
Abstract
A sensitive capillary electrophoresis –laser-induced fluorescence method has been developed for the determination of six fluoroquinolones of human (ofloxacin, lomefloxacin, and norfloxacin) and veterinary use (danofloxacin, enrofloxacin, and sarafloxacin) in different kinds of water. Fluorescence detection was achieved using a He-Cd laser, with a wavelength of 325 nm. Separation was performed in a fused-silica capillary, and conditions were optimized to obtain the most adequate separation and with the best sensitivity. The separation was carried out in a 70-cm-long capillary (75 μm internal diameter, effective length 55 cm) by using a 125 mM phosphoric acid separation buffer at pH 2.8, with 36% of methanol. The water sample pretreatment involved the separation and preconcentration of the analytes by solid phase extraction. Two reverse-phase cartridges have been evaluated, namely Oasis hydrophilic–liphophilic balance and Strata-X; the latter provided the best recoveries for the selected analytes. The method shows very low detection limits (0.3–1.9 ng/L) with acceptable recoveries and precisions and has been successfully applied to the analysis of well and tap water samples.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Quinolones are an important group of antibiotics highly active against a wide spectrum of Gram-negative and Gram-positive bacteria. Their activity is based on the inhibition of bacterial DNA synthesis, and they are widely used in human as well as in veterinary medicine for the treatment of pulmonary, urinary, and digestive infections. The wide application range and the extensive use of quinolones in veterinary and human medicine represent a potential hazard for the environment, because a high percentage of antibiotics consumed are excreted unchanged via urine and feces into domestic sewage and are discharged to wastewater treatment plant effluent into the aquatic environment [1], causing an emerging environmental problem. Thus, it is not possible to exclude the presence of these compounds in aquifers, where manure or sludge is dumped to soils, or where hospital wastewaters is discharged, via a relatively small sewage treatment plant, into the nearest stream [2, 3]. Quinolones have been mainly analyzed by high-performance liquid chromatography (HPLC) and capillary electrophoresis (CE) combined with UV–visible detection, fluorescence, or mass spectrometry (MS). Different sample treatments, including solvent extraction, pressurized liquid extraction, and solid phase extraction (SPE), have been applied depending on the characteristics of the sample and on the different quinolone residues found. Some recent reviews summarize some of these analytical strategies [4, 5].
Concerning the CE analysis of these compounds, the chemical structure of quinolones, with a carboxylic group (acidic quinolones) and in most of the cases with an amino group as well (zwitterionic quinolones), allows their separation in simple buffers used in capillary zone electrophoresis. However, depending on the quinolone mixture to be resolved, it can be necessary to use non-aqueous capillary electrophoresis in order to achieve a complete resolution of signals [6]. Micellar electrokinetic chromatography has also been used to improve sensitivity rather than to improve resolution by means of on-line sweeping [7]. UV–visible is the preferred detection mode for the monitoring of the residues of these compounds by CE [6–14] although fluorescence [15, 16], MS [17–19], amperometry [20], and electrochemiluminescence [21] have also been applied. Taking into account the low concentrations expected in environmental samples, very sensitive methods for their quantification are required. Apart from off-line SPE procedures, several strategies have been used to improve sensitivity in CE for the determination of quinolones, as the on-line coupling of SPE sorbents [19] or the use of an extremely sensitive detection technique, such as laser-induced fluorescence (LIF) detection, considering the native fluorescence of some quinolones. CE-LIF has been only applied for the determination of ofloxacin (OFLO) and its metabolites [22], moxifloxacin [23] and ciprofloxacin and its metabolite [24] in body fluids, and enrofloxacin (ENRO) and ciprofloxacin in chicken muscle [15]. In this paper, we propose a specific and sensitive method based on the use of CE coupled to LIF detection for the simultaneous quantification of residues of six commonly used fluoroquinolones (FQs) of human (OFLO, lomefloxacin (LOME), and norfloxacin (NOR)) and veterinary use (danofloxacin (DANO), ENRO, and sarafloxacin (SARA)) that could be present in water samples. A previous SPE step has been optimized in order to improve selectivity and to achieve very low detection limits. The obtained results demonstrate the possibilities of CE-LIF for the quantification of residues of these compounds at very low concentrations in waters.
Materials and methods
Chemicals
All reagents were of analytical reagent grade. Solvents were HPLC grade and FQs were of analytical standard grade. Ultrapure water (Milli-Q Plus system, Millipore Bedford, MA, USA) was used throughout the work. Acetonitrile (ACN), methanol (MeOH), citric acid monohydrate, ammonium acetate, and tris(hydroxymethyl)–aminomethan (Tris) were purchased from Merck (Darmstadt, Germany). Sodium hydroxide, sodium dodecyl sulfate (SDS), sodium di-hydrogen phosphate monohydrate, and phosphoric acid (85%) were obtained from Panreac-Química (Barcelona, Spain). FQs (OFLO, LOME, NOR, DANO, ENRO, and SARA) and hexadecyltrimethyl-ammonium bromide (CTAB) were obtained from Sigma (St. Louis, MO, USA). Stock standard solutions (100 μg/mL) of each FQ were prepared by dissolving the appropriate amount of each substance in an ACN/water 50:50 (v/v) solution, and they were stored in the dark at 4 °C. Under such conditions, they were stable for at least 1 month. Working standard solutions were freshly prepared by dilution of the stock solutions with ACN/water 50:50 (v/v).
Extraction cartridges containing an Oasis hydrophilic–liphophilic balance (HLB; 200 mg, 6 mL; Waters, Milford, MA, USA) and Strata-X Polymeric Reversed Phase (200 mg, 6 mL; Phenomenex, supplied by Micrón, Madrid, Spain) were used for sample treatment.
A solvent filtration system from Millipore and nylon membranes (0.2 µm, 47 mm; Supelco, Bellefonte, PA, USA) were used for sample filtration, and Acrodisc Nylon membrane syringe filters (0.2 μm, 13 mm, Pall Corp., MI, USA) were used to filter final extract and background electrolyte (BGE) before CE analysis.
Instrumentation
CE experiments were carried out on an HP3D CE instrument (Agilent Technologies, Waldbronn, Germany) coupled to a LIF detector (Zetalif Evolution model LIF UV-01, Picometrics S.A., Ramonville, France), equipped with an He-Cd Laser (excitation length 325 nm). Data were collected using the software provided with the instrument (ChemStation version A.09.01). Separations were performed in an uncoated fused-silica capillary from Polymicro Technologies (Phoenix, AZ, USA).
For pH measurements, a pH meter (Crison model pH 2000, Barcelona, Spain) with a resolution of ±0.01 pH unit was used. SPE was carried out on a Visiprep™ DL vacuum manifold for 12 cartridges (Supelco). A vortex-2 Genie (Scientific Industries, Bohemia, NY, USA) was used for recomposition of the extracts after SPE treatment.
Electrophoretic procedure
Half an hour before starting a series, the laser was shut on. Before the first use, the new capillary (70 cm × 75 μm internal diameter, effective length 55 cm) was conditioned by rinsing with 1 M NaOH at 60 °C for 15 min, then with water at room temperature for 10 min, and finally with the BGE for 25 min, all at 1 bar N2 pressure. Every day, the capillary was prewashed with 0.1 M NaOH for 8 min, water for 1.5 min, and BGE for 15 min, at a N2 pressure of 1bar. After each run, the capillary was washed with 0.1 M NaOH for 2 min, water for 1 min, and BGE for 2 min, to maintain an adequate repeatability of run-to-run injections. At the end of the day, the capillary was cleaned with water for 4 min and finally flushed with air for 4 min.
BGE consisted on an aqueous solution of 125 mM phosphoric acid adjusted to pH 2.8 with 4 M sodium hydroxide and containing 36% of MeOH. The electrophoretic separation was achieved by applying a voltage of 26 kV (normal mode). Sample was injected by hydrodynamic injection using a pressure of 50 mbar for 10 s. The temperature of the capillary (35 cm thermostated plus 35 cm at room temperature) was kept constant at 15 °C. It is recommended to maintain stable the room temperature at a value around 20 °C.
Sample treatment
Two different well waters (a mountain well and an irrigated-land well, both from Alomartes, Granada, Spain) and one tap water (Alomartes, Granada, Spain) were analyzed.
Samples of 250 mL were spiked with appropriates volumes of the FQ standard solutions, and their pH values were adjusted to 6. Then, samples were shacked for 1 min, left in the dark for at least 30 min, and then filtered to remove suspended matter. The samples were then passed at a flow rate of 5 mL/min through a Strata-X cartridge (33 µm polymeric reversed phase, 200 mg, 6 mL; previously conditioned with 2 mL of MeOH plus 2 mL of water at a flow rate of 0.5 mL/min), the cartridge was then dried, and the analytes were eluted with 2 mL of ACN/H2O 75:25 (5% acetic acid) plus 2 mL MeOH/ACN 50:50. This extract was dried under a gentle N2 stream at 35 °C. The extract was recomposed in 0.5 mL of ACN/BGE 25:75, vortexed for approximately 1 min, and filtered.
Results and discussion
Optimization of the CE-LIF experimental conditions
Electrophoretic conditions have been optimized considering the compatibility and requirements of the LIF detection (in terms of sensitivity, resolution, and generated current), namely, pH, BGE nature, BGE concentration, organic modifier, voltage, and temperature. These analytes are zwitterions with two different pK as (see Fig. 1), so pH affects their separation in CE, and it has been stated that better resolutions are achieved at alkaline pH values [25]. However, fluorescence intensities of the selected quinolones are low at alkaline conditions, so for this reason, the pH was studied in the range of 2.0–7.5, using a concentration of 100 mM of different buffers: phosphoric acid and ammonium acetate for acidic pH and phosphate buffer and Tris for neutral pH values. Finally, a value of pH of 2.8 was selected as optimum. At this pH, citric and phosphoric acid solutions were tested, obtaining the best result for the later. Nevertheless, an exact adjustment of the buffer pH is not so critical, because the electrophoretic mobilities of the quinolones hardly change with pH values between 2.0 and 4.0 [9, 25, 26].

Structures and pK as of the selected fluoroquinolones
Then, in order to improve the resolution, the phosphoric acid concentration for the BGE was modified between 100 and 150 mM (adjusting the pH with NaOH), and the best results in terms of resolution, migration time, and current intensity were found at a concentration of 125 mM. The effect of different modifiers in the BGE, such as micellar media (SDS and CTAB), ACN, MeOH, and ethanol was studied. MeOH provided the best results, and therefore its influence was studied in the range of 5–40%. The migration times decreased when increasing the MeOH percentage, obtaining the best resolution and a shorter analysis time with a percentage of 36% of MeOH in the BGE, in agreement with the results found in the literature [27]. Then, the separation voltage was varied from 20 to 28 kV. A voltage of 26 kV was selected as optimum, as it provided the best compromise between resolution, migration time, and electric current. The effect of the temperature on the separation was tested in the range of 15–30 °C, selecting 15 °C as optimum, as this temperature provided the best resolution, and no significant differences in peak areas were observed.
Optimization of the SPE procedure
In order to obtain suitable limits of quantification (LOQs) for the determination of FQs in environmental waters (expected to be at very low concentration levels), a SPE procedure was proposed for preconcentration of the analytes in real samples, using aqueous solutions of the analytes. This alternative has proved to be suitable for the determination of quinolones in different samples such as meat [12, 28], milk [18, 29], biological fluids [30, 31], or waters [16, 32–34]. Based on these applications, we decided to carry out a comparative study for SPE using two polymeric sorbents commonly used (Oasis HLB and Strata-X), which would provide SPE procedures for clean up and preconcentration adequate to obtain low enough limits of detection for water samples. For preconditioning of the cartridges, commercial specifications were used: 2 mL of MeOH plus 2 mL of water at a flow rate of 0.5 mL/min. To achieve a complete analyte elution from the sorbents, four different compositions for the elution solution were tested, in the aqueous standard samples (approx. pH 6), namely, (a) 4 mL of MeOH, (b) 2 mL MeOH/H2O 75:25 plus 2 mL MeOH, (c) 2 mL ACN/H2O 75:25 (5% acetic acid) plus 2 mL ACN/MeOH 50:50, and (d) 2 mL H2O/MeOH 75:25 (5% acetic acid) + 2 mL MeOH. The elution solvent selected was (c) because the highest recoveries were obtained with both cartridges, with no significant differences. The percentage of acetic acid was studied in the range from 0% to 5%, and the best recoveries (>90%) were obtained for 5%. As it has been previously commented, the selected analytes are amphoteric compounds that can be in a cationic, anionic, or zwitterionic form; thus, considering the reversed phase nature of the selected sorbents, the pH of the sample was studied between 3 and 8 (analytes as zwitterions), achieving the highest retention for both cartridges when the pH was comprised between 6 and 8. A pH value of 6 was selected, as it was closer to the original pH of the samples. At this pH, slightly better recoveries were obtained with the Strata-X (92.0–104.4%), in comparison with HLB (70.6–84.9%) cartridge, so it was selected as the sorbent for the SPE procedure.
Once the optimum SPE procedure was applied, the final eluate was dried under a gentle N2 stream. Since the solvent for extract recomposition is an important variable, different solvents were studied, namely, ACN, H2O, BGE, ACN/H2O 50:50, and ACN/BGE 25:75. In all cases, a volume of 0.5 mL was used, and the mixture was vortexed for 1 min. The mixtures ACN/BGE 25:75 and ACN/H2O 50:50 gave the best results, but due to the lower conductivity of the mixture ACN/H2O 50:50, which caused eventual current fall, ACN/BGE 25:75 was selected as optimum recomposition solvent.
Finally, different water sample volumes (50, 100, 250, and 500 mL) were used in the application of the SPE procedure for preconcentration purposes. No significant differences in recovery percentages were obtained, so 250 mL was the selected as optimum volume in order to obtain a highest preconcentration factor (500) with satisfactory recovery in an adequate analysis time.
Validation of the proposed CE-LIF method
Calibration, LOD, and LOQ
The whole analytical method was validated using matrix-calibration curves with irrigated-land well water samples as a representative matrix, free of analytes, and spiked with different concentration levels of analytes (2, 10, 20, 40, and 60 ng/L for DANO and 10, 30, 50, 100, and 150 ng/L for the rest). Each concentration level was injected by triplicate. Considering the preconcentration factor (500), those concentration levels correspond to final concentrations in the test samples of 1, 5, 10, 20, and 30 µg/L for DANO and 5, 15, 25, 50, and 75 µg/L for the rest. A blank sample was also analyzed, and no matrix peaks were found co-migrating with the analytes. Calibration curves were established considering peak areas as a function of the analyte standard concentration. The statistic parameters calculated by least-square regression, limits of detection (LODs) and LOQs of the method are shown in Table 1. LODs and LOQs were considered as the minimum analyte concentration yielding an S/N ratio equal to 3 and 10, respectively. As can be seen, very low LODs were obtained in all cases, making possible the quantification of the analytes at their expected concentrations in these kinds of samples.
Precision study
The precision of the whole method was evaluated in terms of repeatability (intraday precision) and intermediate precision (interday precision), using irrigated-land well water. Repeatability was assessed by means of repetitive application of the whole procedure to five samples (experimental replicates) spiked at two concentration levels (10 and 40 ng/L for DANO and 30 and 100 ng/L for the rest), each one injected by triplicate (instrumental replicates) on the same day. Intermediate precision was assessed with a similar procedure, but the five samples were analyzed in different days. The results, expressed as relative standard deviation (RSD) of peak areas and migration times, are given in Table 2. As can be observed, acceptable precision was obtained in all cases.
Recovery studies
In order to check the trueness of the proposed methodology, recovery experiments were carried out in three different types of water: two different wells water (mountain well and irrigated-land well) and one tap water.
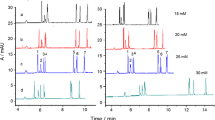
Water samples were spiked with a mixture of the six studied FQs at different levels (10 and 40 ng/L for DANO and 30 and 100 ng/L for all the rest). Each level was prepared in triplicate, submitted to the proposed SPE method, and injected by duplicate. Blank samples were also analyzed, and in none of them, FQ residues were detected. The results are shown in Table 3. Typical electropherograms of a treated blank sample and a spiked water sample are shown in Fig. 2.

Electropherograms at optimum conditions of a blank sample of mountain well water, b spiked mountain well water, c spiked irrigated-land well water, and d spiked tap water. Spiked analyte concentrations are 100 ng/L for 1 OFLO, 2 LOME, 3 NOR, 5 ENRO, and 6 SARA and 40 ng/L for 4 DANO
As can be seen, the proposed method provides satisfactory results in terms of trueness and precision, so method accuracy for the analysis of these samples was demonstrated.
Concluding remarks
A simple, sensitive, and selective method for the determination of six FQs in different water samples has been developed, showing LIF as a very attractive detection technique in CE. LOQs were in the very low ng/L and could be easily improved to pg/L range by just increasing the sample volume or diminishing the volume of recomposition solvent used after SPE process. This method is one of the most sensitive proposed for the analysis of these compounds, being able to determinate six analytes simultaneously. The recoveries and precision obtained are good enough and show the suitability of this procedure for the monitoring of FQ residues in water samples and for providing data of the occurrence of these compounds in the environment.
References
Halling-Sorense B, Nielson SN, Lanzky PE, Ingerslev LF, Holten Lutzhoft HC, Jorgensen SE (1998) Chemosphere 36:357–393
Ellis JB (2006) Environmental Pollut 144:184–189
Cha JM, Yang S, Carlson KH (2006) J Chromatogr A 1115:46–57
Andreu V, Blasco C, Picó Y (2007) Trends Anal Chem 26:534–556
García-Campaña AM, Gámiz-Gracia L, Lara FJ, del Olmo Iruela M, Cruces-Blanco C (2009) Application of capillary electrophoresis to the determination of antibiotics in food and environmental samples. Anal Bioanal Chem 395:967–986. doi:10.1007/s00216-009-2867-9
Hernández M, Borrull F, Calull M (2002) Electrophoresis 23:506–511
Zhao Y, Wang L, Li Y, Han YY, Du GL (2007) Gaodeng Xuexiao Huaxue Xuebao 28:62
Sun H, He P, Lv YK, Liang SX (2007) J Chromatogr B 852:145–151
Barrón D, Jiménez-Lozano E, Cano J, Barbosa J (2001) J Chromatogr B 759:73–79
Barrón D, Jiménez-Lozano E, Bailac S, Barbosa J (2002) J Chromatogr B 767:313–319
Barrón D, Jiménez-Lozano E, Bailac S, Barbosa J (2003) Anal Chim Acta 477:21–27
Beltrán JL, Jiménez-Lozano E, Barrón D, Barbosa J (2004) Anal Chim Acta 501:137–141
Jiménez-Lozano E, Roy D, Barrón D, Barbosa J (2004) Electrophoresis 25:65–73
Sun H, Zhao W, He P (2008) Chromatographia 68:425–429
Horstkötter C, Jiménez-Lozano E, Barrón D, Barbosa J, Blaschke G (2002) Electrophoresis 23:3078–3083
Ferdig M, Kaleta A, Vo TDT, Buchberger W (2004) J Chromatogr A 1047:305–311
Juan-García A, Font G, Picó Y (2006) Electrophoresis 27:2240–2249
Lara FJ, García-Campaña AM, Alés-Barrero F, Bosque-Sendra JM, García-Ayuso LE (2006) Anal Chem 78:7665–7673
Lara FJ, García-Campaña AM, Alés-Barrero F, Bosque-Sendra JM (2008) Electrophoresis 29:2117–2125
Wang L, Wu X, Xie Z (2005) J Sep Sci 28:1143–1148
Zhou X, Xing D, Zhu D, Tang Y, Jia L (2008) Talanta 75:1300–1306
Horstkötter C, Blaschke G (2001) J Chromatogr B 754:169–178
Moller J-G, Stass H, Heinig R, Blaschke G (1998) J Chromatogr B 716:325–334
Bannenfeld KH, Stass H, Blaschke G (1997) J Chromatogr B, 692:453–459
Barrón D, Jiménez-Lozano E, Barbosa J (2000) Anal Chim Acta 415:83–93
Schmitt-Kopplin P, Burhenne J, Freitag D, Spiteller M, Kettrup A (1999) J Chromatogr A 837:253–265
Barrón D, Jimenez-Lozano E, Barbosa J (2001) J Chromatogr A 919:395–406
Bailac S, Ballesteros O, Jiménez-Lozano E, Barrón D, Sanz-Nebot V, Navalón A, Vilchez JL, Barbosa J (2004) J Chromatogr A 1029:145–151
Rodríguez E, Moreno-Bondi MC, Marazuela MD (2008) J Chromatog A 1209:136–144
Hernández M, Borrull F, Calull M (2000) J Chromatogr B 742:255–265
Garcés A, Zerzanova A, Kucera R, Barrón D, Barbosa J (2006) J Chromatogr A 1137:22–29
Golet EM, Alder AC, Hartmann A, Ternes TA, Giger W (2001) Anal Chem 73:3632–3638
Prat MD, Benito J, Compañó R, Hernández-Arteseros JA, Granados M (2004) J Chromatogr A 104:27–33
Reverte S, Borrull F, Pocurull E, Marce RM (2003) J Chromatogr A 1010:225–232
Acknowledgments
The authors thank the financial support from the Ministry of Science and Innovation (Ministerio de Ciencia e Innovación, Project Ref CTM2006-06363-TECNO). Manuel Lombardo Agüí thanks the University of Granada for a predoctoral grant (Plan Propio UGR).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lombardo-Agüí, M., Gámiz-Gracia, L., García-Campaña, A.M. et al. Sensitive determination of fluoroquinolone residues in waters by capillary electrophoresis with laser-induced fluorescence detection. Anal Bioanal Chem 396, 1551–1557 (2010). https://doi.org/10.1007/s00216-009-3309-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-3309-4