
Abstract
Chemiluminescence is a fundamental process of chemistry consisting in the conversion of chemical energy stored in chemical bonds into light. It is used by nature and by man-made technology, being especially relevant in chemical analysis. The understanding of the phenomenon strongly relies in the study of peroxide models such as 1,2-dioxetanones. In the present contribution, the singlet S 2 and the triplet T 2 potential energy surfaces of the unimolecular decomposition of 1,2-dioxetanone have been mapped along the O–O and C–C bond coordinates on the grounds of the multiconfigurational CASPT2//CASSCF approach. Results confirm the energy degeneracy between T 2, T 1, S 1, and S 0 at the TS region, whereas S 2 is unambiguously predicted at higher energies. Triplet-state population is also supported by the spin–orbit couplings between the singlet and triplet states partaking in the process. In particular, the first-principle calculations show that decomposition along the T 2 state is a competitive process, having a small (~3 kcal/mol) energy barrier from the ground-state TS structure. The present findings can explain the higher quantum yield of triplet-state population with respect to the excited singlet states recorded experimentally for the unimolecular decomposition of 1,2-dioxetanone models.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Chemiluminescence is defined as the emission of light induced by a chemical reaction. When occurring in living beings, the phenomenon is called bioluminescence, and it is used for several purposes such as a communication system between different members of a species, for hunting, or also to avoid predators, among many others [1]. On the other hand, bio- and chemiluminescence constitute a powerful tool broadly used in different fields, such as gene expression [2], biosensors for environmental pollutants [3], cancer monitoring [4], food industry [5, 6], and forensic science [7, 8]. The most used chemiluminescent systems are usually based on a high-energy O–O bond, whose thermal rupture promotes the population of excited states. 1,2-dioxetanone is in this scenario the model used to study the chemiluminescence phenomena of larger bioluminescent systems such as the firefly luciferin and coelenterazine (see Fig. 1) [9].

1,2-Dioxetanone as the molecular unit to study chemiluminescent and bioluminescent processes. Atom labeling of the peroxide is also shown
Experiments have been carried out on 1,2-dioxetanones and also on 1,2-dioxetanes, pointing to an inefficient chemiexcitation process and a higher yield of production of triplet excited states [10,11,12,13,14,15,16,17]. For instance, the thermal decomposition of dimethyldioxetanone gives rise to singlet and triplet excited states with yields of 0.05–0.10 and 1.5%, respectively [11, 12]. Since methylation increases the efficiency of the excited-state production, the yields in 1,2-dioxetanone are expected to be lower [18]. To increase the chemiluminescence efficiency, the decomposition reaction has to be catalyzed via an intra- or inter-molecular mechanism in which a fluorophore with a low ionization potential and high fluorescence quantum yield firstly facilitates the dioxetanone decomposition via an electron-density transfer process and secondly becomes excited and emits light [9, 19]. Several chemical aspects of the bio-/chemiluminescence require still further research, such as what is the chemical structure of the light-emitting species [20,21,22,23,24], the role of biradicals or the triplet states [25,26,27,28,29,30], or the differences and similarities between the intra- and inter-molecular mechanisms for the catalyzed peroxide decomposition [31]. This urges the revision of past studies with more sophisticated experimental techniques and more accurate theoretical methodologies and the design of new analyses not carried out so far [19, 31, 32]. In the present study, we revisit the decomposition reaction of 1,2-dioxetanone by multiconfigurational quantum chemistry focusing on the differences between the population mechanisms for the excited singlet and triplet states.
In 2009, Liu et al. [33] documented the thermal decomposition process of 1,2-dioxetanone on the grounds of multistate complete-active-space second-order perturbation theory (MS-CASPT2) calculations. The authors reported the presence of several transition state (TS) structures along the molecular rupture. It was found that the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS structure governs the O3–O5 bond breaking, placed at ~30 kcal/mol above the reactant. The authors also reported that the mentioned TS represents a non-adiabatic crossing area between the ground and the first singlet (S 1) and triplet (T 1) excited states, which are of \( {}^{1,3}\left( {n,\sigma^{*} } \right) \) nature. From this point, and as the system evolves toward decomposition by enlarging the C1–C2 bond, a second \( {}^{1}\left( {n,\sigma^{*} } \right) \)-TS is reached. The latter structure represents in turn another non-adiabatic crossing between the \( {}^{1,3}\left( {n,\sigma^{*} } \right) \) and the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \) electronic states, driving thus the branching toward the ground-state decomposition (no light emission), or the chemiluminescent pathway. A detailed analysis of the conical intersection (CI) branching spaces revealed that the former channel involves a torsion of the O–C–C–O mode, whereas the latter path takes place within motion in the molecular plane. Liu et al. [33] therefore demonstrated that both the lowest-lying singlet (S 1) and triplet (T 1) excited states were accessible from the ground state (S 0). Further studies carried out by Greenman and Mazziotti [34, 35] and using two-electron reduced-density matrix (2RDM) methods supported the description provided by the CASSCF/CASPT2 approach [33].
In contrast to the aforementioned theoretical study, a more recent work using the density functional theory (DFT) method showed the S 1 state to be very high in energy with respect to S 0 along the decomposition reaction coordinate [30], which was used to explain the different chemiexcitation yields for the singlet and triplet excited states. However, the latter theoretical approach was shown to have instabilities of the wave function, which produced an incorrect description of the chemiluminescent mechanism [28, 29]. The proposed concerted dissociation of the O–O and C–C bonds was proved to be invalid by using both accurate multiconfigurational quantum chemistry methods and DFT approaches based on a stable wave function [28, 29]. The mechanism was proved to take place through a biradical region in which only the O–O bond is broken and the C–C bond is still formed. Although the general aspects of the 1,2-dioxetanone chemiluminescence were reported in the work by Liu et al. [33], the higher ratio of triplet versus singlet excited-state production determined experimentally remains unexplained [10,11,12,13,14,15,16,17]. In the present work, we have carried out further accurate theoretical studies, which provide a rationale of such experimental evidence. For such purposes, we have focused here on the role of higher excited states of the molecule (S 2 and T 2), which were not considered in previous studies [33]. As shall be seen below, whereas the S 2 state appears at much higher energies along the dissociation reaction coordinate, T 2 becomes accessible in the biradical region. This implies the presence of two chemiexcitation paths on the triplet manifold versus only one on the singlet excited manifold, which agrees with the experimental observations [10,11,12,13,14,15,16,17].
2 Methods and computational details
The CASSCF method has been extensively demonstrated in the literature to provide a balanced description of the ground and excited states of molecules and correct dimensionalities of the crossing points between several potential energy hypersurfaces (PEHs) [36,37,38,39,40,41,42]. In addition, the wave functions constructed with this method are appropriate for the calculation of several molecular properties such as dipolar moments or spin–orbit couplings (SOCs) [43,44,45,46]. In the present work, the CASSCF wave functions have been built by distributing 16 electrons into the 13 molecular orbitals included in the active space, as described elsewhere [33]. The triple-\( \zeta \) atomic natural orbital basis set including relativistic core correlation (hereafter, ANO-RCC-VTZP basis set) has been used throughout as a reasonable compromise between accuracy and computational cost [47, 48], and four roots have been demanded in the state average (SA)-CASSCF procedure in the computations of both singlet and triplet states. No symmetry restrictions (C 1) have been imposed in the calculations.
Two types of CASSCF optimizations have been performed in the present study. On the one hand, a minimization of the second triplet state T 2 has been conducted relaxing all degrees of freedom of the molecule in order to locate a well-defined minimum. On the other hand, constrained optimizations fixing both C1–C2 and O3–O5 bonds and relaxing the other internal coordinates have been undertaken in order to map the T 2 potential energy surface (PES) along the mentioned coordinates. Thereby, the C1–C2 bond distance has been increased from 1.5 to 2.4 Å in steps of 0.1 Å, whereas the peroxide O3–O5 bond has been changed from 2.5 to 3.0 Å in steps of 0.1 Å. In addition, the topology of the converged geometries has been carefully analyzed in order to detect possible hysteresis between the structures. Thus, all the reported geometries in the present work are connected through vicinal regions of the PES.
The MS-CASPT2 method [49, 50] has been used on top of the CASSCF converged geometries to compute the dynamic electron correlation and provide accurate energies. This protocol, known as MS-CASPT2//CASSCF, was also used in the previous work by Liu et al. [33] on 1,2-dioxetanone. Both MS- and single-state approaches provide similar results, which point to an accurate computation of the dynamical electron correlation. The (MS)-CASPT2//CASSCF protocol has been widely demonstrated in the last decades to provide accurate energies within an average error of ~0.2 eV from the experimental recordings [27, 51,52,53,54,55]. The zeroth-order Hamiltonian as originally implemented has been used in all the computations by setting the ionization-potential electron-affinity parameter to 0.00 au [56], whereas an imaginary level shift of 0.20 au has been used to minimize the effect of weakly intruder states [57]. Unless otherwise specified, all energies reported in the present work are relative to the S 0 energy at the ring-closed minimum [33].
SOCs have been computed with the restricted active space state interaction method [43, 58] using the atomic mean field approximation [59, 60], according to:
where \( \hat{H}_{\text{SO}} \) is the spin–orbit Hamiltonian, and \( T \) and S refer to the triplet- and singlet-state CASSCF wave functions, respectively.
All the electronic structure calculations have been performed with the MOLCAS-7 program [61, 62].
3 Results and discussion
This section is organized as follows: Firstly, the S 2 and T 2 accessibility during the thermal decomposition process of 1,2-dioxetanone is appraised. Secondly, the region of near degeneracy between the lowest-lying singlet and triplet states is analyzed focusing on the T 2 state. This completes the previous analyses carried out for the S 1 and T 1 states [33]. In the third place, the SOC values between the different electronic states at the relevant geometries are discussed. Finally, the dissociation mechanism of 1,2-dioxetanone on the T 2 state is presented.
3.1 S 2 and T 2 accessibilities
The energetics of the S 2 and T 2 states have been studied along the ground-state reaction coordinate computed by Liu et al. [33]. Results are displayed in Fig. 2. It can be readily seen that the two low-lying triplet states T 1 and T 2 and the lowest-lying singlet excited state S 1 become degenerated with S 0 as the O–O bond of 1,2-dioxetanone stretches and the corresponding TS for the bond breaking, \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS is reached. This region allows the population of the excited states through internal conversion (IC) and intersystem crossing (ISC) processes. On the other hand, the S 2 state is about 13 kcal/mol (~0.56 eV) above the degeneracy area, and therefore, its population during the decomposition process can be safely ruled out.

MS-CASPT2//CASSCF potential energy surfaces of the lowest-lying three singlet (S 0, S 1, and S 2) and two triplet (T 1 and T 2) electronic states along the ground-state O3–O5 bond breaking coordinate [33]. Energies are relative to the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-min structure reported in Ref. [33] (see text). Dashed lines connect the electronic state energies at \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS with those at more distorted geometries
3.2 T 2 minimum
The optimization of the second lowest-lying triplet state (T 2), starting from the structure \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS reported by Liu et al. [33], has been carried out with the CASSCF method. The computed geometry, labeled as (T 2)min, is predicted in the close vicinity of the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS region, as concluded from the bond length similarities displayed in Fig. 3. The relative energies of the most relevant electronic states are also shown. Results indicate that (T 2)min is expected to extend the non-adiabatic crossing area computed at the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS zone, increasing the multidimensionality of the near-degeneracy region. Whereas the minima of the S 1 and T 1 states in this region, (S 1)min and (T 1)min, respectively, are characterized by a biradical nature in which the in-plane p orbital of O3 and the perpendicular one of O5 are singly occupied \( \left[ {{}^{1,3}\left( {n,\sigma^{*} } \right)\;{\text{states}}} \right] \), in the (T 2)min both unpaired electrons are in the in-plane p orbitals of O3 and O5 \( \left[ {{}^{3}\left( {\sigma ,\sigma^{*} } \right)\;{\text{state}}} \right] \). Regarding the geometrical parameters, the O–O bond length of the \( {}^{3}\left( {\sigma ,\sigma^{*} } \right) \) minimum, 2.63 Å, is found between that of the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \) equilibrium structure and the distance for the minima of the \( {}^{3}\left( {n,\sigma^{*} } \right) \) and \( {}^{1}\left( {n,\sigma^{*} } \right) \) states, 2.68 and 2.69 Å, respectively.

CASSCF structures and main bond distances (Å) of \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS, retrieved from Ref. [33], and (T 2)min. MS-CASPT2 energies of the lowest-lying three singlet (S 0, S 1, and S 2) and two triplet (T 1 and T 2) states at the (T 2)min and relative to the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-min energy [33] are also shown
3.3 Population of the triplet states
Population transfer between states of different multiplicity is mediated by ISC processes. In general, the following features between the two states of interest are expected to increase the ISC efficiency: (1) energy degeneracy or near degeneracy, (2) well-defined minimum of PEH in order to “trap” the system long enough to produce the ISC process, and (3) large SOCs [63]. In the case of 1,2-dioxetanone, requirement (1) is fulfilled in the biradical region which is characterized by a flat PEH. From there on, the most energetically favorable decomposition channel is the ground-state dissociation, which is in agreement with the low chemiluminescence yields measured in the experiments [10,11,12,13,14,15,16,17]. Nevertheless, decomposition in the excited singlet and triplet states is still feasible.
Table 1 compiles the SOC values computed between the lowest-lying singlet and triplet states of 1,2-dioxetanone. In particular, SOCs have been computed at the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS structure to appraise the triplet-state population across the thermal decomposition of 1,2-dioxetanone. SOCs were not computed in the previous study by Liu et al. [33], and they can provide qualitative information on the population distribution among singlet and triplet states at the degeneracy region. By inspection of the results summarized in Table 1, it becomes apparent that the four possible ISC processes at the \( ^{1} \left( {\sigma ,\sigma^{*} } \right) \)-TS structure have significant SOC values, thus indicating allowed triplet-state population during the thermal decomposition. The largest values are computed for the \( S_{0} \leftrightarrow T_{1} \) and \( S_{1} \leftrightarrow T_{2} \) crossings, which are expected to contribute the most to the population transfer at the TS zone. The differences in the SOC values can be interpreted according to the nature of the singly occupied orbitals in each state. Thus, singlet and triplet states with different character, \( \left( {\sigma ,\sigma^{*} } \right) \) and \( \left( {n,\sigma^{*} } \right) \), have much higher SOCs than those between states of the same nature. In general, even though the analysis performed here only refers to one representative structure, it can be extrapolated to other geometries in the region of near degeneracy between states (biradical region). Crossings and re-crossings are expected here between singlet and triplet states until the system leaves the region toward the complete dissociation into CO2 and formaldehyde. The relevant aspect is that in the excited-state manifold, only one singlet (S 1) is involved in the population distribution while there are two triplets (T 1 and T 2) participating in such process. This increases the probability for producing triplet excited formaldehyde.
3.4 Decomposition of 1,2-dioxetanone in the T 2 state
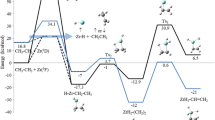
In previous sections, it has been demonstrated that the T 2 state of 1,2-dioxetanone is accessible from the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS structure together with the S 1 and T 1 states, having large SOCs between the singlet and triplet states (see Table 1). Moreover, the T 2 minimum is highly similar to the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS geometry (cf. Fig. 3) visited in the thermal decomposition of the system. Thus, the possible dioxetanone scission on the T 2 surface, leading to excited formaldehyde and carbon dioxide, has been explored by means of relaxed optimizations with O3–O5 and C1–C2 fixed bond distances. As a result, the 2D PEH displayed in Fig. 4 has been obtained. The most favorable channel involves a relatively small barrier of ~3 kcal/mol, demonstrating therefore that the route can be competitive with the S 0, S 1, and T 1 dissociations. The structural parameter that drives the molecular rupture is the elongation of the C1–C2 bond, whereas the O3–O5 distance remains fixed at 2.5 Å. The PEH topology clearly shows that larger O3–O5 elongations significantly increase the molecular energy, leading to a maximum of almost ~45 kcal/mol at O3–O5 and C1–C2 distances of 3.1 and 1.9 Å, respectively. The mechanism shown in Fig. 4 corresponds thus to a stepwise breaking of the O3–O5 bond followed by C1–C2 scission, which takes place in the plane of the molecule. This is similar to the process previously determined for the rupture along the S 1 and T 1 electronic states and differs from the excited-state decomposition predicted for 1,2-dioxetane in which the torsional mode related to the O–C–C′–O′ dihedral angle plays an important role in the production of the excited-state species [26, 64, 65]. As described by Liu et al. [33], the difference might be due to a conjugation effect between the CO2 and formaldehyde moieties, which restricts the torsion around the C–C bond. This might have important consequences in the lifetime of the biradicals since the rotation is expected to trap the molecule in this area and therefore increase the probability of population transfer to the singlet and triplet excited states. This reasoning seems to be in agreement with the experimental observations of lower triplet quantum yields for 1,2-dioxetanones than those for 1,2-dioxetanes [19].

Potential energy hypersurface along the T 2 state relaxed scan of the O3–O5 and C1–C2 coordinates using the MS-CASPT2//CASSCF approach. Energies are relative to the (S 0)min structure
The nature of the T 2 state evolves according to the chemical changes that lead to the molecular rupture. Thus, in the \( {}^{1}\left( {\sigma ,\sigma^{*} } \right) \)-TS vicinity it mainly has \( {}^{3}\left( {\sigma ,\sigma^{*} } \right) \) character localized over the peroxide bond, whereas after decomposition (O3–O5 and C1–C2 distances of 2.5 and 2.4 Å, respectively) the state has marked \( {}^{3}\left( {\pi ,\pi^{*} } \right) \) character and the excitation is mainly centered on the formaldehyde moiety. Therefore, this channel becomes competitive with the decomposition on the T 1 and S 1 excited states, which have \( {}^{1,3}\left( {n,\pi^{*} } \right) \) character. The presence of two triplet channels versus one singlet channel provides a satisfactory explanation for the larger quantum yield of triplet-state population with respect to the singlet state registered experimentally [10,11,12,13,14,15,16,17]. It is important to remark that in spite of the accessibility of the excited states along the thermal decomposition of 1,2-decomposition, low quantum yields of fluorescence and triplet emission are observed in the experiments, which can be attributed to the fact that the ground-state decomposition is much more energetically favorable. Future dynamics simulations shall be helpful to further decipher how the singlet and triplet states interact in the biradical region and how the population is distributed among the S 0, S 1, T 1, and T 2 states.
4 Conclusions
The present contribution tackles the study of the difference between the triplet- and singlet-state population of 1,2-dioxetanone along its thermal decomposition by using multiconfigurational quantum chemistry (MS-CASPT2//CASSCF). The findings obtained and the corresponding analyses complete a previous study performed by Liu et al. [33] with the same methodological approach. Whereas that study focused on the S 0, S 1, and T 1 decomposition channels, the role of higher excited states is studied here. In light of the obtained results, the participation of the S 2 state in the chemiluminescent light emission can be discarded. Meanwhile, the T 2 state is clearly accessible along the decomposition reaction according to the computed energies and the corresponding SOCs between the singlet and triplet states at the transition state of the reaction. The present results support the plausibility of an 1,2-dioxetanone dissociation on the T 2 state yielding H2CO* + CO2 on the triplet manifold. Thus, the present findings point to the existence of two accessible triplet excited pathways versus only one singlet channel along the decomposition reaction of 1,2-dioxetanone, which can explain the higher triplet versus singlet chemiexcitation ratio recorded in early experiments [10,11,12,13,14,15,16,17].
References
Haddock SHD, Moline MA, Case JF (2010) Bioluminescence in the sea. Annu Rev Mar Sci 2:443
Contag CH, Bachmann MH (2002) Advances in vivo bioluminescence imaging of gene expression. Annu Rev Biomed Eng 4:235
Endo G, Yamagata T, Narita M, Huang CC (2003) Bioluminescence biosensor for the detection of organomercury contamination. Acta Biotechnol 23:123
Kim J-B, Urban K, Cochran E, Lee S, Ang A, Rice B, Bata A, Campbell K, Coffee R, Gorodinsky A, Lu Z, Zhou H, Kishimoto TK, Lassota P (2010) Non-invasive detection of a small number of bioluminescent cancer cells in vivo. PLoS ONE 5:e9364
Thakur MS, Ragavan KV (2013) Biosensors in food processing. J Food Sci Technol Mysore 50:625
Stannard CJ, Gibbs PA (1986) Rapid microbiology: applications of bioluminescence in the food industry—a review. J Biolumin Chemilumin 1:3–10
Specht W (1937) The chemiluminescence of hemin: an aid for finding and recognizing blood stains important for forensic purposes. Angew Chem 50:155–157
Quickenden TI, Cooper PD (2001) Increasing the specificity of the forensic luminol test for blood. Luminescence 16:251
Navizet I, Liu Y-J, Ferre N, Roca-Sanjuán D, Lindh R (2011) The chemistry of bioluminescence: an analysis of chemical functionalities. Chem Phys Chem 12:3064–3076
Schmidt SP, Schuster GB (1980) Chemiluminescence of dimethyldioxetanone. Unimolecular generation of excited singlet and triplet acetone. Chemically initiated electron-exchange luminescence, the primary light generating reaction. J Am Chem Soc 102:306–314
Schmidt SP, Schuster GB (1978) Kinetics of unimolecular dioxetanone chemiluminescence. Competitive parallel reaction paths. J Am Chem Soc 100:5559–5561
Adam W, Simpson GA, Yany F (1974) Mechanism of direct and rubrene enhanced chemiluminescence during alpha-peroxylactone decarboxylation. J Phys Chem 78:2559–2569
Adam W, Liu JC (1972) Photooxygenation (singlet oxygen) of tetrathioethylenes. J Am Chem Soc 94:1206–1209
Turro NJ, Chow MF (1980) Chemiluminescent thermolysis of alpha-peroxylactones. J Am Chem Soc 102:5058–5064
Baader WJ, Stevani CV, Bastos EL (2006) Chemiluminescence of organic peroxides. In: Rappoport Z (ed) The chemistry of peroxides. Wiley, Chichester
Adam W (1982) Determination of chemiexcitation yields in the thermal generation of electronic excitation from 1,2-dioxetanes. In: Adam W (ed) Chemical and biological generation of excited states. Academic Press, New York
Adam W, Trofimov AV (2006) Contemporary trends in dioxetane chemistry. In: Rappoport Z (ed) The chemistry of peroxides. Wiley, Chichester
Adam W, Baader WJ (1985) Effects of methylation on the thermal stability and chemiluminescence properties of 1,2-dioxetanes. J Am Chem Soc 107:410–416
Augusto FA, de Souza GA, de Souza Júnior SP, Khalid M, Baader WJ (2013) Efficiency of electron transfer initiated chemiluminescence. Photochem Photobiol 89:1299–1317
Navizet I, Roca-Sanjuán D, Yue L, Liu Y-J, Ferre N, Lindh R (2013) Are the bio- and chemiluminescence states of the firefly oxyluciferin the same as the fluorescence state? Photochem Photobiol 89:319–325
Schramm S, Weiss D, Navizet I, Roca-Sanjuán D, Brandl H, Beckert R, Goerls H (2013) Investigations on the synthesis and chemiluminescence of novel 2-coumaranones. ARKIVOC 3:174–188
Chen S-F, Navizet I, Roca-Sanjuán D, Lindh R, Liu Y-J, Ferre N (2012) Chemiluminescence of coelenterazine and fluorescence of coelenteramide: a systematic theoretical study. J Chem Theor Comput 8:2796–2807
Roca-Sanjuán D, Delcey MG, Navizet I, Ferre N, Liu Y-J, Lindh R (2012) WARNING: the light-emitting molecular structures responsible for the chemiluminescence and fluorescence phenomena are not necessarily the same! Luminescence 27:155–156
Roca-Sanjuán D, Delcey MG, Navizet I, Ferre N, Liu Y-J, Lindh R (2011) Chemiluminescence and fluorescence states of a small model for coelenteramide and cypridina oxyluciferin: a CASSCF/CASPT2 study. J Chem Theor Comput 7:4060–4069
Farahani P, Lundberg M, Lindh R, Roca-Sanjuán D (2014) CASPT2//CASSCF study of the ring-opening mechanism of dewar dioxetane. Luminescence 29:18–19
Farahani P, Roca-Sanjuán D, Zapata F, Lindh R (2013) Revisiting the nonadiabatic process in 1,2-dioxetane. J Chem Theor Comput 9:5404–5411
Aparici-Espert I, Francés-Monerris A, Rodríguez-Muñiz GM, Roca-Sanjuán D, Lhiaubet-Vallet V, Miranda MA (2016) A combined experimental and theoretical approach to the photogeneration of 5,6-dihydropyrimidin-5-yl radicals in nonaqueous media. J Org Chem 81:4031–4038
Roca-Sanjuán D, Lundberg M, Mazziotti DA, Lindh R (2012) Comment on “Density functional theory study of 1,2-dioxetanone decomposition in condensed phase.”. J Comput Chem 33:2124–2126
Yue L, Roca-Sanjuán D, Lindh R, Ferre N, Liu Y-J (2012) Can the closed-shell DFT methods describe the thermolysis of 1,2-dioxetanone? J Chem Theor Comput 8:4359–4363
da Silva LP, Esteves da Silva JC (2012) Density functional theory study of 1,2-dioxetanone decomposition in condensed phase. J Comput Chem 33:2118–2123
Augusto FA, Francés-Monerris A, Fdez. Galván I, Roca-Sanjuán D, Bastos EL, Baader WJ, Lindh R (2017) Mechanism of activated chemiluminescence of cyclic peroxides: 1,2-dioxetane and 1,2-dioxetanone. Phys Chem Chem Phys 19:3955–3962
de Oliveira MA, Bartoloni FH, Augusto FA, Monteiro Leite Ciscato LF, Bastos EL, Baader WJ (2012) Revision of singlet quantum yields in the catalyzed decomposition of cyclic peroxides. J Org Chem 77:10537–10544
Liu F, Liu Y, de Vico L, Lindh R (2009) Theoretical study of the chemiluminescent decomposition of dioxetanone. J Am Chem Soc 131(17):6181
Greenman L, Mazziotti DA (2010) Strong electron correlation in the decomposition reaction of dioxetanone with implications for firefly bioluminescence. J Chem Phys 133:164110
Greenman L, Mazziotti DA (2011) Balancing single- and multi-reference correlation in the chemiluminescent reaction of dioxetanone using the anti-Hermitian contracted Schrödinger equation. J Chem Phys 134:174110
Roca-Sanjuán D, Fdez. Galván I, Lindh R, Ya-Jun L (2015) Recent method developments and applications in computational photochemistry chemiluminescence and bioluminescence. In: Albini A, Fasani E (eds) Photochemistry, vol 42. The Royal Society of Chemistry, London
Serrano-Andrés L, Merchán M, Lindh R (2005) Computation of conical intersections by using perturbation techniques. J Chem Phys 122:104107
Serrano-Pérez JJ, Serrano-Andrés L (2012) Calculation of excited states: molecular photophysics and photochemistry on display. In: Leszczynski J (ed) Handbook of computational chemistry. Springer, Berlin
Serrano-Andrés L, Merchán M (2005) Ab initio methods for excited states. In: Olivucci M (ed) Computational photochemistry. Elsevier, Amsterdam
Roca-Sanjuán D, Francés-Monerris A, Fdez. Galván I, Farahani P, Lindh R, Liu Y-J (2017) Advances in computational photochemistry and chemiluminescence of biological and nanotechnological molecules. In: Albini A, Fasani E (eds) Photochemistry, vol 44. The Royal Society of Chemistry, London
Liu Y-J, Roca-Sanjuán D, Lindh R (2012) Computational photochemistry and photophysics: the state of the art. In: Albini A, Fasani E (eds) Photochemistry, vol 40. The Royal Society of Chemistry, London
Serrano-Andrés L, Roca-Sanjuán D, Olaso-González G (2010) Recent trends in computational photochemistry. In: Albini A (ed) Photochemistry, vol 38. The Royal Society of Chemistry, London
Malmqvist PÅ, Roos BO, Schimmelpfennig B (2002) The restricted active space (RAS) state interaction approach with spin–orbit coupling. Chem Phys Lett 357:230–240
Sergentu D-C, Maurice R, Havenith RWA, Broer R, Roca-Sanjuán D (2014) Computational determination of the dominant triplet population mechanism in photoexcited benzophenone. Phys Chem Chem Phys 16:25393–25403
Marazzi M, Mai S, Roca-Sanjuán D, Delcey MG, Lindh R, González L, Monari A (2016) Benzophenone ultrafast triplet population: revisiting the kinetic model by surface-hopping dynamics. J Phys Chem Lett 7:622–626
Sitkiewicz SP, Oliva JM, Dávalos JZ, Notario R, Saiz-López A, Alcoba DR, Oña OB, Roca-Sanjuán D (2016) Ab initio quantum–chemical computations of the electronic states in HgBr2 and IBr: molecules of interest on the Earth’s atmosphere. J Chem Phys 145:244304
Widmark PO, Malmqvist PÅ, Roos BO (1990) Density-matrix averaged atomic natural orbital (ANO) basis-sets for correlated molecular wave-functions. I. First row atoms. Theor Chim Acta 77:291
Roos BO, Lindh R, Malmqvist PÅ, Veryazov V, Widmark PO (2004) Main group atoms and dimers studied with a new relativistic ANO basis set. J Phys Chem A 108:2851–2858
Andersson K, Malmqvist PÅ, Roos BO (1992) 2nd-order perturbation-theory with a complete active space self-consistent field reference function. J Chem Phys 96:1218–1226
Finley J, Malmqvist PÅ, Roos BO, Serrano-Andrés L (1998) The multi-state CASPT2. Chem Phys Lett 228:299–306
Francés-Monerris A, Merchán M, Roca-Sanjuán D (2013) Electronic UV–Vis transient spectra of the OH reaction products of uracil, thymine, cytosine, and 5,6-dihydrouracil by using the complete active space self-consistent field second-order perturbation (CASPT2//CASSCF) theory. J Chem Phys 139:071101
Giussani A, Segarra-Martí J, Roca-Sanjuán D, Merchán M (2015) Excitation of nucleobases from a computational perspective I: reaction paths. In: Borin AC, Ullrich S, Barbatti M (eds) Photoinduced phenomena in nucleic acids I. Springer, Berlin
Roca-Sanjuán D, Aquilante F, Lindh R (2012) Multiconfiguration second-order perturbation theory approach to strong electron correlation in chemistry and photochemistry. WIREs Comput Mol Sci 2:585–603
Rubio M, Roca-Sanjuán D, Serrano-Andrés L, Merchán M (2009) Determination of the electron-detachment energies of 2′-deoxyguanosine 5′-monophosphate anion: influence of the conformation. J Phys Chem B 113:2451–2457
Segarra-Martí J, Merchán M, Roca-Sanjuán D (2012) Ab initio determination of the ionization potentials of water clusters (H2O)(n) (n = 2–6). J Chem Phys 136:244306
Ghigo G, Roos BO, Malmqvist PÅ (2004) A modified definition of the zeroth-order Hamiltonian in multiconfigurational perturbation theory (CASPT2). Chem Phys Lett 396:142–149
Forsberg N, Malmqvist PÅ (1997) Multiconfiguration perturbation theory with imaginary level shift. Chem Phys Lett 274:196–204
Roos BO, Malmqvist PÅ (2004) Relativistic quantum chemistry: the multiconfigurational approach. Phys Chem Chem Phys 6:2919–2927
Hess BA, Marian CM, Wahlgren U, Gropen O (1996) A mean-field spin–orbit method applicable to correlated wavefunctions. Chem Phys Lett 251:365–371
Christiansen O, Gauss J, Schimmelpfennig B (2000) Spin-orbit coupling constants from coupled-cluster response theory. Phys Chem Chem Phys 2:965–971
Aquilante F, De Vico L, Ferré N, Ghigo G, Malmqvist PÅ, Neogrady P, Pedersen TB, Pitonak M, Reiher M, Roos BO, Serrano-Andrés L, Urban M, Veryazov V, Lindh R (2010) Software news and update MOLCAS 7: the next generation. J Comput Chem 31:224–247
Aquilante F, Pedersen TB, Veryazov V, Lindh R (2013) MOLCAS-a software for multiconfigurational quantum chemistry calculations. WIREs Comput Mol Sci 3:143–149
González-Luque R, Climent T, González-Ramírez I, Merchán M, Serrano-Andrés L (2010) Singlet–triplet states interaction regions in DNA/RNA nucleobase hypersurfaces. J Chem Theor Comput 6:2103–2114
de Vico L, Liu Y-J, Krogh JW, Lindh R (2007) Chemiluminescence of 1,2-dioxetane reaction mechanism uncovered. J Phys Chem A 111:8013–8019
Farahani P, Lundberg M, Lindh R, Roca-Sanjuán D (2015) Theoretical study of the dark photochemistry of 1,3-butadiene via the chemiexcitation of dewar dioxetane. Phys Chem Chem Phys 17:18653–18664
Acknowledgements
This research was supported by Projects CTQ2014-58624-P of the Spanish MINECO/FEDER. D.R.-S. thanks the “Juan de la Cierva” program of the Spanish MINECO (Ref. JCI-2012-13431). A.F.-M. thanks BES-2011-048326 FPI Grant and the EEBB-I-14-08821 mobility program (MINECO). I.F.G. and R.L. acknowledge financial support from the Swedish Research Council (Grant No. 2012-3910), the eSSENCE program, and Uppsala University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Published as part of the special collection of articles derived from the 10th Congress on Electronic Structure: Principles and Applications (ESPA-2016).
Rights and permissions
About this article

Cite this article
Francés-Monerris, A., Fdez. Galván, I., Lindh, R. et al. Triplet versus singlet chemiexcitation mechanism in dioxetanone: a CASSCF/CASPT2 study. Theor Chem Acc 136, 70 (2017). https://doi.org/10.1007/s00214-017-2095-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-017-2095-x