
Abstract
Analysis of biomarkers in exhaled breath condensate (EBC) is a non-invasive method for investigating the effects of different diseases or exposures, on the lungs and airways. N ɛ-carboxymethyllysine (CML) is an important biomarker of advanced glycation end products (AGEs). A method has been developed for simultaneous determination of CML and its precursor, the amino acid lysine, in exhaled breath condensate (EBC). After addition of labelled internal standards (d-4-CML; d-4-lysine), the EBC was concentrated by freeze-drying. Separation and detection of the analytes were performed by hydrophilic-ion liquid chromatography coupled with tandem mass-spectrometric detection (HILIC–MS–MS). The limits of quantification were 10 pg mL−1 EBC and 0.5 ng mL−1 EBC for CML and lysine, respectively. The relative standard deviation of the within-series precision was between 2.8 and 7.8% at spiked concentrations between 40 and 200 pg mL−1 for CML and between 6 and 20 ng mL−1 for lysine. Accuracy for the analytes ranged between 89.5 and 133%. The method was used for the analysis of EBC samples from ten healthy persons from the general population and ten persons receiving dialysis. CML and lysine were detected in all EBC samples with median values of 19 pg mL−1 CML and 11.9 ng mL−1 lysine in EBC of healthy persons and 25 pg mL−1 CML and 9.5 ng mL−1 lysine in EBC of dialysis patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Analysis of exhaled breath condensate (EBC) is a non-invasive method for studying the composition of the airway-lining fluid of the lung. Besides volatile substances, for example carbon monoxide, nitric oxide, or hydrocarbons, exhaled breath also consists of a liquid phase—aerosol droplets containing non-volatile substances [1]. EBC is collected by cooling expired air through a special condenser at −20 °C. It is a unique matrix for analysis of inflammatory markers or markers of oxidative stress and enables study of the early effects of different diseases or exposures on the lung and upper airways [2].
Advanced glycation end products (AGEs) are formed by reaction of reducing sugars (e.g. glucose) or sugar degradation products and the amino groups of proteins (e.g. lysine) or other macromolecules. Further reactions and oxidation during the Maillard reaction in vivo might lead to crosslinks, which can alter the functional properties of different proteins or tissues. As AGEs accumulate in different tissues, this process might be connected with diseases such as diabetic nephropathy, atherosclerosis, or Alzheimer’s disease [3]. In-vitro experiments showed that AGEs might also contribute to the pathogenesis of pulmonary fibrosis, making their analysis in EBC a possible biomarker of pulmonary disease [4, 5].
Nɛ-carboxymethyllysine (CML) is a well-established analytical indicator of AGEs. Increased concentrations of CML have been identified in the blood and tissues of patients with diabetes mellitus, Alzheimer’s disease, atherosclerosis, and liver cirrhosis [5–8]. High serum levels of AGEs have also been found in dialysis patients [9] and recent investigations have even revealed the presence of a DNA glycation adduct in tissues of patients with diabetic nephropathy [3].
Immunochemical assays have usually been used for analysis of CML in human body fluids or tissues [6–10]; only a few research studies have applied instrumental analytical methods, for example GC–MS [11, 12], HPLC coupled with fluorescence detection [13, 14], or tandem mass spectrometry [15].
Our own investigations revealed, for the first time, the presence of CML in the EBC of healthy subjects and patients with diseases such as chronic obstructive pulmonary disease (COPD), diabetes, or pneumonia by use of a highly specific LC–MS–MS method [16]. This method was, however, found to be inappropriate for the determination of lysine as a precursor of CML in EBC. The simultaneous quantification of both compounds would be advantageous to investigate whether the ratio of the concentration of CML to that of its precursor lysine in EBC could be a more specific indicator of AGE formation in the lung and airways.
To achieve simultaneous quantification of CML and lysine we decided to use hydrophilic-interaction liquid chromatography (HILIC). HILIC is a mode of chromatography (similar to normal-phase chromatography) that uses a combination of a hydrophilic stationary phase and a hydrophobic, organic mobile phase [17]. In contrast with classic “normal”-phase chromatography, water-miscible solvents (acetonitrile, methanol) are used for HILIC; the aqueous phase used in gradients is the strongest mobile phase [18]. Hydrophilic interaction, ion-exchange mechanisms, and reversed-phase retention mechanisms result in an order of elution of the analytes from the least to most polar [17, 18]. The retention behaviour of the analytes in HILIC is, therefore, also affected by pH and buffer concentration of the aqueous mobile phase and the type of stationary phase [17]. HILIC has been successfully used for separation of carbohydrates and peptides [17, 19–21]. Further development of different stationary phases has led to increasing application of this technique in analytical chemistry, e.g. in analysis of polar food compounds [22] and amino acids in collagen hydrolysates [23], and for screening for polar metabolites in body fluids [24].
We have therefore developed and validated a very sensitive and reliable method, using HILIC coupled to tandem mass spectrometry, for simultaneous, quantitative determination of the advanced glycation end product N ɛ-carboxymethyllysine and its precursor lysine in exhaled breath condensate. The method has been applied to analysis of the EBC of ten healthy persons of the general population and the EBC of ten patients with diabetic nephropathy receiving dialysis treatment. With this preliminary study we intended to investigate the extent to which high blood levels of CML (as expected in patients with diabetic nephropathy) might also result in high EBC levels of CML, to evaluate possible confounding factors for interpretation of EBC analyses.
Experimental
Reagents and standards
Nɛ-carboxymethyllysine (CML) was purchased from SyMO-Chem (Eindhoven, The Netherlands). l-Lysine monohydrochloride was purchased from Fluka. d-4-l-lysine dihydrochloride [4,4,5,5–2H4] (isotopic purity: ∼90%) was from CDN Isotopes. Isotopically labelled CML was synthesized from d-4-l-lysine as described elsewhere [15, 16].
Ammonium acetate, acetic acid, and formic acid, all of the highest analytical grade available, were supplied by Merck (Darmstadt, Germany). Acetonitrile (HPLC grade) was purchased from J.T. Baker (Germany).
EBC sample collection
Exhaled breath condensate was collected using the Ecoscreen condenser from Jaeger (Höchberg, Germany). The patients, wearing a noseclip to ensure oral breathing, were instructed to breathe tidally through a mouthpiece connected to a condenser until a volume of 200 L exhaled air was achieved (as measured by the EcoVent flow meter). The exhaled air entered and left the condensing chamber (maintained at −20 °C ) through one-way valves, keeping the chamber closed throughout the process. The resulting EBC volumes ranged from 1.2 to 3.7 mL. EBC was divided into portions stored at −80 °C until analysis.
Standard preparation
Two separate starting solutions were prepared containing 1 mg mL−1 CML and lysine in bi-distilled water. The starting solutions of lysine (1 mL) and CML (10 μL) were then placed in a 10-mL volumetric flask and diluted to volume with water. This stock solution contained 100 μg mL−1 lysine and 1 μg mL−1 CML.
This stock solution (100 μL) was placed in a 10-mL volumetric flask and diluted to volume with bi-distilled water to serve as a working solution for preparation of calibration standards in the range of 10–1000 pg mL−1 CML and 1–100 ng L−1 lysine in bidistilled water. The calibration standards were divided into 1.1-mL portions and stored at −80 °C until analysis.
A starting solution of the internal standard containing 1 mg mL−1 of d-4-lysine was prepared. A solution of ∼400 μg mL−1 d-4-CML in water (as verified by LC–MS–MS), from the synthesis of labelled CML, served as starting solution. This starting solution (100 μL) was placed in a 10-mL volumetric flask and diluted to volume with bidistilled water to serve as a stock solution of labelled CML (c∼4 μg mL−1).
The starting solution of d-4-lysine (20 μL) and 200 μL of the stock solution of labelled CML were placed in a 20-mL volumetric flask and diluted to volume with bidistilled water to serve as an internal standards working solution (d-4-lysine 1 μg mL−1; d-4-CML ∼40 ng mL−1). All starting solutions and stock solutions were stored at −20 °C and the working solution of the internal standards was stored at +4 °C.
Sample preparation and calibration
For sample preparation frozen EBC samples or calibration standards were thawed and equilibrated to room temperature. The sample (1 mL) was then transferred to a 1.8-mL crimp-cap vial and 30 μL working solution of the internal standards was added. The sample was stored at −80 °C for approximately 30 min to ensure deep freezing and then placed in a freeze dryer. The freeze-drying process was conducted at −15 °C and at a pressure of approximately 10−2–10−3 mbar.
After complete drying of the samples (this process is preferably performed overnight) the residue was dissolved in 55 μL starting eluent (5 mmol L−1 ammonium acetate buffer, pH 4–acetonitrile, 40:60, v/v). This solution was then transferred to microvials (200 μL) and sealed with crimp caps. This solution (45 μL) was injected into the LC–MS–MS system for quantitative analysis.
Linear calibration plots were obtained by plotting the quotients of the peak areas of the analyte and the corresponding labelled internal standard as a function of the spiked concentration. These plots were used to determine the unknown concentrations of the analytes in EBC samples. A blank value consisting of bidistilled water was also included in every analytical series.
LC–ESI–MS–MS analysis
LC analysis was performed with Hewlett–Packard HP 1100 series equipment including a binary gradient pump, degasser, and autosampler. Chromatographic separation was performed on a ZIC-HILIC column (100 mm × 4.6 mm, particle size 3.5 μm) and a ZIC-HILIC guard column (20 mm × 2.1 mm, particle size 5 μm) from Sequant (Haltern am See, Germany). A precolumn filter (0.5 μm, Supelco) was also installed to protect the column from particulate matter possibly originating from samples.
The solvents used to prepare the mobile phase were 5 mmol L−1 aqueous ammonium acetate buffer, adjusted to pH 4 with acetic acid (component A) and 0.025% (v/v) formic acid in acetonitrile (component B). The initial mobile phase composition was 40% A, which was held for 1 min after injection of the sample. A linear gradient was then started in which the proportion of A was increased from 40% to 80% in 10 min. This composition was held for 5 min before the gradient was restored to the initial conditions within 8 min and held for 5 min. The mobile phase flow rate was constant at 0.5 mL min−1.
Mass spectrometric analysis was performed using a Sciex API 3000 tandem mass spectrometer (MS–MS) with an electrospray ionization (ESI) source working in the positive-ion mode. Instrument control, data acquisition, and evaluation were performed with Analyst 1.3.2 software from Perkin–Elmer. The operating conditions were: applied ESI needle potential +5000 V; nitrogen was used as nebulizer gas (at a pressure of 12 psi = 82.7 hPa), as turbo heater gas (500 °C), and as collision gas. Collision gas and curtain gas were set at 10 and 8 instrument units, respectively. The optimum collision energy and substance-specific mass spectrometric conditions for multiple reaction monitoring (MRM), and the retention times of the compounds, are listed in Table 1. The product-ion mass spectra for CML and lysine are shown in Fig. 1a and b, with the fragmentation patterns for both compounds.

Product-ion mass spectra of CML (a) and lysine (b). Spectra were obtained in ESI positive-ion mode by collision-induced fragmentation of the [M+H]+ precursor ions of m/z = 205 for CML and m/z = 147 for lysine. The spectra were recorded by direct infusion of a solution of 100 ng mL−1 of the analytes into the source of the mass spectrometer at a flow rate of 20 μL min−1
Study subjects
EBC was collected from ten healthy adults (seven males, three females) ranging in age from 34 to 64 years with a median age of 44 years. Four persons reported they were smokers, smoking 1–20 cigarettes per day. EBC was also collected from ten adults with diabetic nephropathy receiving dialysis (eight males, two females), ranging in age from 26 to 73 years with a median age of 48 years. Three persons reported they were smokers, smoking 4–10 cigarettes per day.
Results
Validation of the analytical method
To investigate the effect of the matrix on the calibration of CML and lysine we prepared calibration standards by spiking pooled EBC with CML in the range 10–1000 pg mL−1 and with lysine in the range 1–100 ng L−1, processed these standards together with the aqueous calibration standards, and compared the slopes and linearity of the calibration plots in water and EBC. The calibrations were almost identical; deviation was a few percent only and correlation coefficients were greater than r = 0.995 for both calibration plots. The slopes for CML in water and EBC were y = 0.00083x and y = 0.00086x, respectively. The slopes for lysine in water and EBC were y = 0.082x and y = 0.086x, respectively. The matrix was therefore proved to have no effect on the calibration. Also, because calibration using pooled EBC resulted in quite high background values for both CML (70 pg mL−1) and lysine (80 ng mL−1), accurate quantification at low concentrations would have been hampered.
We therefore decided to prepare quality-control material by spiking water with different amounts of the analytes in question. Because the volume of EBC samples after collection was usually very low (1–3 mL), however, it was almost impossible to collect adequate volumes of real samples for quality-control purposes. Two concentrations were prepared: the low-concentration quality-control material was spiked with 40 pg mL−1 CML and 6 ng mL−1 lysine; the high-concentration quality-control material was spiked with 200 pg mL−1 CML and 20 ng mL−1 lysine, respectively. The spiked quality-control material was divided into 1.1-mL volumes and stored at −80 °C until analysis.
Within-series precision was determined by analysing both quality-control samples eight times in one analytical run. Between-day precision was determined by analysing both quality-control samples within six different analytical series. The accuracy of the method was determined by comparing results from these analyses with the concentrations added.
For further estimation of potential matrix influences we also conducted accuracy experiments using four individual EBC samples from persons of the general population. The EBC samples were analysed unspiked and spiked with 100 pg mL−1 and 10 ng mL−1 CML and lysine, respectively. Individual accuracy was calculated by analysing the spiked and unspiked EBC samples and comparing the results with the amounts of the analytes added.
Recovery of the analytes was calculated by comparing the peak areas of processed aqueous standards (n = 3) containing 40 pg mL−1 CML and 6 ng mL−1 lysine (without addition of labelled internal standards) with injections of solutions of the analytes in starting mobile phase (taking into consideration the concentration factor of the method).
The results of these validation experiments are summarised in Table 2.
The limit of quantification (LOQ), defined as a signal to noise ratio of 5 for the registered ion fragments (Table 1) was estimated to be 10 pg mL−1 for CML. Because the processed blank values contained concentrations of lysine of approximately 1–3 ng mL−1 (see section “Sources of error”, below), the LOQ of lysine was estimated to be 0.5 ng mL−1 (taking into consideration the subtracted blank value). For all processed EBC samples lysine values were well above this level. A blank value for CML was not observed in our study.
Results of EBC analyses
The method presented here was used to examine the concentration of CML and lysine in EBC of healthy subjects and of patients receiving dialysis. The purpose of this preliminary study was to investigate whether higher concentrations of CML, or a higher ratio of CML to its precursor lysine, could be found the patients suffering from diabetic nephropathy than in the control group.
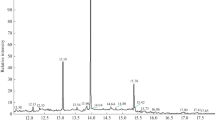
The results of these analyses, and significance levels, are summarized in Table 3. Figure 2 shows a chromatogram obtained from the processed EBC of a person receiving dialysis. The relationship between CML and lysine in EBC is graphically displayed in Fig. 3, including individual data points for both groups. Smoking habits did not significantly effect levels of CML and lysine (or the CML/lysine ratio) either in the individual groups or on the basis of the whole collective (smokers compared with non-smokers, two-sided t-test; CML p = 0.48; lysine p = 0.25; ratio CML/lysine p = 0.75). Sex differences between concentrations of CML and lysine (or the ratio CML/lysine) were not observed. The groups were, however, very small for correct statistical evaluation of these issues.

MRM chromatogram obtained from the processed EBC of a person receiving dialysis, showing the ion transitions for CML (m/z 205→m/z 84), the labelled internal standard d4-CML (m/z 209→m/z 88), lysine (m/z 147→84), and the labelled internal standard d4-lysine (m/z 151→88). The concentration of CML in EBC was 135 pg mL−1 and the concentration of lysine in EBC was 24.2 ng mL−1. The concentration of the spiked internal standards was ∼1 ng mL−1 and 30 ng mL−1 for d-4-CML and d-4-lysine, respectively. Mass spectrometric conditions are given in Table 1

Relationship between CML and lysine in EBC of healthy controls (n = 10) and patients receiving dialysis (n = 10)
Sources of error
The blank analysed in every analytical series usually contained small amounts of lysine (1–3 ng mL−1), because of impurities in the labelled internal standard d-4-lysine (isotopic purity ∼90%). This blank value was subtracted from the values obtained from analysis of unknown samples.
Discussion
Because of their high polarity, analysis of CML and lysine by reversed-phase HPLC without derivatisation (e.g. to fluorescent derivatives) is a challenging task, because both compounds are usually almost unretained on common reversed-phase columns and because of strong disturbing peaks or ion-suppression effects (depending on the matrix). In our previous work we managed to achieve good retention of CML on a reversed-phase C8 column by using nonafluoropentanoic acid (NFPA) as ion-pairing reagent [16]. This procedure failed for determination of the precursor lysine, however. Furthermore, despite its good compatibility with LC–MS, the high concentration of NFPA used led to a mobile phase of very low pH (approx. 2), which resulted in rapid deterioration of the reversed-phase column.
To avoid derivatisation in the determination of both CML and lysine while still using tandem mass spectrometry as the most specific and sensitive means of detection, we decided to use hydrophilic-ion liquid chromatography (HILIC) as an alternative. The zwitterionic properties of the ZIC-HILIC column used in our study make it especially suitable for determination of amino acids. Drawbacks of this technique are comparatively long chromatographic runs, because the manufacturer of the column recommends that gradient elution should not exceed 5% per min and re-equilibration of the column should be sufficient before the next injection, resulting in a total run-time of 29 min.
The column used for this study proved very stable and robust, with no remarkable shift of retention times or deterioration of the separation. It has already been successfully used for determination of other biomarkers in EBC.
Analysis of EBC samples was complicated by the small sample volume available for analysis coupled with the need to detect very low concentrations. Because the sample volume is limited, freeze drying of EBC was chosen as the only step of sample preparation and concentration. Because EBC contains little matrix and few disturbing compounds, further clean-up of the samples was not necessary. The peaks of the analytes were clearly resolved from other compounds, as shown in Fig. 2. The good recovery of both analytes documented in Table 2 reveals this procedure results in no significant analyte losses and is a very gentle and easy mode of sample concentration.
As shown in Table 3, the precision and accuracy of the method can be described as excellent. All possible analyte losses and matrix effects were efficiently compensated by use of labelled internal standards for both analytes. The intraday variation, expressed as relative standard deviation was less than 8% and the interday variation was shown to be less than 5%. The quality-control material was used over the method-development and application period, which lasted approximately 2 months, and no tendency of the analyte to degrade was observed. Three EBC samples were, furthermore, analysed twice over a period of 2 months and there was no significant decrease in analyte concentrations—deviations were within the range of method characteristics (data not shown).
The LOQ of our method, 10 pg mL−1 for CML, is well below the LOD of other published instrumental methods for determination of CML in body fluids. Petrovic et al. analysed CML and its structural analogue N-ɛ-carboxyethyllysine (CEL) in human urine by use of GC–MS and achieved an LOD of 20 ng mL−1 [12]. Friess et al. used HPLC with fluorescence detection for the same purpose and achieved an LOD of 5 ng mL−1 [13]. HPLC with fluorescence detection was used by van de Merbel et al. for determination of CML in plasma; the LOD was 200 ng mL−1 [14]. Detection of CML in plasma by LC–MS–MS resulted in an LOD of 4 ng mL−1 [15]. It must, however, be stated that exhaled breath condensate is a sample with low matrix effects that is not comparable with plasma or urine, which require extensive sample clean-up. Consequently, the LODs achieved in the different methods listed above are hard to compare. Our previous method achieved an LOD of 5 pg mL−1 for determination of CML in EBC which is in good accordance with the LOQ of our current method.
When our method was used for analysis of EBC samples of healthy controls and patients receiving dialysis we were able to determine CML and its precursor lysine in all of the EBC samples analysed. As far as we are aware this is the first time the amino acid lysine has been quantified in exhaled breath condensate.
The results showed there were no significant differences between CML and lysine levels in the two groups (Table 3). This is in agreement with results from our first, preliminary study, in which elevated levels of CML were not observed in patients with pulmonary diseases–exception for a patient suffering from pneumonia, for whom the level of CML was extremely high [16].
It is worthy of note that our results for both CML and lysine vary over a comparatively wide concentration range, both in controls and patients. Dilution by water vapour of the droplets generated in the respiratory tract varies substantially, as already observed by other authors [25]. There is, consequently, an urgent need to adjust results from EBC analysis to take this effect into consideration. Non-volatile substances, for example amino acids or peptides, which are certainly present in the airway-lining fluid of the lung, would be very suitable for correcting for the extent of dilution of the lining fluid in the collected EBC. There is, therefore, strong evidence that accurate quantification of the amino acid lysine may be a powerful tool for compensating for different dilution effects that have hitherto hampered interpretation of results from EBC analysis.
We therefore examined the relationship between CML and its precursor lysine in the analysed EBC samples. A trend toward higher CML/lysine ratios is evident for patients undergoing dialysis, although this trend does not reach the 5% level of significance (Table 3). With regard to the relationship between both markers (Fig. 3), CML values in EBC tend to be lower for control persons, with the exception of one person (58 years, f, non-smoker), for whom the CML level was extraordinarily high. An explanation of these results could not be found in the anamnestic data collected in the course of our study. In contrast, the amount of CML in the EBC of a person with diabetic nephropathy (43 years, m, non-smoker) was quite low, despite quite high lysine levels. Because smoking habits could not explain these observations, other exogenous factors might be the reason.
This indicates that further investigations, with more subjects, in combination with thorough acquisition of anamnestic data with regard to individual life-style habits (beyond smoking habits) may be necessary to evaluate the viability of CML in EBC as a biomarker of pulmonary oxidative stress.
Conclusions
In summary, we have developed and validated a new LC–MS–MS method for simultaneous specific determination of the advanced glycation end product N ɛ-carboxymethyllysine and its precursor lysine in exhaled breath condensate. The method presented here is precise, accurate, and very sensitive. The data for accuracy and precision can be described as excellent, even at very low concentrations. Matrix effects were proved not to affect the results. Because of its simple and rapid sample preparation, the method is very practicable and suitable even for examination of larger series. The limit of quantification of 10 pg mL−1 for CML and 0.5 ng mL−1 for lysine was sufficiently low for determination of the analytes in all samples investigated so far.
The application of this method has provided the first data so far on CML and lysine levels in exhaled breath condensate of healthy subjects and patients receiving dialysis. Simultaneous determination of the amino acid lysine in EBC might be an appropriate means of compensating for different dilution effects that occur during collection of EBC samples.
References
Montuschi P (2005) Clin Chim Acta 356:22–34
Mutlu GM, Garey KW, Robbins RA, Danziger LH, Rubinstein I (2001) Am J Respir Crit Care Med 164:731–737
Li H, Nakamura S, Miyazaki S, Morita T, Suzuki M, Pischetsrieder M, Niwa T (2006) Kidney Int 69:388–392
Kasper M, Schinzel R, Niwa T, Munch G, Witt M, Fehrenbach H, Wilsch-Bräuninger M, Pehlke K, Hofer A, Funk RH (1999) Biochem Biophys Res Commun 261:175–182
Matsuse T, Ohga E, Teramoto S, Fukayama M, Nagai R, Horiuchi S, Ouchi Y (1998) J Clin Pathol 51:515–519
Hwang JS, Shin CH, Yang SW (2005) Diabetes Obes Metab 7:263–267
Kaufmann E, Boehm BO, Süssmuth SD, Kientsch-Engel R, Sperfeld A, Ludolph AC, Tumani H (2004) Neurosci Lett 371:226–229
Nerlich AG, Schleicher ED (1999) Atherosclerosis 144:41–47
Zhang X, Frischmann M, Kientsch-Engel R, Steinmann K, Stopper H, Niwa T, Pischetsrieder M (2005) Clin Chem Lab Med 43:503–511
Yagmur E, Tacke F, Weiss C, Lahme B, Manns MP, Kiefer P, Trautwein C, Gressner AM (2006) Clin Biochem 39:39–45
Ahmed MU, Brinkmann Frye E, Degenhardt TP, Thorpe SR, Baynes JW (1997) Biochem J 324:565–570
Petrovic R, Futas J, Chandoga J, Jakus V (2005) Biomed Chromatogr 19:649–654
Friess U, Waldner M, Wahl HG, Lehmann R, Haring HU, Voelter W, Schleicher E (2003) J Chromatogr B 794:273–280
van de Merbel NC, Mentink CJAL, Hendiks G, Wolffenbuttel BHR (2004) J Chromatogr B 808:163–168
Teerlink T, Barto R, Ten Brink HJ, Schalkwijk CG (2004) Clin Chem 50:1222–1228
Gonzalez-Reche LM, Kucharczyk A, Musiol AK, Kraus T (2006) Rapid Commun Mass Spectrom 20:2747–2752
Alpert AJ (1990) J Chromatogr 449:177–196
Grumbach ES, Wagrowski-Diehl DM, Mazzeo JR, Alden B, Iraneta PC (2004) LC–GC North America 22:1010–1023
Alpert AJ, Shukla M, Shukla AK, Zieske LR, Yuen SW, Ferguson MAJ, Mehlert A, Pauly M, Orlando R (1994) J Chromatogr A 676:191–202
Lindner H, Sarg B, Meraner C, Helliger W (1996) J Chromatogr A 743:137–144
Lindner H, Sarg B, Helliger W (1997) J Chromatogr A 782:55–62
Schlichtherle-Cerny H, Affolter M, Cerny C (2003) Anal Chem 75:2349–2354
Langrock T, Czihal P, Hoffmann R (2006) Amino Acids 30:291–297
Godejohann M (2006) J Chromatogr A DOI 10.1016/j.chroma.2006.10.053
Effros RM, Hoagland KW, Bosbous M, Castillo D, Foss B, Dunning M, Gare M, Lin W, Sun F (2002) Am J Respir Crit Care Med 165:663–669
Acknowledgements
The authors would like to thank Dr. Mariano Gonzalez-Reche and Anna Kucharczyk for their valuable work and for providing the standards of CML and d-4-CML. We would also like to thank Kathy Bischof for the kind review of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schettgen, T., Tings, A., Brodowsky, C. et al. Simultaneous determination of the advanced glycation end product N ɛ-carboxymethyllysine and its precursor, lysine, in exhaled breath condensate using isotope-dilution–hydrophilic-interaction liquid chromatography coupled to tandem mass spectrometry. Anal Bioanal Chem 387, 2783–2791 (2007). https://doi.org/10.1007/s00216-007-1163-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-007-1163-9