CRM-FDMT1 to demonstrate matrix effects lipophilic toxins. Serial two- and fourfold dilutions were prepared and analysed on an QTRAP 4000 instrument in SRM mode, with separation using neutral mobile phase and a MaxRP C12 LC column. Error bars represent ±1 SD (n = 3)
To alleviate matrix effects a clean-up step is sometimes incorporated into the sample preparation procedures for LC-MS. Partitioning of extracts with solvents such as hexane, chloroform and ethyl acetate has been demonstrated to have variable effectiveness when applied to shellfish toxin analysis [16, 20]. The use of solid phase extraction (SPE) has also been examined extensively by other groups [15, 21], with certain applications proving more successful than others. Sample clean-ups can help with the elimination of matrix effects, but they are not always effective for complete elimination of the problem and they frequently result in less than complete recovery of analyte. Therefore, while sample clean-ups such as solvent partitioning and SPE can be effective in a monitoring operation, they are not entirely appropriate for CRM work where high recovery and accuracy of the determination is of utmost importance. A very effective solution for matrix effects is the use of stable isotope labelled internal standards. Unfortunately, these are currently unavailable for shellfish toxins so this leaves two practical approaches that can be used for certification of a matrix CRM: (a) standard addition and (b) MMC.
Standard addition has been applied successfully to test and compensate for matrix effects in shellfish toxin determinations [12, 13, 22]. The principle is that toxin spiked into the sample will be suppressed to the same extent as toxin naturally present. By spiking the sample with known quantities of pure toxin at different levels and demonstrating linearity, the response plot can be used to determine the analyte concentration in a sample extract by extrapolating the x-axis intercept, which defines the true concentration independent of matrix effects. Spiking solutions containing OA, YTX, PTX2 and AZA1 were prepared at levels suitable for spiking CRM-FDMT1 extracts at approximately 1.5, 2, 2.5 and 3 times the analyte concentrations. Preliminary information on CRM-FDMT1 concentrations was obtained from the dilution test samples in order to design the standard addition experiments. Comparing slopes for the spikes prepared in MeOH and in CRM-FDMT1 extracts, matrix effects were observed for all four toxins investigated. Figure 4 illustrates the results observed for AZA1 and PTX2. Matrix effects are shown by comparing the response in spiked CRM-FDMT1 extracts with the response obtained for MeOH spikes. Good linearity was observed for the different toxins at four spike levels with correlation coefficients all >0.993. For all four of the lipophilic toxins investigated ion suppression was observed, ranging from 10–30% suppression, with PTX2 being the most significantly affected. A confounding characteristic of matrix effects is that the extent of the problem can vary significantly between analytes, sample matrices and methods. The dilution study reported here, as well as separate studies on matrix effects in the authors’ laboratory using different LC-MS conditions, have highlighted matrix enhancement as a problem for the OA group toxins (data not shown). Regardless of the type of matrix effect being observed, standard addition seems to be effective in dealing with the problem. However, while the standard addition procedure is effective for handling matrix effects in complex biological samples, it is not generally applicable for routine shellfish toxin monitoring due to the large of amounts of pure toxin required. Additionally, the need for some preliminary analyses of the sample being tested (in an ideal situation) does not lend the method to routine monitoring application where sample turnaround times are of high importance.

Standard addition experiments to test for matrix effects on AZA1 (a) and PTX2 (b). Calibration curves for methanolic solutions of standards are compared with spiked extracts of CRM-FDMT1 and CRM-Zero-Mus. Slopes for AZA1 in CRM-FDMT1, CRM-Zero-Mus and MeOH are 0.816, 0.833 and 1.00, while slopes for PTX2 are 0.653, 0.491 and 1.00, respectively. R 2 values were >0.998 for all curves
MMC is an alternative approach for compensation of matrix effects that has been reported for shellfish toxin analysis [17]. To perform MMC, it is necessary to a have “blank” shellfish tissue that contains none of the toxins being quantified. A mussel homogenate from CRMP (NRC CRM-Zero-Mus), containing non-detectable levels of the major lipophilic toxins, was tested for MMC of CRM-FDMT1. Using the same spike solutions prepared for the standard addition experiment an extract of CRM-Zero-Mus was spiked at the four concentration levels (Fig. 4). The slope of AZA1 in the CRM-FDMT1 and CRM-Zero-Mus extracts are equivalent within the uncertainty of the analysis. This indicates that there is an equivalent level of matrix effects for AZA1 in both matrices, and therefore, shows that CRM-Zero-Mus is a suitable matrix for preparation of AZA1 matrix-matched calibrants for accurate quantitation of CRM-FDMT1. The complexity of the matrix effect issue is again demonstrated as the equivalent plot for PTX2 shows that slopes are different in CRM-FDMT1 and CRM-Zero-Mus, therefore CRM-Zero-Mus is not suitable for preparing matrix-matched calibrants for PTX2, and an alternative matrix must be sourced. These results highlight not only the importance of the matrix being “blank”, but also that care should be taken to ensure that the matrix is a “match” for the samples being analysed. CRM-Zero-Mus was found to be a suitable matrix match for OA in CRM-FDMT1; however, variable results were obtained for YTX (data not shown).
Performance and application of method
The LC-MS method satisfied the criteria for speed and selectivity set at the beginning of this work. Good peak shapes were observed for all the toxins examined. Retention time reproducibility was satisfactory over a 24 hour sequence in a run analysing mussel tissue extracts (ranging from ±0.01 to ±0.04 min). Other performance characteristics are reported in Table 3. Calibration curves prepared for OA, YTX, PTX2, AZA1 and 13-desMe-SPX-C showed correlation coefficients >0.995 for all toxins, thus demonstrating good linearity of the method. A low concentration range was used for 13-desMe-SPX C as we have observed some non-linearity for SPXs at high concentrations using various LC-MS methods. Limits of detection determined from low level MeOH calibrants were 13, 50, 72, 381 and 538 pg/mL for SPX-13-desMe-C, AZA1, PTX2, YTX and OA, respectively (calculated at S/N = 3). Similar to previously reported multi-toxin methods, LODs were lower for toxins analysed in positive ionisation mode [5]. Limits of quantitation (LOQ) from the same standard spiked in blank mussel extract provide more than adequate sensitivity for quantitation well below the current regulatory limit of 160 μg/kg for OA and AZA groups toxins [1].
A chromatogram of the finalised extraction and LC-MS methods applied to CRM-FDMT1 is shown in Fig. 5. The methods will be applied in homogeneity and stability testing, commutability studies and general characterisation work on the material.

LC-MS analysis of lipophilic toxins in an exhaustive extract of CRM-FDMT1 using the MaxRP C12 column (2.5 μm, 2 × 50 mm) and neutral mobile phase (5 mM NH4Ac). Minor isomeric toxins are marked with an asterisk (*). Degradation products of AZA3 from heat treatment of stock tissues are marked with a double asterisk (**)
The method is also suitable for the detailed analysis of individual toxin groups. Figure 6 shows the SPX toxin profile in an extract of CRM-FDMT1 showing that 13-desMe-SPX C is dominant with other SPX analogues present at lower levels. Although SPXs are presently an unregulated class of toxins, the concentration of 13-desMe-SPX C will be certified in CRM-FDMT1.

Major SPX analogues present in CRM-FDMT1 analysed using the MaxRP LC-MS method. The peak marked with section sign (§) in the 694 > 164 transition is due to the 13C isotope signal from the 13-desMe-SPX C. Spirolide of unknown structure marked with an asterisk (*)
While the primary focus of this exercise was selection of methods to be used for the multi-toxin CRM, the methods also have a possible wider application for toxin monitoring exercises. A naturally contaminated mussel sample was extracted using a simple single step extraction procedure and analysed using a fast resolution version of the MaxRP method. Figure 7 shows the determination of OA, DTX2 and ten AZA analogues in the contaminated mussel sample. The gradient was shortened to 3.5 min, and the flow rate was increased to 600 μL/min. All toxins eluted within 4 min for a total run time of 7 min, including re-equilibration. A chromatogram of CRM-FDMT1 analysed using the rapid format of the method is included in the Electronic Supplementary Material. Under these conditions, back pressures remained under 300 bar showing that a fast resolution can be performed with the MaxRP column using conventional LC equipment not capable of withstanding the high back pressures typically associated ultra performance liquid chromatography. An interesting point to note in regard to this LC system is that it enables a separation of some previously unreported AZA isomers that are un-resolved using acidic or basic mobile phases. These isomers appear to be present for all the major AZA analogues and they occur at varying levels depending on the sample and treatment applied prior to analysis (unpublished information).

Analysis of a mussel sample naturally contaminated with OA, DTX2 and AZA1-10 using fast resolution LC-MS: MaxRP C12 column; 5 mM NH4Ac binary gradient at 25–100% B over 3.5 min; flow rate 600 μL/min. Asterisk (*) represents isomeric toxins. Peaks due to 13C isotopes indicated with section sign (§). DP and CE was 60 V and 65 eV, respectively, for AZA4, -5 and 7–10, while other analytes were measured using conditions in Table 1
OA and DTXs are known to form a variety of acyl esters in shellfish [23]. Some of these are present in CRM-FDMT1, but the levels of individual esters will not be certified due to limited availability of calibrants. However, information values will be provided for “total” ester concentrations determined following hydrolysis [24, 25]. The individual ester profile will also be characterised by a separate LC-MS analyses following previously reported methods [25, 26].
Conclusions
A multi-toxin LC-MS method was developed following testing of LC columns with a range of mobile phase pH values to achieve a satisfactory separation. An extraction procedure with high recovery of the major lipophilic toxins was selected. The extent of matrix effects for CRM-FDMT1 was assessed by dilution experiments and approaches to deal with the issue in LC-MS analysis were tested. Standard addition proved highly effective, while MMC testing demonstrated the importance of verifying the suitability of the blank matrix. The methodology developed will be important for the homogeneity and stability testing of CRM-FDMT1 and will be vital for the accurate analyses necessary for final certification and uncertainty assignment of the reference material. The methods should also be of interest to regulatory laboratories involved in routine testing for shellfish toxins.
References
Anonymous (2004) Regulation (EC) No 853/2004 of the European parliament and of the council of 29 April 2004 laying down specific hygiene rules for food of animal origin. Official Journal of the European Union L 139 of 30 April 2004
McCarron P, Emteborg H, Nulty C, Rundberget T, Loader JI, Miles CO, Teipel K, Quilliam MA, Hess P (2011) A mussel tissue certified reference material for multiple toxins. Part 1: design and preparation. Anal Bioanal Chem. doi:10.1007/s00216-011-4786-9
Quilliam MA, Hess P, Dell'Aversano C (2001) Recent developments in the analysis of phycotoxins by liquid chromatography-mass spectrometry. De Koe WJ, Samson RA, van Egmond HP, Gilbert J, Sabino M (Eds) Proceedings of the Xth International IUPAC Symposium on Mycotoxins and Phycotoxins, 21–25 May 2000, Guaruja (Brazil), pp 383–391
McNabb P, Selwood AI, Holland PT, Aasen J, Aune T, Eaglesham G, Hess P, Igarishi M, Quilliam M, Slattery D, van de Riet J, van Egmond HP, Van den Top H, Yasumoto T (2005) Multiresidue method for determination of algal toxins in shellfish: single-laboratory validation and interlaboratory study. J AOAC Int 88:761–772
Fux E, McMillan D, Bire R, Hess P (2007) Development of an ultra-performance liquid chromatography-mass spectrometry method for the detection of lipophilic marine toxins. J Chromatogr A 1157:273–280
Stobo LA, Lacaze JP, Scott AC, Gallacher S, Smith EA, Quilliam MA (2005) Liquid chromatography with mass spectrometry—detection of lipophilic shellfish toxins. J AOAC Int 88:1371–1382
Gerssen A, Mulder PPJ, McElhinney MA, de Boer J (2009) Liquid chromatography–tandem mass spectrometry method for the detection of marine lipophilic toxins under alkaline conditions. J Chromatogr A 1216:1421–1430
Hess P, Nguyen L, Aasen J, Keogh M, Kilcoyne J, McCarron P, Aune T (2005) Tissue distribution, effects of cooking and parameters affecting the extraction of azaspiracids from mussels, prior to analysis by liquid chromatography coupled to mass spectrometry. Toxicon 46:62–71
Quilliam MA (1995) Analysis of diarrhetic shellfish poisoning toxins in shellfish tissue by liquid chromatography with fluorometric and mass spectrometric detection. J AOAC Int 78:555–570
Rodrigues SM, Vale P, Botelho MJ, Gomes SS (2009) Extraction recoveries and stability of diarrhetic shellfish poisoning (DSP) toxins in naturally contaminated samples. Food Addit Contam A 26:229–235
McCarron P, Emteborg H, Hess P (2007) Freeze-drying for the stabilisation of shellfish toxins in mussel tissue (Mytilus edulis) reference materials. Anal Bioanal Chem 387:2475–2486
Ito S, Tsukada K (2002) Matrix effect and correction by standard addition in quantitative liquid chromatographic-mass spectrometric analysis of diarrhetic shellfish poisoning toxins. J Chromatogr A 943:39–46
Fux E, Rode D, Bire R, Hess P (2008) Approaches to the evaluation of matrix effects in the liquid chromatography-mass spectrometry (LC-MS) analysis of three regulated lipophilic toxin groups in mussel matrix (Mytilus edulis). Food Addit Contam A 25:1024–1032
King R, Bonfiglio R, Fernandez-Metzler C, Miller-Stein C, Olah T (2000) Mechanistic investigation of ionization suppression in electrospray ionization. J Am Soc Mass Spectrom 11:942–950
These A, Scholz J, Preiss-Weigert A (2009) Sensitive method for the determination of lipophilic marine biotoxins in extracts of mussels and processed shellfish by high-performance liquid chromatography–tandem mass spectrometry based on enrichment by solid-phase extraction. J Chromatogr A 1216:4529–4538
McElhinney MA (2007) Matrix effects. development of clean-up and LC-techniques contributing towards a reference LCMS method for the analysis of lipophilic marine toxins. MPhil Thesis, Dublin Institute of Technology
Gerssen A, Olst EHW, Mulder PPJ, Boer J (2010) In-house validation of a liquid chromatography tandem mass spectrometry method for the analysis of lipophilic marine toxins in shellfish using matrix-matched calibration. Anal Bioanal Chem 397:3079–3088
NRC-CNRC (2002) Certified calibration solution for the spiromine toxins, 13-desmethyl spirolide C. Certificate of analysis, Institute for Marine Biosciences, Halifax, NS, Canada
Yasumoto T, Takizawa A (1997) Fluorometric measurement of yessotoxins in shellfish by high-pressure liquid chromatography. Biosci Biotechnol Biochem 61:1775–1777
Kilcoyne J, Fux E (2010) Strategies for the elimination of matrix effects in the liquid chromatography tandem mass spectrometry analysis of the lipophilic toxins okadaic acid and azaspiracid-1 in molluscan shellfish. J Chromatogr A 1217:7123–7130
Gerssen A, McElhinney MA, Mulder PPJ, Bire R, Hess P, Boer J (2009) Solid phase extraction for removal of matrix effects in lipophilic marine toxin analysis by liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem 394:1213–1226
McCarron P, Kilcoyne J, Miles CO, Hess P (2009) Formation of azaspiracids-3, -4, -6, and -9 via decarboxylation of carboxyazaspiracid metabolites from shellfish. J Agric Food Chem 57:160–169
Yasumoto T, Murata M, Oshima Y, Sano M, Matsumoto GK, Clardy J (1985) Diarrhetic shellfish toxins. Tetrahedron 41:1019–1025
Mountfort DO, Suzuki T, Truman P (2001) Protein phosphatase inhibition adapted for determination of total DSP in contaminated mussel. Toxicon 39:383–390
Doucet E, Ross N, Quilliam MA (2007) Enzymatic hydrolysis of esterified diarrhetic shellfish poisoning toxins and pectenotoxins. Anal Bioanal Chem 389:335–342
Torgersen T, Wilkins AL, Rundberget T, Miles CO (2008) Characterization of fatty acid esters of okadaic acid and related toxins in blue mussels (Mytilus edulis) from Norway. Rapid Commun Mass Spectrom 22:1127–1136
Acknowledgements
Technical assistance from Elliott Wright is gratefully acknowledged. Dr. Jeremy Melanson is thanked for reviewing this manuscript. This is NRCC manuscript number 51796.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Electronic supplementary material (PDF 70.4 kb)
Rights and permissions
About this article
Cite this article
McCarron, P., Giddings, S.D. & Quilliam, M.A. A mussel tissue certified reference material for multiple phycotoxins. Part 2: liquid chromatography–mass spectrometry, sample extraction and quantitation procedures. Anal Bioanal Chem 400, 835–846 (2011). https://doi.org/10.1007/s00216-011-4803-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-4803-z