
Abstract
Assessing dietary intake of vitamins from all sources, including foods, dietary supplements, and fortified foods, would be aided considerably by having analytical methodologies that are capable of simultaneous determination of several vitamins. Vitamins naturally present in foods may occur in different chemical forms, with levels ranging over several orders of magnitude. Vitamins in dietary supplements and fortified foods, however, are typically added in a single chemical form, and matrix issues are usually not as complex. These sources should thus be relatively amenable to approaches that aim for simultaneous determination of multiple vitamins. Our recent work has focused on development of liquid chromatography (LC)–UV/fluorescence and LC–tandem mass spectrometry methods for the simultaneous determination of water-soluble vitamins (thiamine, niacin, pyridoxine, pantothenic acid, folic acid, biotin, and riboflavin) in dietary supplement tablets and fortified foods, such as formula powders and breakfast cereals. As part of the validation of our methods and collaboration in characterization of a new NIST SRM 1849 Infant/Adult Nutritional Formula powder, we report data on SRM 1849 using isotope dilution mass spectrometric methods. Use of available NIST Standard Reference Materials® as test matrices in our method development and validation gives a benchmark for future application of these methods. We compare three chromatographic approaches and provide data on stability of vitamin standard solutions for LC-based multiple vitamin determinations.

Extracted ion chromatograms of seven vitamins using RP chromatography treatment
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Vitamins are commonly divided into two broad classes: the water- and the fat-soluble vitamins. The water-soluble vitamins include thiamine (B1), riboflavin (B2), niacin (B3), pantothenic acid (B5), pyridoxine and related compounds (B6), biotin, folic acid, cyanocobalamine and related compounds (B12), and ascorbic acid (C). Measurements of vitamin levels in foods and other matrices have long been of interest to the health and nutritional fields and to the food industry [1, 2]. Established methods of vitamin analysis are mostly designed to measure one vitamin at a time, and the procedures tend to be time consuming and based on older technology. Thus, for example, the AOAC Official Methods of Analysis [3, 4] for the B vitamins are mostly single-vitamin microbiological methods that require numerous determinations and up to several days to complete. Literature references [1, 2] summarize various alternative methods for vitamin analysis, including some high-performance liquid chromatography (HPLC) methods. The HPLC methods in the literature mostly specify UV detection and many call for the use of ion-pairing agents in the mobile phase, which improve retention and peak shape for some of the vitamins. In general, these HPLC methods are not validated as AOAC methods. Most of the methods are optimized for a single vitamin and may contain columns and equipment that are no longer current. Although often there may be interest in measurement of only a single vitamin, in other cases, it is desirable to know the levels of multiple vitamins or all vitamins in a sample. Microbiological and some HPLC methods for the B vitamins are also given in the USP Dietary Supplements Compendium [5], which is derived from the US Pharmacopeia-National Formulary and the Food Chemicals Codex. (It should be noted that the USP methods are for pharmaceutical components or finished products such as multivitamin supplements.) In part because of the popularity of dietary supplements, there is renewed interest in vitamin measurement. Dietary supplements account for a significant portion of total vitamin intake for many people, along with vitamin-fortified foods and natural food sources of vitamins. Public health research related to vitamins would be aided considerably by having accurate, quantitative information about the total dietary intake of vitamins from all sources, including dietary supplements and fortified foods. Obtaining such information would be greatly facilitated by having rapid and accurate analytical methodologies that are capable of simultaneous determination of several vitamins.
Development of such methodologies is challenging for several reasons, including the unique solubility characteristics and susceptibility to degradation of the individual vitamins. Solubilities for the water-soluble vitamins range from about 1 g in 1 g of water for niacinamide to about 100 ppm for riboflavin. [1, 2]. In solution, some of the water-soluble vitamins are sensitive to light, and stability may depend on pH. Vitamins naturally present in foods may occur in a variety of different chemical forms, often bound to other food components. Digestion methods successful in freeing one or more vitamins may destroy others. The levels of the different vitamins may range over several orders of magnitude in a given food item, and food matrices may present a variety of complicating factors. Vitamins in dietary supplements and fortified foods, however, are usually added in a single chemical form, levels are generally higher than naturally occurring levels, and matrix issues are usually not as complex. There is typically no need for digestion methods to obtain vitamins from these sources, as the free vitamins can be successfully obtained by extraction. These sources should thus be more amenable to approaches that aim for simultaneous determination of several vitamins, though even here it will often be necessary to make compromises in the analytical conditions in order to successfully determine all of the desired vitamins.
In the last several years, several HPLC methods for simultaneous determination of multiple water-soluble vitamins have been reported, though few have been rigorously validated [6-15]. Due to the difficulties noted above, these efforts focus almost exclusively on free vitamins such as those in fortified foods or dietary supplements. UV detection, although generally not selective enough for analysis of vitamins from natural sources, is often used in these multivitamin analyses [6-11, 13, 15] and has the advantage of being widely available and relatively simple to use. UV detection is compatible with ion-pairing agents that can help to improve the peak shape of some of the vitamins. Pantothenic acid and biotin do not have chromophores and so are not well-suited for UV detection, but they can be determined at wavelengths of 210 nm or less at sufficiently high levels and with sufficient chromatographic separation from interferents [10]. Fluorescence detection is more selective and sensitive, but except for naturally fluorescent pyridoxine and riboflavin, requires derivatization or other chemical modification of target vitamins [11, 15]. Mass spectrometry (MS), and especially tandem MS, though not compatible with ion-pairing agents, is otherwise well suited as a detector for multivitamin analysis [11-15].
The Agricultural Research Service (ARS) USDA has been involved over the past several years in establishing a Dietary Supplement Ingredient Database (DSID) under joint development by ARS and the Office of Dietary Supplements (ODS), National Institutes of Health (NIH) [16]. Vitamins are among the “highest priority” and “high priority” components to be included in the DSID. The Food Composition Method Development Laboratory (FCMDL) was involved in helping to characterize a new SRM 3280—Multivitamin/Multielement Tablets [17] developed (for use in DSID contract analysis) by the National Institute of Standards and Technology (NIST) with support of the Office of Dietary Supplements (ODS), National Institute of Health. In the process of characterizing this SRM, it was necessary for NIST to obtain, by customized synthesis, samples of isotopically labeled vitamins. Some of this labeled material was made available to FCMDL to assist in characterization of SRM 3280 by isotope dilution mass spectrometry (IDMS) techniques. Since the basic mission of FCMDL of method development for food components extends beyond dietary supplements to fortified foods and ultimately to natural levels of vitamins in foods, the next logical step was to extend the IDMS technology developed in the dietary supplement studies to our projects involved in the next phase of fortified foods. As part of this process, we were collaboratively involved in the characterization of SRM 1849—Infant/Adult Nutritional Formula vitamin-fortified milk based powder released by the NIST in July 2009 [18].
In this paper, we report analytical results on the levels of seven water-soluble vitamins (thiamine, niacinamide, pyridoxine, pantothenic acid, folic acid, riboflavin, and biotin) in SRM 1849 as obtained by IDMS. While due to cost and complexity IDMS methods may generally not be suited for routine analysis, it is an excellent approach for establishing reference values and can be useful in investigating and validating methods. Because of the difficulties noted above, multivitamin methods involve making compromises in chromatographic or other conditions, as one cannot optimize for all vitamins simultaneously. There is no single, best approach to a multivitamin determination, and in practice methods used may depend on the available chromatographic and detection systems, on the matrix, and on the vitamins of interest, among other considerations. A flexible approach should be advantageous, as long as the final method one uses can be demonstrated to be valid.
Herein we examine three chromatographic approaches for simultaneous multiple water-soluble vitamin determinations. Two of them use a C18 column and reversed phase (RP) conditions: One uses the common electrospray LC-MS mobile phase of water and acetonitrile with 0.1% formic acid, and the other replaces the 0.1% formic acid with a formic acid/ammonium formate buffer for operation at a higher pH. The third is a hydrophilic interaction chromatography (HILIC) approach, which would be expected to retain the more hydrophilic vitamins longer than RP chromatography. We also provide data on the stability of our vitamin standard solutions, which we hope will be of some practical benefit to those who may perform multivitamin analysis. Accurate quantitation obviously depends on accurately characterized levels of vitamin standards, and the preparation of such standards is one of the most time consuming and laborious aspects of LC-based multiple vitamin analysis. Finally, we present our estimates of the vitamin levels of SRM 1849 and examine the sources of the associated uncertainties in some detail. The analysis of uncertainty follows NIST Technical Note 1297 [19], which recommends expressing certain measurement results as an expanded uncertainty that includes identifiable sources of uncertainty that are not reflected in the sample standard deviation.
Materials and methods
Vitamin standards, isotopically labeled vitamins, and analytical samples
Vitamin reference standards were obtained from U.S. Pharmacopeia (Rockville, MD, USA), and all quantitative determinations relied on gravimetrically prepared solutions of these standards. Isotopically labeled versions of the vitamins used in IDMS were obtained as follows: 4,5,4-methyl-13C3-thiamine chloride and 4,5-bis(hydroxymethyl)-13C4-pyridoxine hydrochloride from Cambridge Isotope Labs, Inc., Andover, MA, USA; 2,4,5,6-d 4-niacinamide from CDN Isotopes, Quebec, Canada; calcium pantothenate-13C 156 N2, biotin-2H2, and 13C 154 N2-riboflavin from Isosciences, King of Prussia, PA, USA; folic acid-13C5 from Merck Eprova AG, Schaffhausen, Switzerland. As mentioned above, most of these labeled vitamin compounds were obtained through the NIST which previously had them custom-synthesized from the above sources with support from the Office of Dietary Supplements, NIH. SRM 1849—Infant/Adult Nutritional Formula was obtained from the National Institute of Standards and Technology (Gaithersburg, MD, USA) prior to its public release in July 2009.
Reagents
Acetonitrile was Fisher Scientific (Fair Lawn, NJ, USA) Optima Grade. Water was either Fisher Optima grade or in house from a Millipore (Bedford, MA, USA) Milli-Q Plus system. Mass spectrometry grade formic acid was from Fluka/Sigma-Aldrich Chemie GMBH (Steinheim, Germany). Ammonium hydroxide was Optima Grade from Fisher Scientific. Reagent grade phosphoric acid was from J. T. Baker Chemical Company (Phillipsburg, NJ, USA). A.C.S. reagent grade sodium phosphate dibasic was from Fischer Scientific. Hydrochloric acid, 6 M, was from G. Frederick Smith Chemical Co. (Columbus, OH, USA). A.C.S. reagent grade sodium acetate trihydrate was from J.T. Baker Chemical Co. A.C.S. Reagent grade acetic acid was from Aldrich Chemical Co. (Milwaukee, WI, USA). Beta-mercaptoethanol (BME) was from Sigma Chemical Co. (St. Louis, MO, USA).
Preparation of vitamin solutions and samples
USP standard and labeled vitamin solutions were prepared as follows: Thiamine hydrochloride and riboflavin were prepared using 0.1 M pH 2.1 phosphate buffer with 0.1% BME as a preservative; calcium pantothenate solutions were prepared using 0.1 M pH 5.6 acetate buffer with 0.1% BME; biotin and folic acid solutions were prepared using 0.1 M pH 8.6 phosphate buffer with 0.1% BME; niacinamide and pyridoxine hydrochloride solutions were prepared using 0.1 M HCl with no added BME. Vitamin solutions were prepared using low-actinic glassware and were stored at 4 °C. Initial USP vitamin stock solutions were prepared at a level of 300 ppm or higher (over 30 mg of vitamin weighed), with appropriate dilutions made to prepare working solutions. Riboflavin, due to its low water solubility, was prepared by weighing 30 to 40 mg in 1,000 mL with no subsequent dilution. Mass measurements were made using either a Mettler Toledo (Columbus, OH, USA) AX205 balance or an Ohaus (Pine Brook, NJ, USA) GA200D balance. Except for thiamine hydrochloride, moisture and other appropriate corrections were made according to the directions of the accompanying USP certificates. We were not equipped to do a moisture determination by titration for thiamine hydrochloride, as suggested on its certificate, but assigned it a level of 2.6% moisture, equivalent to 0.5 mol of water per mole of thiamine hydrochloride, after discussions with USP and other investigations. Note, however, that the USP thiamin hydrochloride is presumed to be in the so-called monohydrate form, which may contain from 0 to 0.9 mol of water per mole of thiamine hydrochloride [20]. Because they were calibrated against the USP standard solutions, weighing uncertainty was less of a concern for the labeled vitamin solutions. They were prepared at levels of 200 ppm or less, with appropriate dilutions made as needed. Calibration solutions containing USP standard and labeled vitamins were prepared at levels ranging from 500 ppb for biotin and folic acid to 2 ppm for niacinamide and pantothenic acid. The calibration solutions contained all seven vitamins and were diluted to final mass using 0.1 M pH 2.1 phosphate buffer. Calibration solutions were prepared within 1 day of the preparation for analysis of SRM 1849 samples. Preparation of SRM 1849 samples consisted of weighing an appropriate amount (typically 0.2 to 0.5 g) and extracting with 25 to 50 g of 0.1 M pH2 phosphate buffer. Labeled vitamin spikes were added at the time of weighing. Samples were shaken, centrifuged, and filtered through a 0.45 μm filter before injection into the LC system.
Chromatography and mass spectrometry
The LC-UV-MS system was a Waters (Milford, MA, USA) Quattromicro triple quad mass spectrometer in combination with an Agilent (Santa Clara, CA, USA) 1100 LC system. For all work reported here, the MS was operated in multiple reaction monitoring (MRM) mode (MS/MS), using positive electrospray ionization. MRM transitions monitored for the natural and labeled versions of the vitamins are listed in Table 1. A Phenomenex (Torrance, CA, USA) Synergi HydroRP (250 × 2 mm, 4 um particle size) column was used for RP chromatography. The typical programmed gradient mobile phase was: 0 –5 min: 95% A/B (A: 0.1% formic acid in water; B: 0.1% formic acid in acetonitrile); 5–15 min: linear gradient to 25:75 A/B; 15–18 min: hold at 25:75 A/B; 18–21 min: linear gradient to 95:5 A/B; 21–27 min: hold at 95:5 A/B. A second gradient was used in some experiments with the Phenomenex HydroRP column, as described in the “Results” section. In this case, solvent A was aqueous 20 mM pH 3.7 formic acid/ammonium formate, and solvent B was acetonitrile with 0.1% formic acid. The gradient was 0–2 min: 98:2 A/B; 2–10 min: linear gradient to 90:10 A/B; 10–18 min: linear gradient to 25:75 A/B; 18–19 min: hold at 25:75 A/B; 19–20 min: linear gradient to 98:2 A/B; 20–32 min: hold at 98:2 A/B. HILIC chromatography was performed using a SeQuant (Umea, Sweden) ZIC-HILIC column (150 × 2 mm, 3.5 um particle size). The HILIC gradient was 0–2 min: isocratic 20:80 A/B (20 mM pH 3.7 formic acid/ammonium formate:0.025% formic acid in acetonitrile); 2–15 min: linear gradient to 70:30 A/B; 15–20 min: hold at 70:30 A/B; 20–23 min: linear gradient to 20:80 A/B; 23–32 min: isocratic 20:80 A/B.
IDMS calculations
In IDMS, the analyte level is determined by measuring the isotopic abundance ratio for a sample containing a known amount of an isotopically enriched analogue of the analyte [21, 22]. The ratio can be defined in terms of the relevant isotopic abundances and the amounts of the natural and isotopic spike materials in the sample [23]. In cases where there is no contribution by the natural material to the signal monitored for the spike and vice versa, the ratio equation simplifies to
where X nat and X sp are the amounts of the natural and spike materials, respectively, in the sample, R is the experimentally determined isotopic abundance ratio (i.e., peak area ratio), and C is a factor introduced to correct for instrument and other sources of bias, determined by measuring the abundance ratio for a standard containing both the natural and isotopically labeled forms of the analyte. With the exception of biotin, this simplification is appropriate for the vitamins considered here. Sulfur containing natural biotin has about a 5% isotopic abundance at the mass monitored for the isotopically labeled analogue, 2H2-biotin. We account for this contribution in the results presented here, although using the simplification causes only about a 1% to 2% overestimation of biotin levels.
Results
Chromatographic treatments
In most reported electrospray LC-MS work on multiple vitamins, determinations are made using C18 columns and RP chromatography with mobile phases of water and acetonitrile with 0.1% formic acid. Figures 1, 2, and 3 show LC-MS/MS chromatograms for seven water-soluble vitamins (USP standards) using three chromatographic approaches, the second and third of which differ from the usual approach. In Fig. 1, chromatograms were obtained using the Phenomenex Hydro RP C18 column and RP chromatography, as described in the “Materials and methods” section. In comparison to the gradient reported in Chen et al. [13], which had a 100% aqueous initial mobile phase, the 95:5 water/acetonitrile starting conditions here resulted in sharper peaks for all vitamins except for pantothenic acid, but with niacinamide and pyridoxine not resolved when viewed in a total ion chromatogram and with a tendency for splitting of the thiamine standard peaks (about 5% of the total thiamine peak area in the smaller, earlier eluting part), though not for the thiamine peaks for SRM 1849. Lack of resolution of the niacinamide and pyridoxine peaks does not present a problem for quantitation with the MS/MS approach used here, but it would be an issue when using UV detection. The splitting of the thiamine standard peak is assumed to contribute to the variance of the thiamine determination, but does not appear to affect the estimated levels otherwise; the area of the smaller portion is included in peak area estimates for the thiamine standards.

Extracted ion chromatograms of seven vitamins using RP chromatography treatment

Extracted ion chromatograms of seven vitamins using RP pH 3.7 chromatography treatment

Extracted ion chromatograms of seven vitamins using HILIC chromatography treatment
All seven vitamins can be reliably determined using this RP approach; nevertheless, alternative approaches are worth exploring and may offer some advantages, especially when using detection methods less selective than MS/MS. Thiamin can be problematic using RP chromatography, as it is barely retained under typical RP conditions and can exhibit considerable tailing or peak splitting. In Fig. 1, thiamine elutes at barely more than one column volume, and it is retained only slightly better under 100% aqueous starting conditions. Pyridoxine and niacinamide are a little better retained under typical RP conditions, but they can also exhibit significant tailing. RP chromatography normally works very well for pantothenic acid, folic acid, riboflavin, and biotin. In Fig. 2, the Phenomenex Hydro RP column was again used, but mobile phase component A was changed to 20 mM pH 3.7 formic acid/ammonium formate (component B remained acetonitrile with 0.1% formic acid). The gradient used to obtain the chromatograms in Fig. 2 is described in the “Materials and methods” section. The elution order remains the same as in Fig. 1, but all peaks are retained longer, with the biggest effect occurring for niacinamide. The peaks for thiamine, pyridoxine, and niacinamide are wider than in Fig. 1, and although the thiamine peak shape is not symmetric, it is not completely split. The thiamine signal level is also significantly greater here than under the conditions in Fig. 1; the peak areas were about 50 times larger for SRM 1849 test samples. Peak areas for the other vitamins are similar to those observed under the RP conditions of Fig. 1. The signal enhancement for thiamine appears to be due to a pH effect, as the organic content of the mobile phase at the time of its elution is even lower (2%) than in the RP case (5%). It may be relevant that thiamine differs from the other vitamins in that it does not require addition of a proton to be detected by ESI-MS, due to the positive charge on the nitrogen in the thiazole ring. The degree of tailing for the niacinamide, pyridoxine, and thiamine peaks in Fig. 2 is similar to that in Fig. 1. The pantothenic acid peak is narrower, and those for folic acid, riboflavin, and biotin are similar to their shapes in Fig. 1.
Figure 3 shows chromatograms obtained using the SeQuant ZIC-HILIC column and the HILIC gradient as described in the “Materials and methods” section. The elution order is obviously quite different from that obtained under RP conditions, with biotin being poorly retained and thiamine the most strongly retained. The thiamine, pyridoxine, and niacinamide peaks are wider than in Fig. 1, but tailing is no worse, and the signal levels are greater. Using SRM 1849 test samples, the thiamine peak areas are about 20 times greater using HILIC as compared to RP, pyridoxine about four times greater, and niacinamide about 1.3 times greater. These signal enhancements are probably due to the higher percentage of organic component in the mobile phase at the time of elution. Although it is not a major issue when using MS/MS detection, it should be noted that there is incomplete resolution of biotin, pantothenic acid, niacinamide, and riboflavin and of folic acid and pyridoxine. Reliable quantitative results can be obtained for all seven vitamins using this HILIC-IDMS approach. The HILIC method is a good alternate choice for the three more hydrophilic vitamins, though it offers no advantages for the other four vitamins.
Stability of vitamin standard solutions
Accuracy of the vitamin measurements by IDMS depends on accurately knowing the levels of the vitamin standard solutions. Reliability of mass measurements when preparing the standards is of course one important consideration. Another is the stability of the vitamin standard solutions. Besides its importance for accuracy, if stability of the standard solutions under conditions of storage can be demonstrated, a considerable amount of time and effort can be saved by reducing the need for preparation of fresh solutions. Vitamin standard solutions were prepared and stored as described in the “Materials and methods” section, based on guidance from Refs. [1] and [2]. Solutions were prepared at intervals over a period of more than 1 year, as indicated in Table 2, and were tested against freshly made vitamin standard solutions by LC-IDMS. Two samples of each of the older vitamin solutions were tested. Uncertainties listed in Table 2 for the levels by IDMS are 95% confidence limits (CL) determined by using pooled estimates of the sample standard deviations. Uncertainties listed for the gravimetrically determined levels are 95% CL due to weighing uncertainty, with an estimated standard deviation of the mass measurements of 0.0002 g.
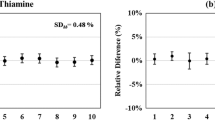
For six of the seven vitamins tested (niacinamide, pyridoxine, thiamine, pantothenic acid, riboflavin, and biotin), there is no convincing evidence that significant degradation has occurred, even for solutions over 1 year old. In one case, the pantothenic acid solution over 1 year old, the experimentally determined level was about 6% higher than the gravimetrically assigned level, but this is likely due to an error in the initial gravimetric data; determinations of pantothenic acid in SRM 1849 using this standard (not reported) were about 6% lower than determinations using the newer standards. In the case of biotin, even though not statistically significant, one might suspect a small amount of degradation, as the experimentally determined levels are all nominally lower than the gravimetric levels; however, even in the worst case, the solution over 1 year old, the amount of possible degradation is not more than about 6%. Degradation was observed for folic acid. Each of the three older folic acid solutions was determined at a level about 10% lower than the corresponding gravimetric level. Further experiments are needed to better characterize the time characteristics of the folic acid degradation. As it does not get worse in the period from 6 months to 1 year, it may be due to some component of the pH 8.6 buffer solution which, when depleted, causes no further problems. The stability data for biotin and folic acid are presented graphically in Fig. 4, where the standard solution vitamin levels as determined by LC-IDMS, expressed as percentage of the relevant gravimetric level, are plotted against age of solution.

Vitamin levels of biotin and folic acid in standard solutions as determined by LC-IDMS, expressed as percentage of gravimetric level, versus age of solution. Error bars are 95% confidence limits
Determination of vitamin levels in SRM 1849
Vitamin levels in SRM 1849 were determined by LC-IDMS on three occasions, several months apart, using six samples for each set. The three chromatographic protocols described above were used as indicated in Table 3. Sets I and II were determined using RP chromatography, with additional runs using HILIC for determination of niacinamide, thiamine, and pyridoxine. Similar runs were made for set III, and an additional run, using the same RP column (Phenomenex Synergi Hydro RP), but with 20 mM pH 3.7 formic acid/ammonium formate as mobile phase A in place of aqueous 0.1% formic acid, was also made. The uncertainties in Table 3 are 95% CL based on the sample variance associated with each individual set of six samples. Although ANOVA indicates that in some cases the results from the three sets of data are not in agreement at a 95% CL, it is obvious that the results for each vitamin are very similar across the data sets and across the different chromatographic treatments. The widest range among the means, both in absolute and relative terms, is for the RP treatment of niacinamide, where it is about 6 ppm, or 6%. One-way ANOVA of the RP data indicates that the three sets of niacinamide results are not in agreement at a 95% CL, but that for the other six vitamins, they are. One-way ANOVA of the HILIC data indicates that for none of the three vitamins (niacinamide, thiamine, and pyridoxine) can the three sets of results be considered in agreement at a 95% CL. Note, however, that the precision of the HILIC data is better than the corresponding RP data, probably because of the usually better HILIC peak shapes for these three vitamins. Thus, the HILIC data may be sensitive to statistical differences that are not detectable with the RP treatment. Relative standard deviations for the RP treatment are about 3% or less for niacinamide, pyridoxine, and pantothenic acid, and about 5% or less for riboflavin and biotin. For thiamin and folic acid, they are mostly over 5%. For thiamin, the relatively higher uncertainty is probably related to the poorer peak shape, and for folic acid, relatively weaker signal level probably contributes. Although there is only one data set using the pH 3.7 RP treatment, it appears to provide a similar level of precision as the regular RP treatment. In the case of folic acid, the pH 3.7 RP treatment appears to give much worse precision, but that is due to a significantly lower signal level, in comparison to the RP data, obtained for folic acid when the samples were run, probably due to a storage issue and a several day delay in running the samples after they were prepared.
The “between group” discrepancies revealed by ANOVA are probably related to sample preparation or instrumental conditions that varied over the time of the experiments, although it should be noted that IDMS should correct for most sources of such bias. Where they are statistically significant, the between group discrepancies will affect the estimates of variance associated with estimates of the vitamin levels. These situations can be treated by using variance component analysis and in particular by treating time (date of analysis) as a random blocking effect [24, 25]. Blocking here simply refers to treating related observations as a group (each set of six observations giving rise to the entries in Table 3 can be considered a block), and random refers to the effect not being fixed by the experimenter. Values of a random effect are assumed to arise from a normal distribution, and so each block is assumed to have a different sampling of such effects. In addition to the random error detected by ANOVA, there are other sources of uncertainty that should be considered before giving estimates of the vitamin levels, and these sources are discussed below.
Our final estimates of the vitamin levels in SRM 1849 were obtained by pooling the data used for Table 3 and are given in Table 4. Although any of the three chromatographic approaches could be used to obtain good estimates for all seven vitamins, based on precision, we have used the HILIC data for niacinamide, thiamine, and pyridoxine and the RP data for pantothenic acid, riboflavin, biotin, and folic acid. The NIST-certified levels are given in column 2 [18]. Our estimated levels in column 3 give an expanded uncertainty of coverage factor k = 2, which roughly corresponds to a 95% CL. Standard errors of the means in the fourth column are based on the three relevant sets of data from Table 3 for each vitamin, and, in the cases of niacinamide, thiamine, and pyridoxine, account for the uncertainty associated with random block effects. In these three cases, the estimates of the standard errors of the means were obtained using SAS Proc Mixed (SAS version 9.2, SAS Institute Inc., Cary, NC, USA). For pantothenic acid, riboflavin, biotin, and folic acid, estimates of the standard errors of the means were determined using pooled sample variances from the appropriate three sets of data. The mean USP standard deviations in the fifth column are estimates of the uncertainty in the mean vitamin level estimates due to uncertainty in the assigned values of the relevant USP standard solutions. The two significant sources of the latter uncertainty are uncertainty in the mass measurements of the solid USP vitamin used to prepare the standard solutions and uncertainty in the purity of the USP vitamins after applying the recommended moisture and other corrections. A series of test measurements gave an estimated standard deviation of 0.0002 g for individual mass measurements, which resulted in a relative standard deviation of less than 1% in most cases in the vitamin masses used. The USP standards, with one exception, were assumed to have a ±1% purity uncertainty, at a 95% CL, after the appropriate corrections. This estimate is in recognition that although USP does devote some effort to their characterization, these materials are certified only for uses related to compliance with USP-NF monograph standards. Thiamine was assumed to have a total purity uncertainty of ±2.6%, due to the additional uncertainty related to its moisture content. The mass and purity uncertainties were combined to determine estimated variances for the USP stock solutions, and contributions to the uncertainty of the estimated SRM 1849 vitamin levels were determined by a propagation of uncertainty treatment using the common first-order Taylor series approximation. The sixth column in Table 4 gives an estimate of uncertainty due to the correction factors, C, used in the IDMS calculations (see “Materials and methods” section). The correction factors used in calculating the vitamin levels were mean values obtained from multiple determinations of spiked standards, and so the standard errors of the means in column 4 do not reflect this source of uncertainty. The s values in the sixth column were determined using pooled sample variances and reflect the range of values obtained for C in the individual runs of the standards. Contributions to the uncertainty of the SRM 1849 vitamin levels were again obtained using a first-order Taylor series approximation for the propagation of errors. The variances associated with columns 4, 5, and 6 were combined according to \( {S^2}_{\text{overall}} = {S^2}_{{\text{std}}\,{\text{err}}\,{\text{of}}\,{\text{mean}}} + {S^2}_{{\text{mean}}\,{\text{USP}}} + {S^2}_{{\text{mean}}\,C} \) to give an estimate of the expanded uncertainty for the vitamin levels, which, when multiplied by a coverage factor of k = 2, define the limits in column 3.
The estimates are consistent with the NIST-certified levels in all cases. The limits of uncertainty are, in most cases, smaller than those of the NIST estimates, but it should be recognized that the NIST levels are derived from results obtained by several different methods and from several sources and may include sources of uncertainty not recognized in this work [18]. For our results, the uncertainty in the assigned vitamin standard levels, which has roughly equal contributions from the mass and purity uncertainties, is a significant source of uncertainty in the estimated vitamin levels. It makes the largest contribution of the three sources for thiamine, pantothenic acid, and biotin, and it contributes an amount similar to that of the standard error of the mean in the other cases. Although the estimate of the purity uncertainty for the vitamin standards could be overly conservative, it would not be an easy task to determine a better, quantitatively based estimate of the purity uncertainty for these materials. The uncertainty in the mass measurements could, of course, be reduced by weighing a larger amount of the standards. The uncertainty in the correction factor contributes a relatively small, but not negligible, amount to the total uncertainty in the vitamin estimates. The standard errors of the means primarily reflect the uncertainty due to the LC-IDMS method, including sample preparation. Random block effects can have a significant influence on this uncertainty, but are often not accounted for when results of chemical measurements are reported. More careful monitoring of experimental conditions over time may reveal the source of such effects, which could then perhaps be addressed and reduced.
Conclusions
Although estimates of vitamin levels should be able to be obtained more precisely by a “one vitamin at a time” approach, good estimates can be obtained for vitamins in fortified foods and dietary supplements by HPLC methods designed to measure several vitamins simultaneously. Several different chromatographic treatments have been shown to be adequate for such determinations, so that it should be possible to adjust conditions to fit available systems and most any matrix under consideration. Under appropriate conditions of preparation and storage, most vitamin solutions are stable even up to 1 year. Under the conditions reported here, stability problems were noted for folic acid, and stability was questionable for biotin, but with further investigation, appropriate conditions can perhaps also be determined for them. Demonstration of stability has practical benefit for those involved in frequent vitamin analysis, as it reduces the need for preparation of fresh solutions. LC-IDMS is an appropriate method to use for the determination of vitamin levels in reference materials. Our estimates using LC-IDMS are consistent with the NIST-certified levels for SRM 1849, though it should be noted that some of our results were among those used by NIST in establishing those levels. An important source of uncertainty in this work was that associated with the assigned vitamin standard levels. Reducing the uncertainty associated with the relevant mass measurements is straightforward, but there remains the uncertainty associated with the purity of vitamin standards. Random block effects, though often not accounted for in analytical determinations, can also be a significant source of total uncertainty.
References
Eitenmiller RR, Landen WO Jr (1999) Vitamin analysis for the health and food sciences. CRC, Boca Raton
Ball GFM (2006) Vitamins in foods. CRC, Boca Raton
Sullivan DM, Carpenter DE (1993) Methods of analysis for nutritional labeling. AOAC International, Gaithersburg
AOAC International (2006) Official methods of analysis, 18th edn. AOAC International, Gaithersburg
USP Dietary Supplements Compendium (2009) United States Pharmacopeial Convention Rockville, MD
Moreno P, Salvado V (2000) J Chromatogr 870:207–215
Vinas P, Lopez-Erroz C, Balsalobre N, Hernandez-Cordoba M (2003) J Chromatogr 1007:77–84
Kadakal C, Poyrazoglu ES, Artik N, Nas S (2004) J Food Qual 27:171–180
Lombardi-Boccia G, Lanzi S, Aguzzi A (2005) J Food Compos Anal 18:39–46
Heudi O, Kilinc T, Fontannaz P (2005) J Chromatogr 1070:49–56
Leporati A, Catellani D, Suman M, Andreoli R, Manini P, Niessan WMA (2005) Anal Chim Acta 531:87–95
Gentili A, Caretti F, D"Ascenzo G, Marchese S, Perret D, DiCorcia D, Rocca LM (2008) Rapid Commun Mass Spectrom 22:2029–2043
Chen P, Wolf WR (2006) Anal Bioanal Chem 387:2441–2448
Chen P, Ozcan M, Wolf WR (2006) Anal Bioanal Chem 389:343–347
Chen P, Atkinson R, Wolf WR (2009) J AOAC Intl 92:680–687
Dwyer JT, Picciano MF, Betz JM, Fisher KD, Saldanha LG, Yetley EA, Coates PM, Radimer K, Bindewald B, Sharpless KE, Holden J, Andrews K, Zhao C, Harnly J, Wolf WR, Perry CR (2006) J Food Compos Anal 19:S108–S114
NIST (2009) Certificate of Analysis, SRM 3280—Multivitamin/Multielement Tablets. https://www-s.nist.gov/srmors/view_cert.cfm?srm=3280
NIST (2009) Certificate of Analysis, SRM 1849 - Infant/Adult Nutritional Formula. https://www-s.nist.gov/srmors/view_detail.cfm?srm=1849
Taylor BN, Kuyatt CE (1994) Guideline for evaluating and expressing the uncertainty of NIST measurement results, NIST technical note 1297, U.S. Government Printing Office, Washington, D.C.
Griesser UJ (2004) Crystal forms of thiamine hydrochloride (vitamin B1): new analytical data of a compound with an intriguing solid state history. PhandTA8: 8th International Conference on the Application of Physical Chemistry in Pharmacy. http://www.eurostarscience.org/conferences/abstrsph8/Griesser.pdf.
Fassett JD, Paulsen PJ (1989) Anal Chem 61:643A–649A
Webb KS, Carter D, Barwick VJ (1999) Metrologia 36:89–99
Goldschmidt RJ, Wolf WR (2007) J AOAC Intl 90:1084–1089
Box GEP, Hunter WG, Hunter JS (1978) Statistics for experimenters. Wiley, New York
Hinkelmann K, Kempthorne O (2008) Design and analysis of experiments, volume 1, 2nd edn. Wiley, Hoboken
Acknowledgments
The authors would like to thank Mary Camp and Matthew Kramer of the USDA Beltsville Agricultural Research Center Biometrical Consulting Service for consultations on statistical treatments and for assistance with SAS.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Goldschmidt, R.J., Wolf, W.R. Simultaneous determination of water-soluble vitamins in SRM 1849 Infant/Adult Nutritional Formula powder by liquid chromatography–isotope dilution mass spectrometry. Anal Bioanal Chem 397, 471–481 (2010). https://doi.org/10.1007/s00216-009-3373-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-3373-9