
Abstract
A semi-automated liquid chromatography–tandem mass spectrometry (LC/MS/MS) method was developed for the simultaneous quantification of the antifungal drug itraconazole (ITZ) and its coactive metabolite hydroxyitraconazole (OH-ITZ) in human plasma. The plasma samples underwent liquid–liquid extraction (LLE) in 2.2 mL 96 deepwell plates. ITZ, OH-ITZ and the internal standard (IS) R51012 were extracted from plasma, using a mixture of acetonitrile (ACN) and methyl t-butyl ether (MTBE) as the organic solvent. This specific mixture, due to its composition, had a significant impact on the performance of the assay. All liquid transfer steps, including preparation of calibration standards and quality control samples as well as the addition of the IS, were performed automatically using robotic liquid handling workstations for parallel sample processing. After vortexing, centrifugation and freezing, the supernatant organic solvent was evaporated. The analytes and IS were dissolved in a small volume of a reconstitution solution, an aliquot of which was analyzed by combined reversed phase LC/MS/MS, with positive ion electrospray ionization and a TurboIonSpray interface, using multiple reactions monitoring (MRM). The method was shown to be sensitive and specific to both ITZ and OH-ITZ, it revealed excellent linearity for the range of concentrations 2–500 ng mL−1 for ITZ and 4–1000 ng mL−1 for OH-ITZ, it was very accurate and it gave very good inter- and intra-day precisions. The proposed high-throughput method was employed in a bioequivalence study after per os administration of two 100 mg tablets of ITZ, and it allowed this study to be completed in under four days.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
ITZ is an oral triazole antifungal agent with a broad spectrum of activity against most human fungal pathogens. It acts by inhibiting the biosynthesis of ergosterol, a major component of the cell membrane of yeast and fungal cells. The drug is extensively metabolized in the liver via a saturable cytochrome P-450 (CYP)-mediated process. Among the large number of metabolites of ITZ, OH-ITZ, formed by side-chain hydroxylation of ITZ, is biologically active and typical plasma concentrations are usually higher than those of ITZ.
Numerous HPLC methods [1–4] have been described for the quantitative determination of ITZ and its hydroxy metabolite in human plasma. Most of them employ traditional analysis techniques, namely LC-UV or LC-fluorescence, which often lack the combined sensitivity and selectivity needed to analyze complex mixtures. Liquid chromatography coupled with tandem mass spectrometry is currently the method of choice for the quantitative determination of drugs [5–7] in complex biological matrices such as biological fluids and tissues because of its specificity, sensitivity and high throughput. Few methods have been reported for the determination of both ITZ and OH-ITZ employing mass spectrometric detectors. Two of them [8, 9] use single-stage quadrupole instruments. Recently, Vogeser et al [10] have reported a LC/MS/MS method for the determination of both ITZ and its active metabolite with automated solid-phase extraction.
Plasma protein precipitation (PPP), liquid–liquid extraction (LLE) and solid phase extraction (SPE) are the sample preparation techniques most commonly used to process plasma and tissue samples. All three of them have been employed for the treatment of biological samples prior to the determination of ITZ. Protein precipitation has the potential to be significantly less time-consuming. SPE is the technique most amenable to automation since it is commercially available in the 96-well plate format and can be also adapted for direct injection. However, LLE generally provides cleaner extracts than the previously mentioned techniques, as evidenced by fewer matrix effects and less tendency for backpressure build-up in the chromatographic column as more samples are injected [11]. Moreover, it can be quickly developed and applied to most categories of pharmaceutical compounds. On the other hand, the drawback of this technique is that it requires a large amount of organic solvent and is less amenable to automation. Cross-well leaking occurs during mixing when 96-well plates are used, while the use of low volumes of sample and solvent may result in inadequate sensitivity. As is evident from the above, automation of LLE processes in order to match the throughput of LC/MS/MS analysis constitutes a challenge. The introduction of several liquid handling workstations for the parallel processing of samples has greatly facilitated this task, resulting in the development of several automated methodologies for the determination of drugs and their metabolites in biological fluids over the last few years [12–17].
In the present study we report the development and validation of the first automated high-throughput method for the determination of ITZ and its active metabolite, OH-ITZ. All liquid transfer steps, including preparation of calibration standards and quality control samples (QCs), transfer of study samples and addition of internal standard were performed automatically on the Multiprobe II EX-HT. Tomtec Quadra 96 was programmed to perform the addition of the organic solvent as well as the transfer of the supernatant organic layer after extraction of the analytes into a new 96-well plate. Following the example of Xue et al [18], a small amount of acetonitrile containing the internal standard was added prior to the addition of methyl t-butyl ether, which was used as the extraction solvent as well as for protein precipitation. The extracted samples, after evaporation and reconstitution with mobile phase, were injected onto a reversed phase LC system and analyzed using a mass spectrometer interfaced to a Turbo Ion Spray inlet with multiple reaction monitoring (MRM) in the positive ionization mode. The proposed method enabled the automated high-throughput and reliable determination of ITZ and OH-ITZ in a bioequivalence study after per os administration of two 100 mg tablets in 28 healthy volunteers.
Experimental
Chemicals and reagents
ITZ, OH-ITZ and the compound R51012 (a dimethyl homolog of itraconazole; (±)-cis-4-[4-[4-[4-[[2-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]-methoxy]phenyl]-1-piperazinyl]phenyl]-2,4-dihydro-2-(1-methylpropyl)-3H-1,2,4-triazol-3-one]), used as the IS, were obtained from HELP (Athens, Greece). Methanol, acetonitrile (both HPLC grade), methyl t-butyl ether (analysis grade) were obtained from Sigma-Aldrich (Athens, Greece). Glacial acetic acid (analysis grade) was purchased from Metrolab (Athens, Greece). All aqueous solutions and buffers were prepared using deionized and doubly distilled water (Resistivity >18 MΩ) from a Millipore Milli-Q Plus System (Malva, Athens, Greece). Pooled human control plasma (heparinized) was kindly donated from Ippokrateio Hospital (Athens, Greece).
Instrumentation
A PerkinElmer Multiprobe II HT-EX workstation (PerkinElmer, Downers Grove, IL, USA) equipped with an eight-tip robotic arm that can be moved along the x–, y– and z–axes was employed to transfer the plasma samples from 2 mL Eppendorf microfuge tubes (Lab Supplies, Athens, Greece) into 2.2 mL square 96 deep well plates (Sigma-Aldrich, Athens, Greece) as well as for the addition of the internal standard. The workstation was controlled by WinPrep Software. Conductive disposable tip-boxes (200 μL) were purchased from E&K Scientific Products (Cambell, CA, USA). A tipchute, reagent troughs and a tip flush/wash station were purchased from PerkinElmer. A Tomtec Quadra 96 model 320 robotic liquid handling system equipped with a 96-tip pipetting head (Bidservice, NJ, USA) was used to add the extraction solvent as well as to transfer the supernatant organic layer, after extraction, into a new 2.2 mL 96 deep well plate and for reconstitution of the samples after evaporation. The latter procedure was performed in a Zymark TurboVap 96-well format plate evaporator (Malva, Athens, Greece). Solvent evaporation occurs through nitrogen application, produced by an Agilent Nitrogen Generator (Duratec, Hockenheim, Germany), that receives air from a SF4 Air Compressor (Atlas Copco, Athens, Greece). An Eppendorf 5810 R (Bacakos, Athens, Greece) centrifuge that could accommodate 96-well plates as well as Eppendorf microfuge tubes was also utilized during sample preparation. The HPLC system included one Agilent 1100 series binary pump, a degasser and a column oven/cooler (Hellamco, Athens, Greece). The CTC PAL autosampler (Hellamco, Athens, Greece) could accommodate six 96 deep well plates, allowing automated measurement of a large number of samples. A PE Sciex API 3000 triple quadrupole mass spectrometer (AB/MDS-Sciex, Concord, Ontario, Canada) interfaced with the HPLC via a turbo ionspray source was used for the mass analysis and detection, operating under Analyst 1.4 software. Eppendorf deepwell mats used to cover the 96-well plates were purchased from Sigma-Aldrich. Finally, a 96-well plate vortex-mixer (MS1 Minishaker) was obtained from Metrolab (Athens, Greece).
Chromatographic and mass spectrometric conditions
The isocratic HPLC elution mobile phase was composed of 80% acetonitrile and 20% 10 mM ammonium acetate (v/v). The pH of the 10 mM ammonium acetate solution was adjusted to 3.5 by adding glacial acetic acid. A flow rate of 0.6 mL min−1 was used for sample analysis on a YMC-Pack ODS-A (C18) (Schermbeck, Germany) analytical column (50 mm×4.0 mm i.d). The column was maintained at ambient temperature (∼23 °C) whilst the autosampler temperature was set at 10 °C. The injection volume was 50 μl and the total run time was set at 2.0 min.
The turbo ionspray of the mass spectrometer was operated in the positive ionization mode. The tuning parameters were optimized for ITZ, OH-ITZ and R51012 by infusing a 100 ng mL−1 standard solution containing all three compounds in mobile phase at 20 μL min−1 via an external syringe pump (Harvard 11 plus) directly connected to the mass spectrometer. The turbo ionspray source temperature was maintained at 400 °C and the turbo ionspray voltage was set at 5000 V. The curtain gas was set at 11 (arbitrary units), and the declustering potential (DP) at 51 V for ITZ and its hydroxy metabolite and 46 V for the IS. The nebulizer gas (GS1) was set at 8 (arbitrary units) while the turbo ionspray gas (GS2) was set at 7 L min−1. The analytes were detected by monitoring the precursor→product ion transition using the multiple reaction monitoring (MRM) scan mode with 150 ms dwell time and 5 ms pause time for each transition. The MRM was performed at m/z 705.3→392.1 for ITZ, 721.3→408.2 for OH-ITZ and 733.3→460.3 for the IS (R51012). The collision-induced dissociation (CID) gas was set at 9 (arbitrary units) and the collision energy was set at 51, 51 and 47 V for ITZ, OH-ITZ and R51012 respectively. Data were acquired using Analyst 1.4 software.
Preparation of standard and quality control / method validation samples
Initially, separate stock solutions of ITZ and OH-ITZ were prepared by dissolving the accurately weighed respective reference compounds in methanol/water 50/50 (v/v) to yield final concentrations of 100 μg mL−1 (SI1, stock ITZ 1 and SOH-I1, stock OH-ITZ 1) and 2000 ng mL−1 (SI2 and SOH-I2). These solutions were then serially diluted with the same diluent mixture to achieve standard working solutions containing both ITZ and OH-ITZ at concentrations of 40, 100, 200, 1000, 2000, 4000 and 10,000 ng mL−1 for ITZ and 80, 200, 400, 2000, 4000, 10,000 and 20,000 ng mL−1 for OH-ITZ, respectively. A 100 μg mL−1 internal standard (IS1) working solution was also prepared in the same solvent mixture. Further dilution of IS1 by a factor of 200 with acetonitrile resulted in the IS2 working solution (500 ng mL−1).
Quality control/method validation (QC/MV) stock solutions (100 μg mL−1 and 2000 ng mL−1) were prepared from a separate weighing. Dilutions were used to prepare four levels of QC/MV working solutions at 40, 120, 1200 and 8000 ng mL−1 for ITZ and 80, 240, 2400 and 16,000 ng mL−1 for OH-ITZ, respectively. All of these solutions were stored at 4 °C and were brought to room temperature before use. All working solutions were transferred into 2 mL Eppendorf microfuge tubes. Calibration standards and QC/MV samples were prepared in the same biological matrix (human plasma) as the samples to be analyzed, utilizing the Multiprobe II HT-EX workstation, according to the following procedures:
-
Transfer 950 μL of human control plasma from trough to empty, properly labeled, Eppendorf microfuge tubes twice, to reach a final volume of 1900 μL.
-
Spike 100 μL of standard and QC/MV working solutions into the corresponding tubes containing the control plasma.
All Eppendorf tubes were capped and stored at –20 °C until required for assay.
The calibration curve consisted of a blank sample (matrix sample processed without IS), a zero sample (matrix sample processed with IS), and seven nonzero standards covering the expected range of concentrations to be quantified. The above spiking procedure resulted in final concentrations for the calibration standards of 2, 5, 10, 50, 100, 200 and 500 ng mL−1 for ITZ and 4, 10, 20, 100, 200, 500 and 1000 ng mL−1 for OH-ITZ. Similarly, the following concentration levels of QC/MV samples were achieved: MVL (2 ng mL−1 ITZ and 4 ng mL−1 OH-ITZ), MV1/QC1 (6 ng mL−1 ITZ and 12 ng mL−1 OH-ITZ), MV2/QC2 (60 ng mL−1 ITZ and 120 ng mL−1 OH-ITZ) and MV3/QC3 (400 ng mL−1 ITZ and 800 ng mL−1 OH-ITZ). QC and MV samples have two distinct purposes: the results for QC samples provide the basis for accepting or rejecting analytical runs, while the results for MV samples are used to calculate the bias and precision of the assay methodology.
Standard curves and acceptance criteria
To test the specificity of the method, blank samples of human plasma were obtained from six individual sources from Aretaieio hospital. Each blank sample was tested for interference using the proposed extraction procedure and chromatographic and mass spectrometric conditions. No significant interferences at the retention times of the analytes or IS were observed.
To define the relationship between concentration and response, a calibration curve containing seven nonzero standards, ranging from 2 to 500 ng mL−1 and from 4 to 1000 ng mL−1 for ITZ and OH-ITZ respectively, was prepared for each analytical run. This range was suitable for the bioequivalence study after per os administration of a single 100 mg capsule of ITZ. In addition, a zero and a blank plasma sample were also analyzed to confirm the absence of probable interferences. Peak area ratios of ITZ and OH-ITZ to IS were used for regression analysis. The calculated concentrations were determined from linear regression using 1/x 2 weighting and the equation y=a+bx, where x is the concentration of ITZ and OH-ITZ, b is the slope; a is the intercept and y corresponds to the ratio of peak areas. Individual standard curve data from five runs met all of the preset criteria:
-
<20% deviation from nominal concentration at the limit of quantitation (LOQ), which was defined as the lowest standard.
-
<15% deviation of nominal concentrations of standards from their back-calculated concentrations, except for the case of LOQ (lowest standard concentration=limit of quantitation), where 20% deviation is acceptable.
-
At least five out of seven nonzero standards of each nominal concentration should meet the above criteria, including the LOQ and the calibration standard at the highest concentration.
Sample extraction and preparation
Initially, all plasma samples were thawed at room temperature, vortexed and centrifuged at 3500 rpm for 5 min at approximately 4 °C. The Eppendorf tubes were decapped and placed into 24-position microfuge racks on the deck of the Multiprobe. 2.2 mL 96 deep well plates were also placed on the deck of the workstation, as well as reagent troughs containing the IS solution. The instrument was programmed to transfer 150 μL of each of the calibration, quality control and subject samples from the Eppendorf tubes into the appropriate wells of a 96-well plate. Then, to each well, 200 μL of IS2 solution was added. The deep well plates were removed from the Multiprobe, vortex-mixed for 10 min, and placed, one at a time, onto the deck of the Tomtec workstation. The deck also contained a reservoir with MTBE and a rack with 96 disposable tips. 1300 μL of MTBE, in four aliquots of 325 μL, were transferred from the reservoir into all wells of each plate; the plates were covered with a mat and vortex-mixed for 25 min. Next, the samples were centrifuged for 15 min, at 3500 rpm and 4 °C, and then frozen for 20 min at –30 °C. The mats were carefully removed and 800 μL of the supernatant organic layer, in two aliquots of 400 μL, were transferred from the original sample plates into the respective positions of new 2.2 mL 96 deep well plates. The height of the transfer stage was carefully chosen so that only the organic layer was aspirated. The plates were then placed into the Zymark TurboVap 96-well format plate evaporator, and the organic extracts were evaporated to dryness. The time required for complete evaporation ranged between 15 and 20 min when a flow rate of approximately 40 (arbitrary units) was employed. Reconstitution of the dry residue was achieved by addition of 300 μL of mobile phase. Finally, the plates were vortex mixed for 10 min and transferred into the autosampler for analysis.
Results and discussion
Our goal, in the present study, was to develop and validate an improved semi-automated method for the rapid and reliable determination of ITZ and its hydroxy metabolite in human plasma, and then apply it to a bioequivalence study.
The utilization of the 96-well format was the critical step in the automation process that permitted the LLE procedure to be conducted in a batch-wise fashion. Consequently, the addition of MTBE as well as the transfer of the supernatant, after the extraction, into a clean 96-well plate was performed extremely rapidly, resulting in a significant boost in method throughput. Moreover, the use of the 96-well format plate evaporator allowed the rapid evaporation of 192 samples at the same time (two 96-well plates). The only steps that could not be automated during the sample clean-up procedure were vortexing and centrifuging. Nevertheless, the proposed method’s throughput is remarkably high considering that in only five hours time, 400 samples were ready to be submitted for analysis versus 250 when the procedure was carried out manually.
Under the chromatographic conditions utilized in the present study, the retention times were about 1.50 min for ITZ, 1.13 min for OH-ITZ and 1.68 min for IS, with a total runtime of 2 min, while other methods require 3–5 min. To avoid carry-over effects from the autosampler, two pre- and two post-injection washes of the syringe and the six-port valve, with 100 μL each of two different wash solvents, were performed.
LLE was performed according to a previously proposed methodology [18]. A mixture of ACN and MTBE was used to perform the analyte extraction from the biological matrix. The addition of 200 μL of ACN to 150 μL of plasma, followed by the addition of 1300 μL MTBE, had the following advantages over conventional methodologies:
-
ACN served as both the IS solution and the protein precipitation reagent, so only one addition was required.
-
ACN resulted in the elimination of the irregular emulsion that is observed when MTBE is employed for the extraction of analytes from plasma.
-
ACN mixing with MTBE is satisfactory as long as the ACN/MTBE ratio remains above 1:3. In our case the ratio was approximately 1:7 and no separation of ACN with MTBE was observed.
-
The addition of ACN did not increase the time required for evaporation.
-
No buffer addition was necessary, as the polarity of the organic phase was adjusted adequately due to the presence of ACN. This is confirmed by the fact that buffer addition did not improve the extraction process.
Freezing enabled the organic solvent to be removed more easily, while it reduced the possibility of transferring plasma sample components.
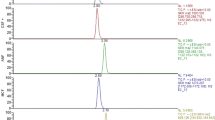
Along with the extraction procedure, the LC/MS/MS system was used to separate and monitor ITZ, OH-ITZ and IS from the extracted samples. The MS spectra for the molecules of interest are dominated by the [M+H]+ ions: m/z 704.7 for ITZ, 721.3 for OH-ITZ and 732.9 for IS, while the MS/MS daughter ion spectra of the protonated molecules produced major product ions at m/z 392.3 for ITZ, 408.1 for OH-ITZ and 460.3 for IS, respectively (Fig. 1). A representative MRM LC/MS/MS chromatogram is shown in Fig. 2.

Product ion spectra of ITZ, OH-ITZ and IS generated by collision-induced dissociation of the corresponding parent ions

Representative MRM chromatograms of ITZ, OH-ITZ and IS obtained from a MV2 sample
Standard curves
After the extraction procedure and the LC/MS/MS conditions had been defined, a full validation was performed by our GLP-compliant laboratory, according to currently presented US Food and Drug Administration (FDA) bioanalytical method validation guidance [19], which was presented above.
The regression coefficients (R 2) for the five runs were greater than 0.998 for both analytes; the average linear slope was 0.964 (S b=0.006) for ITZ and 0.940 (S b=0.009) for OH-ITZ while the average intercept was 1.072 (S a=1.231) for ITZ and 5.122 (S a=.872) for OH-ITZ, respectively. The experimental values for the F-test (Mandel) were smaller than 0.691 for ITZ and 0.222 for OH-ITZ, when the (theoretical) threshold value of F-distribution (5%, one-sided) was 4.170. Based on the presented data, it was concluded that the calibration curves used in this method were linear over the operating range used. The test for proportionality was also successful; the t-test experimental values of 0.871 and 1.323 for ITZ and OH-ITZ, respectively, were smaller than the theoretical value of 2.042 (5%, two-sided). In conclusion, the 96-well liquid–liquid extraction procedure employed in this method was capable of producing satisfactory concentration data for ITZ and OH-ITZ standard samples.
Accuracy and precision
When validating the method, the precision and accuracy were also assessed by analyzing MV samples, as defined above, in five runs on three separate days. The % accuracy was determined by calculating the deviations of the predicted concentrations from their nominal values. In all cases the values were within the acceptable range. The intra-assay precision was assessed by analyzing six replicates at each MV level, while inter-assay precision was determined over five runs conducted over three days, by analyzing 30 samples. Data for both types of accuracy and precision (expressed as CV%) are presented in Tables 1 and 2 for ITZ and OH-ITZ, respectively.
Stability studies
As a part of the method validation, data were also generated to ensure that ITZ and OH-ITZ were stable at distinct timing and temperature conditions, as well as to check the stability of the analytes in stock solution. Plasma samples containing two concentration levels of ITZ and OH-ITZ were used for the stability experiments: Low-medium (S l), 10 ng mL−1 for ITZ and 20 ng mL−1 for OH-ITZ, and medium-high (S h) 100 ng mL−1 for ITZ and 200 ng mL−1 for OH-ITZ.
To evaluate freeze-thaw stability, a freeze and thaw cycle was defined as the storage of S l and S h samples at – 20 °C followed by thawing at room temperature. Samples were analyzed after the fourth cycle, along with fresh reference samples of the same concentration. Table 3 shows the corresponding results (back-calculated concentrations) of four freeze-thaw cycles vs fresh ones, which should not vary more than 10%.
To evaluate short-term stability, six aliquots of S l and S h were maintained, immediately after preparation, at room temperature for six hours, which exceeds the time period that samples would remain at room temperature before analysis. As for long-term stability, aliquots of the two sample-types were initially frozen at–20 °C for 30 days, thawed and analyzed. The 30 day period is more than adequate for the completion of a bioequivalence study, since the current method allows the measurement of 400 samples per day. The results shown in Tables 4 and 5 indicate that both analytes are stable under the conditions described above, as the mean variation is below 10%.
The stabilities of stock and working solutions (stored at 4 °C) were estimated by comparing fresh and old dilutions in mobile phase. The measurements proved that ITZ and OH-ITZ concentrations in stock solution remain stable. Autosampler stability was another part of the method validation that should have been investigated. It was assessed by comparing QC samples included at the beginning, at halfway and upon completion of each of the five analytical runs. Results for the stability of the analytes in the autosampler (10 °C) are presented in Table 6, and were within the acceptance criteria, which established that the mean result at completion of the runs should be ≥90% of the mean result at the beginning of the runs for at least two-thirds of the levels tested.
Extraction recovery and matrix effect
The extraction efficiency of the analytical method was assessed using data from five runs. LLE recoveries were determined by comparing the absolute peak areas of the analytes, spiked before the extraction in the blank control matrix and spiked after the LLE into the extracts of blank control matrix (post-spiked) at three QC concentrations, (QC1, QC2 and QC3). Mean values of extraction recovery for ITZ in QC1, QC2 and QC3 were 54.5%, 66.31% and 58.5%, respectively, while for OH-ITZ the corresponding values were 57.5%, 59.3% and 58.1%. Mean extraction recovery for the IS was 66.5%. The above recovery values were consistent and more than adequate for the specific concentration range.
Endogenous matrix components may change the efficiency of droplet formation or droplet evaporation, which in turn affects the amount of charged ion in the gas phase that ultimately reaches the detector [20, 21]. These compounds are probably the main cause of ion suppression effects during electrospray ionization. The extent of this phenomenon is dependent upon the sample extraction procedure and it is also compound-dependent. In order to evaluate matrix effect in the present study, two sets of test samples were prepared by directly spiking the analytes into reconstitution solution with and without the presence of residue extracted from pooled control plasma. Ion suppression was assessed at three QC concentrations by comparing the mean analyte peak areas obtained from these two sets of testing samples. The results obtained from these experiments are shown in Table 7. Despite the presence of analyte ion suppression, the quantitative performance of the proposed method did not deteriorate significantly, as evidenced by the accuracy and precision results mentioned above.
Application of the assay to a bioequivalence study
The present method was utilized for the analysis of plasma samples obtained from 28 healthy volunteers after the administration of a 100 mg capsule of ITZ, as part of a bioequivalence study. The protocol of this study was in accordance with the international standards for these studies. The concentration–time profiles of ITZ and OH-ITZ in these volunteers are represented in Fig. 3, and they show that the proposed method is suitable for pharmacokinetic studies of ITZ in human plasma.

Mean plasma concentration–time curves from 28 subjects for ITR and OH-ITR
Conclusions
We have presented a semi-automated 96-well LLE, LC/MS/MS method for the simultaneous determination of ITZ and its coactive metabolite, hydroxy-itraconazole, in human plasma. The application of two liquid-handling robotic workstations greatly simplified the extraction process, resulting in significant advantages over existing methods with manual sample treatment in terms of throughput and efficiency. Another advantage over these methods was the relatively small quantity of human plasma used for analysis. Moreover, the mixture of ACN/MTBE used as the extraction solvent, aside from eliminating the formation of the irregular emulsion observed when only MTBE was employed, managed to adequately adjust the polarity of the organic solvent. The proposed method was applied to a bioequivalence study of ITZ, and it allowed the study to be completed in just 4 days. The described high-throughput method possessed excellent precision and accuracy and proved to be reliable. It is expected that this approach could be applied to the extraction and analysis of other pharmaceutical compounds from biological samples.
References
Wong JW, Nisar Ur-Rand, Yuen KH (2003) J Chromatogr B 798:355–360
Srivatsan V, Dasgupta AK, Kale P, Datla RR, Soni D, Patel M, Patel R, Mavadhiya C (2004) J Chromatogr A 1031:307–313
Gubbins PO, Gurley BJ, Bowman J (1998) J Pharm Biomed Anal 16:1005–1012.
Compas D, Touw DJ, De Goede PNFC (1996) J Chromatogr B 687:453–456
Zhong L, Eisenhandler R, Yeh KC (2001) J Mass Spectrom 36:736–741
Chen X, Zhong D, Liu D, Wang Y, Han Y, Gu J (2003) Rapid Commun Mass Spectrom 17:2459–2463
Dotsikas Y, Kousoulos C, Tsatsou G, Loukas YL (2005) Rapid Commun Mass Spectrom 19:2055–2061
Carrier A, Parent J (2000) J Chromatogr B 745:413–420
Yao M, Chen L, Srinivas LR (2001) J Chromatogr B 752:9–16
Vogeser M, Spöhrer U, Schiel X (2003) Chem Lab Med 41:915–920
Jemal M (2000) Biomed Chromatogr 14:422–429
Basileo G, Breda M, Fonte G, Pisano R, James CA (2003) J Pharm Biomed Anal 32:591–600
Kitchen CJ, Wang AQ, Musson DG, Yang AY, Fisher AL (2003) J Pharm Biomed Anal 31:647–654
Zhang N, Yang A, Rogers JD, Zhao JJ (2004) J Pharm Biomed Anal 34:175–187
Barattè S, Sarati S, Frigerio E, James CA, Ye C, Zhang Q (2004) J Chromatogr A 1024:87–94
Zhang J, Zeng W, Kitchen C, Wang AQ, Musson DG (2004) J Chromatogr B 806:167–175
Zhang N, Hoffman KL, Li W, Rossi DT (2000) J Pharm Biomed Anal 22:131–138
Xue YJ, Pursley J, Arnold ME (2004) J Pharm Biomed Anal 34:369–378
CDER (2001) Guidance for industry: Bioanalytical method validation. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Rockville, MD
Bonfiglio R, King RC, Olah TV, Merkle K (1999) Rapid Commun Mass Spectrom 13:1175–1185
Annesley TM (2003) Clin Chem 49:1041–1044
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kousoulos, C., Tsatsou, G., Apostolou, C. et al. Development of a high-throughput method for the determination of itraconazole and its hydroxy metabolite in human plasma, employing automated liquid–liquid extraction based on 96-well format plates and LC/MS/MS. Anal Bioanal Chem 384, 199–207 (2006). https://doi.org/10.1007/s00216-005-0159-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-005-0159-6