
Abstract
Porous silicon carbide offers a great potential as a sensor material for applications in medicine and energetics; however, the theoretical chemical characterization of its surface is almost nonexistent, and a correct understanding of its chemical properties could lead to the development of better applications of this nanostructure. Hence, a study of the effects of different passivation agents on the structure and electronic properties of porous silicon carbide by means of density functional theory and the supercell technique was developed. The porous structures were modeled by removing columns of atoms of an otherwise perfect SiC crystal in the [001] direction, so that the porous structure exhibits a surface exclusively composed of Si atoms (Si-rich) using different surface passivation agents, such as hydrogen (H), fluoride (F) and chloride (Cl). The results demonstrate that all of the passivation schemes exhibit an irregular band gap energy evolution due to a hybridization change of the surface. The structural analysis shows a great dependence of the bond characteristics on the electronegativity of the bonded atoms, and all of the structural and electronic changes could be explained due to steric effects. These results could be important in the characterization of pSiC because they provide insight into the most stable surface configurations and their electronic structures.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In recent years, the need for the development of alternative energy solutions has issued an increasing interest in the investigation of new materials for devices such as solar and fuel cells. One of the most attractive alternatives to enhance the performance of solar cells is the use of nanomaterials, specially the nanoporous materials, which can be used as a backside reflector, or for an overall better performance [1, 2]; furthermore, one of the main concerns of the renewable energy sources is the performance of storage devices such as batteries and super capacitors, which can also be improved using porous materials as electrodes [3, 4]. On the other hand, silicon carbide (SiC) is a binary compound, which has been identified as an alternative semiconductor material for power electronics, because the SiC exhibits some excellent chemical and physical properties, such as their high strength and hardness, low expansion coefficient, chemical and thermal stability at elevated temperatures, good thermal shock resistance and thermal conductivity, high resistance to corrosion, high Young’s modulus, ability to withstand high temperatures and mechanical stresses and wide band gap [5]. In particular, the β-SiC polytype can be synthesized in a variety of nanostructures, such as nanoporous, which is especially interesting since this kind of nanostructure provides a large highly reactive internal surface area, leading to its potential application as a fast-response hydrogen-sensing material [6], which would be fundamental for applications where the H is used as an alternative to fossil fuels. For these applications, it is essential to understand the effects of surface modifications on the electronic band structure of porous silicon carbide (pSiC). Other interesting applications of pSiC can be found in the biotechnology field, where pSiC could be used as a membrane in implantable biosensors because it exhibits less protein adhesion than porous silicon [7]. Additionally, pSiC exhibits highly efficient blue-to-violet photoluminescence at room temperature [8], which makes it suitable for optoelectronic applications.
Motivated by recent theoretical studies and experimental developments on the synthesis and characterization of pSiC [9], we have carried out a study of the effects of different passivation agents on the structure and electronic properties by means of density functional theory (DFT) using the generalized gradient approximation (GGA), based on the pseudopotential plane-wave approach with the supercell technique.
The changes of the pSiC electronic states using different surface passivation agents, such as hydrogen (H), fluoride (F), and chloride (Cl), were analyzed.
The results demonstrate that the electronic properties of pSiC are greatly influenced by the surface passivation of the porous structure. The changes of the band gap that arise due to surface pore chemistries lead to the possibility of band gap engineering.
2 Model and calculation scheme
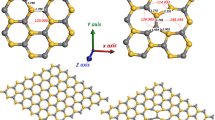
The nanopores were modeled using the supercell scheme [10, 11], by removing columns of atoms of Si and C in the [001] direction of an otherwise perfect 3C-SiC crystal. To this end, a 32-atom supercell with lattice parameters of A = B = 8.6 Å and C = 4.3 Å was chosen. Due to the binary nature of SiC, there are multiple surface configurations that the porous structures could give rise. In this work, we focus on a pSiC surface composed exclusively composed of Si atoms (Si phase) at the surface, as shown in Fig. 1. This configuration was chosen to evaluate the viability of this kind of surface since the majority of the experimental evidence suggests that the surface of pSiC is mainly composed of a C-rich phase [12]. This approach to the supercell was chosen due to current limitations of a typical DFT calculation, which becomes too computationally expensive with a large number of atoms, to model a porous structure with more semblance to experimental results. A large supercell has to be constructed to achieve various porous configurations and morphologies; thus, the model is limited to a highly periodical and ordered one, which would be hard to achieve experimentally, since the majority of the porous materials have irregular pore morphologies and distributions [13, 14] with multiple surface configurations. However, it is worth studying the proposed Si phase as a limit case of a Si enriched surface, which would cause diverse modifications to the electronic structure of these porous materials; besides, this approach has two advantages: it is simple and emphasizes the interconnection feature of the system, in contrast to the quantum wire model [15]. This pore morphology was achieved by removing 13 atoms from the supercell as illustrated in Fig. 1.

Top view of a 32-atom supercell 3C-SiC bulk crystal (dotted line), b full H passivated Si-rich porous SiC
All surface dangling bonds were passivated with H atoms. To study the effects of different chemical passivation agents on the electronic and structural properties of pSiC, four H atoms were replaced with four F (H + F) and Cl (H + Cl) atoms. In order to evaluate the most probable sites for this atoms to bond to the pSiC surface, two positions were chosen for the inclusion of the four F or Cl atoms; one at the center of the pore walls and the second on the corners of the pores, with the main difference being the distance and position of the neighboring H atoms.
The electronic band structure and density of states (DOS) of pSiC were calculated using the first-principles density functional theory scheme based on a generalized gradient approximation with a revised version of the Perdew–Burke–Ernzerhof (RPBE) [16] exchange–correlation functional and norm-conserving pseudopotentials [17] as implemented in the CASTEP code [18]. All structures were optimized using the BFGS scheme [19] to obtain their minimum energy configuration, the cutoff energy used was 850 eV, and finally a highly converged set of k points was employed, with grids up to 3 × 3 × 5 in size, according to the Monkhorst–Pack scheme [20].
3 Results and discussion
In Fig. 2, the electronic band structures of pSiC with different chemical passivation schemes, full H, and partially fluorinated (H + F) and chlorinated (H + Cl) saturations (Fig. 2a–c, respectively) are shown. It can be seen that each of the cases features a direct band gap, which would enhance the optical activity on this structures. The full H case has a lower band gap energy compared to that of the H + F and H + Cl passivation. Also, it can be observed that the H + F and H + Cl cases exhibit a similar band gap energy and behavior such as a trap-like state near the conduction band minimum. The increased band gap energy of the H + F and H + Cl cases compared to the full H could be attributed to the charge transfer that occurs on the surfaces of the structures due to the presence of the F and Cl atoms, respectively (especially by Si–F and Si–Cl bonds [21, 22]), which in turn enhances the charge transfer between Si and C, thus creating a greater band gap. It is worth noting that especially the H + Cl case has semi-flat states around the maximum valence band energy, which may be due to highly localized orbitals around the Cl and H atoms.

Electronic band structure of the Si-phase pSiC with a full H, b H + F and c H + Cl passivation at the surface. The respective atomic structure models are shown in the top panel
To further analyze the electronic structures of the pSiC, we calculated their atom-resolved electronic density of states (DOS). The results are shown in Fig. 3. In this figure, the top panel (Fig. 3a) shows the DOS for the H + F case. The low panel (Fig. 3b) represents the DOS for H + Cl case. It is clear that the valence band states in the H + F pSiC are dominated by contributions from the C atoms, while the principal contributor to the conduction band states is Si. This behavior suggests that a donor–acceptor system is operating in the structure, where Si is the electron donor and C is the acceptor of the electrons that fill most of the electronic states of the C atoms in the valence band, thus suggesting a highly ionic bond nature. Different behaviors are observed for the H + Cl case, in which the main contributors to the valence band are C and Cl atoms and, to a lesser degree, the H atoms. Thus, the donor–acceptor system of the H + F is different from that of the H + Cl pSiC. This difference can be explained since the chloride is more polarizable than fluorine. The bonds of Si–Cl involve considerable covalency–ionicity fluctuations [23]. This may be due to steric effects and the high electronegativity of the Cl atoms, which strongly attract electrons, thus reducing the electronic charge that C can attract from Si.

Atom-resolved densities of states (DOS) for a H + F and b H + Cl passivated pSiC. The orange, blue, red, black and green lines represent the DOS of Si, F, H, C and Cl atoms, respectively
Figure 4a, b shows the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) isosurfaces of the H + F and H + Cl. For the H + F (Fig. 4a), the HOMO is principally located on a C atom and around of the Si–F and Si–H bonds. The LUMO is located primarily around Si atoms, near to the C atoms and around to the F atoms. For the H + Cl (Fig. 4b) case, a similar behavior is observed, where the HOMO orbital is principally located on a C atoms and around of the Si–Cl and Si–H bonds. The LUMO is located primarily around Si atoms, near to the C atoms and around to the Cl atoms. Both observations are consistent with the information obtained from the DOS analysis in the previous section. It can also be seen that the trap-like state observed in the electronic band structure arises from highly localized orbitals around the F and Cl.

HOMO and LUMO of the a H + F and b H + ClpSiC surfaces with 40.6 % porosity are depicted. The blue (lower panels) and green (higher panels) isosurfaces represent the HOMO and LUMO orbitals, respectively, with an isovalue of 0.6 for the HOMO surface and 0.18 for the LUMO
Finally, with the previous analysis the general effects of F and Cl have been studied. However, due to the complexity of the pore surface some interesting effects arise in the pore corners, which modify the structure and electronic properties of pSiC. We compared the electronic band structure of the H + F and H + Cl in the corners and wall positions schemes.
In Fig. 5a for H + F corners case, the highest energy valence band has a wide dispersion, especially at k = R (0.5b 1, 0.5b 2, 0.5b 3, where b x are the reciprocal lattice vectors of the supercell), which could be caused by the multiple effects that arose in the geometry optimization, such as the formation of H2 molecules and a surface reconstruction of the Si groups in the pore walls, creating H–Si–Si–H bonds. Compared to Fig. 5a, the passivation with F in the pore walls presents a large band gap energy, which could be attributed to the absence of the surface reconstruction and dangling bonds, which are present in the corner passivation, and this particular behavior could be caused by steric effects due to the proximity of H and the limited space in the pore corners.

Electronic band structure of the Si-phase pSiC with a H + F corners case, b H + F centers case, c H + Cl corners case and d H + Cl wall case passivation at the surface. The respective atomic structure models are shown in the top panel
Furthermore, in Fig. 5c the band structure of H + Cl corner case shows an accentuated crossing of the Fermi energy compared to Fig. 5a, which can be explained by the results of the geometry optimization, where besides the H molecules the Cl atoms are unbounded from the pore surface and concentrate on the pore cavity, and this could be explained by the interaction of the Si–Cl generating an inductive effect, distributing a larger positive charge on the Si initially bonded to the Cl and thus favoring the polarization of the Si–Cl bond and its subsequent departure. This effect produced trap-likes states near the conduction band minimum and the valence band maximum. Cl atoms in the structure are polarized and gain a partial negative charge (as a result of the Si–H bonds), so that in this region a charge repulsion remains, and the steric effects result in the expulsion of all Cl atoms to the pore center positions [24].
Figure 5d shows the case of H + Cl passivation located in the pore walls, where the chemical environment is the most favorable, and since there are no steric repulsions effects involving the Si–Cl bond, the Si atoms gain all the positive charge, and the Si bonded to Cl acquires slightly more positive charge since the Si–Cl single bond can be described as having certain ionic character (Si + Cl−) that corresponds to the difference in electronegativity between Si and Cl. A Si–Cl bond distance of 2.01 Å has been reported for several molecules, whereas we calculated 2.096 Å. The bond length appears to depend on the difference between the electronegativities of the bonded atoms; the bond is shorter if the difference is large, as is the case for the walled Si–Cl [25]; however, this distance could be affected by steric effects as seen in Fig. 5c. The highly electronegative Cl generates a high polarity in the Si–Cl bond.
To verify this observation, calculations of the Mulliken charges of each atom were carried out, due to the large quantity of data, and the results can be seen in Table 1, and the guides to the atom labeling are seen in Figs. 1 and 4. In all cases, the Si atoms always maintains a positive charge, and in H + F and H + Cl wall cases, the Si3, Si6, Si9 and Si12 atoms have the highest positive charge +1.53 and +1.13e, respectively. The larger positive charge in the H + F case may be due to the nature of the passivating element, because the F is more electronegative than Cl [26].
Analyzing the structures where the surface was passivated at the corners comparing the atoms with either F or Cl bonded, the Si atoms (Si1, Si4, Si7 and Si10) in the H + F passivated pore, the charge increased by +0.03e compared with Si atoms (Si3, Si6, Si9 and Si12) of the H + F wall case structure. The difference may be that in the corner model each Si atom is surrounded by three C atoms causing a decrease in charge of Si by an inductive effect of these three C attached to Si [27]. The results of the Mulliken charge analysis sustain the observations made at the surface optimization results. The results show that the Si-rich surface could be stable depending on the passivation agent in its surface.
4 Conclusions
In summary, the electronic properties of porous silicon carbide with a Si-rich surface were studied using the density functional theory scheme. Results show that the electronic properties are highly dependent of the surface passivation of the structure, having the F and Cl lower energy band gap due to trap-like states induced by the high electronegativity of F and Cl which limits the charge transfer between Si and C. Also, it could be observed that the environment around Cl and F could dramatically change the properties of the structure since, when in a proximity of various H atoms, the F and specially Cl become repelled from the structure inducing a surface reconstruction and metallic states within the electronic band structure. These results could be useful to determine the most probable locations for other species other than H in the pore surface, and also the orbital analysis suggests that the wall bonds are stable.
References
Garralaga Rojas E, Hensen J, Baur C, Brendel R (2011) Sintering and reorganization of electrochemically etched mesoporous germanium layers in various atmospheres. Sol Energy Mater Sol Cells 95(1):292–295. doi:10.1016/j.solmat.2010.04.042
Lima FAS, Vasconcelos IF, Lira-Cantu M (2015) Electrochemically synthesized mesoporous thin films of ZnO for highly efficient dye sensitized solar cells. Ceram Int 41(8):9314–9320. doi:10.1016/j.ceramint.2015.03.271
Ge M, Fang X, Rong J, Zhou C (2013) Review of porous silicon preparation and its application for lithium-ion battery anodes. Nanotechnology 24(42):422001. doi:10.1088/0957-4484/24/42/422001
Kim M, Oh I, Kim J (2015) Effects of different electrolytes on the electrochemical and dynamic behavior of electric double layer capacitors based on a porous silicon carbide electrode. Phys Chem Chem Phys 17(25):16367–16374. doi:10.1039/C5CP01728A
Lee S-H, Yun S-M, Kim S, Park S-J, Lee Y-S (2010) Characterization of nanoporous β-SiC fiber complex prepared by electrospinning and carbothermal reduction. Res Chem Intermed 36(6–7):731–742. doi:10.1007/s11164-010-0175-9
Naderi N, Hashim MR, Saron KMA, Rouhi J (2013) Enhanced optical performance of electrochemically etched porous silicon carbide. Semicond Sci Technol 28(2):025011. doi:10.1088/0268-1242/28/2/025011
Kim K-S, Chung G-S (2011) Fast response hydrogen sensors based on palladium and platinum/porous 3C-SiC Schottky diodes. Sens Actuators, B 160(1):1232–1236. doi:10.1016/j.snb.2011.09.054
Naderi N, Hashim MR (2013) Visible-blind ultraviolet photodetectors on porous silicon carbide substrates. Mater Res Bull 48(6):2406–2408. doi:10.1016/j.materresbull.2013.02.078
Kim K-S, Chung G-S (2011) Characterization of porous cubic silicon carbide deposited with Pd and Pt nanoparticles as a hydrogen sensor. Sens Actuators, B 157(2):482–487. doi:10.1016/j.snb.2011.05.004
Trejo A, Cruz-Irisson M (2013) Computational modeling of the size effects on the optical vibrational modes of H-terminated Ge nanostructures. Molecules 18(4):4776. doi:10.3390/molecules18044776
Calvino M, Trejo A, Iturrios MI, Crisóstomo MC, Carvajal E, Cruz-Irisson M (2014) DFT study of the electronic structure of cubic-SiC nanopores with a C-terminated surface. Journal of Nanomaterials 2014:7. doi:10.1155/2014/471351
Shin W, Seo W, Takai O, Koumoto K (1998) Surface chemistry of porous silicon carbide. J Electron Mater 27(4):304–307. doi:10.1007/s11664-998-0405-8
Shishkin Y, Choyke WJ, Devaty RP (2004) Photoelectrochemical etching of n-type 4H silicon carbide. J Appl Phys 96(4):2311–2322. doi:10.1063/1.1768612
Fang C, Föll H, Carstensen J (2006) Electrochemical pore etching in germanium. J Electroanal Chem 589(2):259–288. doi:10.1016/j.jelechem.2006.02.021
Miranda A, Trejo A, Canadell E, Rurali R, Cruz-Irisson M (2012) Interconnection effects on the electronic and optical properties of Ge nanostructures: a semi-empirical approach. Physica E 44(7–8):1230–1235. doi:10.1016/j.physe.2012.01.017
Hammer B, Hansen LB, Nørskov JK (1999) Improved adsorption energetics within density-functional theory using revised Perdew–Burke–Ernzerhof functionals. Phys Rev B 59(11):7413–7421. doi:10.1103/PhysRevB.59.7413
Hamann DR, Schlüter M, Chiang C (1979) Norm-conserving pseudopotentials. Phys Rev Lett 43(20):1494–1497. doi:10.1103/PhysRevLett.43.1494
Clark SJ, Segall MD, Pickard CJ, Hasnip PJ, Probert MI, Refson K, Payne MC (2005) First principles methods using CASTEP. Z Kristallogr 220:567–570. doi:10.1524/zkri.220.5.567.65075
Pfrommer BG, Côté M, Louie SG, Cohen ML (1997) Relaxation of crystals with the quasi-Newton method. J Comput Phys 131(1):233–240. doi:10.1006/jcph.1996.5612
Monkhorst HJ, Pack JD (1976) Special points for Brillouin-zone integrations. Phys Rev B 13(12):5188–5192. doi:10.1103/PhysRevB.13.5188
Tossell JA (1987) Quantum mechanical studies of Si–O and Si–F bonds in molecules and minerals. Phys Chem Miner 14(4):320–326. doi:10.1007/BF00309804
Shaik S, Danovich D, Silvi B, Lauvergnat DL, Hiberty PC (2005) Charge-shift bonding—a class of electron-pair bonds that emerges from valence bond theory and is supported by the electron localization function approach. Chem Eur J 11(21):6358–6371. doi:10.1002/chem.200500265
Lommerse JPM, Stone AJ, Taylor R, Allen FH (1996) The nature and geometry of intermolecular interactions between halogens and oxygen or nitrogen. J Am Chem Soc 118(13):3108–3116. doi:10.1021/ja953281x
Gillespie RJ, Robinson EA (2005) Models of molecular geometry. Chem Soc Rev 34(5):396–407. doi:10.1039/B405359C
Wang W, Wong N-B, Zheng W, Tian A (2004) Theoretical study on the blueshifting halogen bond. J Phys Chem A 108(10):1799–1805. doi:10.1021/jp036769q
Lauvergnat D, Hiberty PC, Danovich D, Shaik S (1996) Comparison of C–Cl and Si–Cl bonds. A valence bond study. J Phys Chem 100(14):5715–5720. doi:10.1021/jp960145l
Lambert JB, Wang G-T (1988) Participation of beta carbon-silicon bonds in the development of positive charge in five-membered rings. J Phys Org Chem 1(3):169–178. doi:10.1002/poc.610010307
Acknowledgments
This work was partially supported by Project 252749 from CONACyT and multidisciplinary Projects SIP-IPN 2014-1640 y 2014-1641 from InstitutoPolitécnico Nacional.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published as part of the special collection of articles “CHITEL 2015 - Torino - Italy”.
Rights and permissions
About this article

Cite this article
Calvino, M., Trejo, A., Crisóstomo, M.C. et al. Modeling the effects of Si-X (X = F, Cl) bonds on the chemical and electronic properties of Si-surface terminated porous 3C-SiC. Theor Chem Acc 135, 104 (2016). https://doi.org/10.1007/s00214-016-1861-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-016-1861-5