
Abstract
The hydration of NaCl has been widely studied and believed to be important for understanding the mechanisms of salt dissolution in water and the formation of ice nucleus, cloud, and atmospheric aerosols. However, understanding on the poly-NaCl ion pair interacting with water is very limited. Here, we investigated the adsorption of water molecules on (NaCl)3, using both theoretical calculations and anion photoelectron spectroscopy measurements. The calculated vertical detachment energies and the experimental ones agree well with each other. Furthermore, we found that, for neutral (NaCl)3(H2O) n (n = 2–7) clusters, the water-doped cuboid and structures formed by adding water molecules on the Na–Cl edges of the cuboid are energetically favored; water molecules preferentially bind to the Na–Cl edge if the NaCl ion pair has larger partial charges than others. We also found the anionic structures are more various compared with neutral ones, and the Na+ and Cl− ions are hydrated more easily in the anionic clusters than in the corresponding neutrals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Salt ions can strongly affect the water/air surface tension and the solubility of proteins [1–5]. They are also related to the formation and reactivity of sea salt aerosols [6–8]. Thus, one challenge is to understand the ion–water and water–water interactions at the molecular level and to obtain the detailed solvation mechanisms of ions and/or ion pairs in water [9–11]. Molecular clusters are considered to play important roles in providing the connection between the properties of isolated molecules/atoms and those of the condensed phase systems. In particular, due to their significance in many fields, small clusters of sodium chloride and water molecules NaCl(H2O) n have called attention from both experimental [11–16] and theoretical [17–33] groups.
In contrast to the large number of studies on the clusters formed by a single sodium chloride ion pair and water molecules, i.e., NaCl(H2O) n , studies on (NaCl) x (with x > 1) are rare. There do exist several studies on sodium chloride clusters [34–37]. For example, Sunil and Jordan [34] performed theoretical calculations on negative (NaCl)2–4 clusters and discussed the properties of the excess electron binding. This topic was revisited by Anusiewicz et al. [35] and they argued that there are two types of anions in this system, dipole-bound anions and solvated-electron species based on their high-level calculations. Iwona et al. [36] reported results on (NaCl)1–6 calculations in which various isomers with different bonding forms are presented. Ayuela et al. [37] also theoretically predicted the structures of (NaCl) n (n ≤ 19) clusters, using the ab initio perturbed-ion model. It is noted that these two studies [36, 37] actually gave two similar minimal structures of (NaCl)3. However, these studies do not include water molecules in the clusters, and thus, the detailed interactions of (NaCl) x with water remain unexplored. Motivated by the potential importance of (NaCl)3(H2O) n clusters in the formation of doped crystals [38], atmospheric processes at the ocean surface [39, 40], and salt dissolution processes [41], we herein investigated the initial hydration of (NaCl)3 by using theoretical calculations and photoelectron spectroscopy experiments, for clusters (NaCl)3(H2O) n (n = 0–6). The calculations were performed for the clusters both in the anionic and neutral states.
Photoelectron spectroscopy in combination with quantum calculations is a very mature tool to study clusters [42]. However, computational studies on the clusters with relatively large size are hindered by the outstanding difficulty in covering the large configuration space of the clusters. There exist many local minima on the potential energy surfaces; thus, it is computationally challenging to locate all the stable isomers. In the current study, to overcome this difficulty, we used our well-established integrated tempering sampling (ITS) molecular dynamics [43] to generate structures of low configuration energies. ITS allows efficient search of structures by sampling over a broad energy range and thus permits fast identification of low-energy structures and transition states. The low-energy structures obtained from the ITS calculations are then optimized using both classical force field and DFT functional. This procedure was proven effective and efficient in structure search for (H2O) n , LiI(H2O) n , CsI(H2O) n [44], and NaCl(H2O) n [45] with n being up to 20 in our previous studies. We note here that other global minimization methods besides ITS can be efficient and effective for searching the low-energy structures. However, through ITS simulation, one can readily obtain the various thermodynamic properties in addition to stable structures as well [44].
The rest of this account is organized as follows: In Sect. 2, we described the experimental details and theoretical methodologies; experimental spectra and computational results of both anions and neutrals and discussions are arranged in Sect. 3; and Sect. 4 is the concluding remarks.
2 Experimental details and theoretical methodologies
In this part, we will first describe the details of the anion photoelectron spectroscopy experiments. Then, a brief introduction of the basic principles of the integrated tempering sampling (ITS) method will be discussed. The theoretical details, including the classical MD (and ITS) simulations and the DFT calculations, are described in the last part.
2.1 Photoelectron spectroscopy
The experiments were conducted on a homebuilt apparatus consisting of a time-of-flight (TOF) mass spectrometer and a magnetic-bottle photoelectron spectrometer, which has been described previously [46]. Briefly, the (NaCl) −3 (H2O)n clusters were produced in a laser vaporization source, by laser ablating a rotating and translating NaCl disk target with the second harmonic (532 nm) light pulses from a Nd:YAG laser, while helium carrier gas with ~4 atm backing pressure seeded with water vapor was allowed to expand through a pulsed valve to generate and cool the hydrated (NaCl) −3 clusters. The cluster anions were mass-analyzed by the TOF mass spectrometer. The (NaCl) −3 (H2O) n (n = 0–6) clusters were each mass-selected and decelerated before being photodetached by the 532 nm (2.331 eV) photons from another Nd:YAG laser. The photodetached electrons were energy-analyzed by the magnetic-bottle photoelectron spectrometer. The instrumental resolution was approximately 40 meV for electrons with 1 eV kinetic energy.
2.2 Integrated tempering sampling (ITS) method
Briefly, in the ITS simulation, a modified potential energy function is obtained from the original potential by making use of an energy distribution function with the form of a summation of Boltzmann factors over a series of temperatures:
where \(\beta = 1/k_{B} T\), k B is the Boltzmann constant, T the temperature, and U the potential energy of the system.
The sampled potential energy range by such a method is largely expanded, to both low and high potential energies, when compared to standard MD simulations. In the current study, we used 300 discrete temperatures even spaced in the range of 100–250 K. In the ITS simulation, the system is governed with a biased potential function in the form of Eq. (1). An equilibrium simulation with ITS yields a biased distribution function as a function of the potential energy of the system,
where \(Q^{{\prime }} = \mathop \smallint \nolimits e^{{ - \beta {\prime }U\left( r \right)}} {\text{d}}r\). In ITS, since \(U^{{\prime }} \left( r \right)\) is a known function of \(U(r)\), the distribution function \(\rho (r)\) for the system with the unbiased potential can be recovered back from \(\rho^{{\prime }} ({\text{r}})\) as:
where \(Q = \int {e^{ - \beta U\left( r \right)} {\text{d}}r}\). More details can be found in our previous publications [43, 47].
2.3 Simulation details
We used SANDER module of AMBER 9.0 program package [48] to perform MD simulations of (NaCl)3(H2O) n (n = 0–7), in which the ions 08 parameters [49] and single-point-charge/extended (SPC/E) water model [50] were utilized. The ion-water cross terms were calculated with the Lorentz Berthelot combination rules. These models are popular and well established in AMBER program package, and they give convincing bulk solution properties comparing to experimental data [49, 50]. One may expect more reliable thermodynamic properties in the classical simulations when more accurate models, such as polarizable force field, are utilized to these systems, but our earlier studies did show that the classical force field used in this study provides reasonable description of the cluster structures compared with the quantum calculations [44]. The initial configurations used for MD were generated randomly and followed by an energy minimization. To avoid the molecules to escape from the clusters to the vacuum, a spherical potential is applied to the center of mass of every atom. The restricting force of the added potential has the form of:
where r is the spatial vector of an atom, ɛ is the maximal restrict force [here, we set = 5 kcal/(mol Å)], r 0 is the radius of sphere (here, we set r 0 = 7 Å), and is the flexibility constant of the sphere shell (we set = 5). In this way, the molecules are confined in a spherical space with a radius of 7 Å.
We collected a 100-ns-long ITS simulation trajectory for each system of (NaCl)3(H2O) n . Hundreds of different low-energy configurations were chosen from simulated trajectory as the initial structures for structure optimization using classical force field. These structures then converged to several dozens of local minimal structures, and then, re-optimizations by DFT calculations were performed. The DFT calculations were performed using GAUSSIAN 09 program package [51]. The structures of (NaCl)3(H2O) n were fully optimized with DFT employing the dispersion-corrected hybrid functional ωB97XD [52]. The Pople’s all-electron basis set 6-311++G(d,p) was used for all the atoms. The ωB97XD functional has been shown to perform extraordinarily well on describing dispersion interaction [53], which is the main consideration for such weak-interaction cluster systems. As the energetics and geometries of one electron-bound anion clusters will be evaluated, we add the diffuse functions to all atoms. After these structures were fully optimized, harmonic vibrational frequencies were calculated to confirm that they are indeed local minima and to obtain the zero-point energy corrections and the Gibbs free energies. Zero-point vibrational energy corrections were incorporated to the sum of the electron energies.
3 Results and discussions
3.1 The experimental and theoretical vertical detachment energies
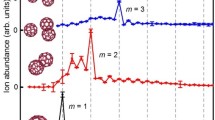
The photoelectron spectra of (NaCl) −3 (H2O) n (n = 0–6) clusters recorded at 532 nm are shown in Fig. 1. In general, all the spectral features of the clusters are broad, and their maxima are about 0.99, 1.04, 1.09, 1.11, 1.03, 1.05, and 1.07 eV for n = 0–6, respectively. Specifically, for (NaCl) −3 (H2O), there is an additional small feature centered at about 1.51 eV. Figures 2 and 3 present the typical low-energy structures for each cluster size along with their relative energies and calculated VDEs (the geometries and energetics are discussed below). The calculated VDEs of the lowest energy structures of (NaCl) −3 (H2O) n (n = 0, 1, and 3–6) are in good accord with the experimental values. While for (NaCl) −3 (H2O)2, the theoretical VDE of the lowest energy isomer, which is a distorted cuboid structure, is 0.54 eV and much lower than the experimental value of 1.09 eV. The VDE value is also 0.54 eV when calculated at the MP2/aug-cc-pvtz level of theory. These results at both ωB97XD and MP2 levels suggest that the structure 2A− is unlikely the structure observed in the experiments. By examining the Gibbs free energies of these structures, it is found that structure 2C− has the lowest Gibbs free energy and its theoretical VDE is 1.05 eV, consistent with the experimental measurement of 1.09 eV very well. For (NaCl) −3 (H2O) n (n = 0, 1, and 3–6) clusters, we also checked their Gibbs free energies and found that the lowest electronic energy structures also have the lowest Gibbs free energies. These results suggest that the photoelectron spectra may relate to the structures of relative large stability in Gibbs free energies. The good performance of ωB97XD/6-311++G(d,p) on the (NaCl) −3 (H2O) n clusters shown above provides the credence of the following discussions on the corresponding neutral clusters.

Photoelectron spectra of the (NaCl) −3 (H2O) n (n = 0–6) clusters taken with 532 nm photons

Typical minimal energy structures of (NaCl) −3 . The relative energies to the corresponding smallest values (first line) and the theoretical VDEs (second line) are presented. All the energies are in eV

Typical minimal energy structures of (NaCl) −3 (H2O) n (n = 1–6). They are presented in the similar way as those in Fig. 2
3.2 Structures of (NaCl) −3 (H2O) n
3.2.1 Typical minimal energy structures of (NaCl) −3
The typical minimal energy structures of (NaCl) −3 are displayed in Fig. 2. As discussed later, the structures of salt–water molecules being adsorbed on (NaCl) −3 mostly evolve from these pure salt structures. The global minimum of the anion (0A− in Fig. 2) is a “Λ-shaped” structure, similar to that reported in the previous studies [34–37]. The VDE of this structure is calculated to be 1.045 eV, in good agreement with the experimental measured value of 0.99 eV. Structure 0B− is higher in energy by only 0.001 eV than the structure 0A−. To the best of our knowledge, this “fish-shaped” C2V-symmetric structure (structure 0B−) has not been reported so far for either anionic or neutral forms. The kite-shaped 0C−and a little distorted kite-shaped 0F−, which are optimized from the initial configuration of by changing the positions of Na+ and Cl− of structure 0C−, are higher in energy than 0A− by 0.170 and 0.519 eV, respectively. The linear 0D− and Y-shaped 0E− structures are much less stable than others identified here. In summary, most of the structures found in the current study are very similar to those reported previously. Therefore, we followed Anusiewicz et al. [35] for the naming of these structures. Besides, the current study also identifies a new low-energy “fish-shaped” structure (0B−), which has not been reported before. Furthermore, the excess electron substantially increases the complexity of the potential energy surface, which will be shown by the richness of the anionic structures in comparison with the corresponding neutral ones.
3.2.2 Typical minimal energy structures of (NaCl) −3 (H2O) n (n = 1–6)
Figure 3 shows the typical minimal energy structures of (NaCl) −3 (H2O) n (n = 1–6). For n = 1, five of the six lowest energy structure are formed through adsorbing one water molecule to the “Λ-shaped” structure of (NaCl) −3 (see 0A− in Fig. 2). In structure 1A−, the O atom of the water interacts with two Na atoms, and the two H atoms of the water are dangled. Due to the excess electron, it can be seen that the two dangling H atoms forming two O–H···e− bonds (see Fig. S1 in Supporting Information), which have been suggested to be strongly favored in the liquid phase of methanol [54].
For n = 2, the global minimum structure 2A− is a slightly distorted cuboid. As mentioned above, the calculated VDE of this configuration is 0.54 eV. Yang et al. [55] have previously found that the neutral (NaCl)4 forms a particularly stable cuboid and should have relatively low electron affinity. The other “Λ-shaped”-based structures are formed by re-orientation of the water molecule to interact with Na or Cl ions in different ways for both n = 1 and 2. In addition to these structures, the 1D− and 2C− structures are derived from the newly identified “fish-shaped” structure (0B− in Fig. 2).
For (NaCl) −3 (H2O) n (n = 3–6) clusters, the excess electron causes a significant perturbation to the neutral structures. It can be seen that all the local minimal structures are derived from the distorted cuboid (NaCl) −3 (H2O)2 (4A− and 6A−) or much more distorted structures (3A− and 5A−). For these clusters with larger number of water molecules, the cuboid-based structures no longer dominate. At the same time, the Na+ and Cl− ions are much more hydrated in the anions (such as 3A−, 3C−, and 3D−) than in the corresponding neutrals, which can be seen later. It therefore appears that the excess electron causes enhanced hydration of Na and Cl ions. For the rest of the large number of anionic clusters not discussed in details here, we provided their Cartesian coordinates, corresponding relative energies, and theoretical VDEs in Supporting Information.
3.3 Structures of (NaCl)3(H2O) n
3.3.1 Typical minimal energy structures of (NaCl)3(H2O) n (n = 0–2)
To understand salt–water interactions, it is certainly more interesting to study the neutral salt–water clusters. Figure 4 shows the typical minimal energy structures for (NaCl)3(H2O) n (n = 0–2) clusters. For (NaCl)3, the previous studies [34–37] identified and discussed two minimal energy structures, i.e., the global minimum 0A, which is a 2-dimensional (2D) plane structure and the D3h-symmetric 0B structure as shown in Fig. 4. In the current study, a third minimal energy structure (“fish-shaped” 0C in Fig. 4) was also identified though it is relatively high in energy (0.174 eV higher than 0A). It is also noted here that the D3h-symmetric structure is not found to be stable in the anionic form. The optimization from 0B in the anionic state leads the structure into the arrow-shaped 0D− structure. These findings are consistent with previous studies [35] and could be a result of the high symmetry of 0B, whose dipole moment is zero, and thus, its binding of an excess electron is too weak to form a stable anion.

Typical minimal energy structures of (NaCl)3(H2O) n (n = 0–2). The relative energies to the corresponding smallest values are presented. All the energies are in eV
Among the (NaCl)3(H2O) structures, 1A is resulted from adsorbing the water molecule on the edge of Na–Cl of the structure 0A of (NaCl)3. Adsorption of one water leads to a structure transition from planar (NaCl)3 (“arrow-shaped” 0A) to structure 1B, which is higher in energy than 1A by 0.034 eV. In structure 1B, (NaCl)3 is actually very similar to the “Λ-shaped” structure, which is the global minimum of the anionic state (0A− in Fig. 2). The other local minimum structures with much higher energies than the global minimum structure can be seen to arise from the cyclic (1C and 1D), “fish-shaped” (1E), or “arrow-shaped” (1F) (NaCl)3 structures.
It is well known that the lowest energy structure of the (NaCl)4 cluster is a Td-symmetric cuboid [36, 37]. This study finds that the global minimum structure of (NaCl)3(H2O)2 cluster is a distorted cuboid (2A in Fig. 4). In structure 2A, two water molecules occupy the sites of Na+ and Cl− of (NaCl)4, respectively, with one water molecule interacting with the other two Na+ through its O atom, and another one connecting to two Cl− via hydrogen bonds. One hydrogen bond also forms between the two water molecules, and one hydrogen atom of the Na-coordinated water is dangling. Other structures such as 2B, 2C, 2D, 2E, and 2F can all be regarded as the distorted forms of the 2A structure. In these structures, the positions and orientations of the two water molecules deviate from the cuboid 2A structure. 2G is a derivative of the planar arrow-shaped structure 0A and is much higher in energy than others. Different from (NaCl)3(H2O), most of the seven low-energy structures of the (NaCl)3(H2O)2 cluster are non-planar. From the above analyses, we can see that the adsorption of one/two water molecule(s) or one electron causes a (NaCl)3 structure transition from planar to non-planar.
3.3.2 Typical minimal energy structures of (NaCl)3(H2O) n (n = 3–7)
We show the typical low-energy structures of (NaCl)3(H2O) n (n = 3–7) clusters in Fig. 5. Interestingly, the cuboid formed by (NaCl)3(H2O)2 is maintained in many low-energy structures. A chart of possible structure evolution is given in Fig. 5.

Cuboid structure evolution in the clusters of (NaCl)3(H2O) n (n = 2–7). The relative energies to the corresponding smallest values are presented. All the energies are in eV
For (NaCl)3(H2O)3, 3A and 3B are almost degenerate in energy with the latter being higher by only 0.001 eV. The water molecule that does not belong to the cuboid, as mentioned above, binds to the Na–Cl edges. By examining the Mülliken charge of the atoms in structure 2A, it is found that water molecules prefer to bind to the Na–Cl edges, for which the Na and Cl atoms have the largest sum of partial charges (SPCs) (see the Supporting Information for the Mülliken charges in Table S1). Interestingly, when the SPC values of the two atoms are both small, binding of the water molecule to a Na–Cl edge causes the separation of Na+ and Cl− (see 3C in Fig. 5 and the Na–Cl distance in Table S2 in Supporting Information). Adsorption on the Na–Cl edge of medium SPC values results in two structures much higher in energy (3D and 3E in Fig. 5). In these two structures, Na+ and Cl− remain in contact.
The lowest energy structure of (NaCl)3(H2O)4 cluster 4A is resulted from adsorbing the other two water molecules on the two Na–Cl edges, and it is noted that the SPCs of these two edges are the highest and second highest values in structure 3A. When adsorption of water is on the other Na–Cl edges (4B to 4G), the resulted structures are of higher energies. A penta-prism structure 4H, higher in energy than 4A by 0.074 eV, was also found for the (NaCl)3(H2O)4 cluster. In this structure, the four water molecules form a four-member ring, and this ring further connects to the Λ-shaped-like (NaCl)3 structure.
The lowest energy structure of (NaCl)3(H2O)5 cluster 5A can be constructed from 4A with the newly added water molecule interacting with two Cl− and one Na+ ions. The dipole of the water molecule is almost parallel to the plane composed of Cl–Na–Cl. The binding pattern of the water molecule in this cluster is similar to that on NaCl(001) surface [56]. The adsorbed water molecules, as mentioned above, preferentially interact with Na and Cl ions rather than other water molecules. In the latter case, less stable structures form, such as structures 5D and 5F in Fig. 5.
The low-energy structures of (NaCl)3(H2O)6 also remain the basic cuboid form. The adsorbed water molecules on the NaCl ions can be seen in 6A, 6B, and 6C. Interestingly, a fused cuboidal structure 6D, which can be seen as two cuboid units sharing a face formed by (NaCl)(H2O)2 fragment, is also obtained. Although the water molecules form more water–water hydrogen bonds in 6D than in 6A, 6B, and 6C, it is still less stable than 6A by 0.023 eV.
Different from (NaCl)3(H2O) n (n = 2–6), the low-energy structures of (NaCl)3(H2O)7 are derivatives of the fused face-sharing cuboid 6D. The seventh water molecule can be adsorbed on the different Na–Cl edges (7A, 7B, 7C, 7D, and 7F). As can be predicted from the partial charge analysis of structure 6D, the most stable structure of (NaCl)3(H2O)7 is formed by adding water molecule on the Na–Cl edge of the largest SPC value, which is the edge composed of “1Cl” and “5Na” (see the label in Table S1 in Supporting Information). Two structures 7E and 7G, maintaining only one cuboid consisting of (NaCl)3(H2O)2 fragment, also appear as local minimal structures on the energy surface, and they are higher in energy than 7A by 0.056 and 0.069 eV, respectively.
By using enhanced sampling simulations followed by optimization using ωB97XD density functional, we also obtained a larger number of structures of the non-cuboid nature, including the structures formed by adsorbing water molecule(s) on the several basic structures of (NaCl)3 and other structures with Na+ and Cl− being hydrated. These structures are not shown or discussed here, and the Cartesian coordinates and energetics of all the fully optimized cluster structures are provided in Supporting Information.
3.4 Water binding energies of the lowest energy structures
To examine the stability of the cluster as a function of cluster size, we define the water binding energy (WBE) using Eq. (5):
In the above equation, E[(NaCl)3(H2O) n ] and E[(NaCl)3(H2O) n−1] are the energy of the clusters with the number of water molecules being n and n − 1, respectively; E(H2O) is the energy of water monomer (zero-point energy correction is included) and the value is −76.410660 atomic unit. WBE characterizes the absolute value of the energy decrease when a water molecule binds on the cluster. Figure 6 shows the WBEs as a function of the number of water molecules n in the lowest energy structures for both neutrals and anions. The binding energies for (NaCl)3(H2O) n (n = 1–7) are 0.644, 0.765, 0.610, 0.597, 0.565, 0.502, and 0.563 eV, respectively. The largest WBE of 0.765 eV is responsible for the second water molecule binding to structure 1A to form structure 2A. When n changes from 3 to 6, the WBE slightly decreases, which means that the further binding of water molecules to the clusters stabilizes the cluster less than the second water molecule does. When n changes from 6 to 7, a small increase of the WBE can also be found, which, as reflected from the structures changes, is due to that the fused face-sharing cuboid structure (7A) becomes the lowest energy structure in (NaCl)3(H2O)7. The two maxima of the WBE in Fig. 6 are in accord with the first and second cuboid structures being the lowest energy ones when n = 2 and 7.

The change of water binding energies (WBEs) versus the number of water molecules n for the most stable isomers of (NaCl)3(H2O) n
For the anions, the maximum WBE is 0.741 eV when n = 1, corresponding to the energy difference when the structure changes from 0A− to 1A−. As can be seen in Figs. 2 and S1 and as mentioned earlier, the two dangling O–H⋯e− bonds likely stabilize the cluster; the binding of the second water molecule results in the cuboidal 2A− structure, but the WBE of 0.613 eV is smaller than when the first water is added. For n = 2 to 6, the WBEs are all smaller than the corresponding value for n = 1. A maximum of WBE occurs at n = 3 and a slight increase exists when n changes from 5 to 6. However, these sudden increases in WBE cannot be apparently indicated by the structural changes because, as mentioned in Sect. 3.2, the anionic structures are largely perturbed and Na+ and Cl− are more hydrated than the corresponding neutral ones.
4 Concluding remarks
Water adsorption on (NaCl)3 in both anionic and neutral states is investigated in this study by photoelectron spectroscopy experiments and DFT calculations. The possible observed structures in experiments are assigned according to the DFT results. The calculated vertical detachment energies (VDEs) and the experimental ones agree well with each other. The enhanced sampling method, ITS, was employed to ensure that the low-energy structures of the clusters are fully sampled. The efficiency of this approach has been shown in finding new minimal energy structures of (H2O)12 and (H2O)20 clusters [44], which were missed by other methods. The effectiveness of this method was again demonstrated in the present study. For instance, we identified a new “fish-shaped” C2V-symmetric structure in both anionic and neutral states, which has not been reported before [35–37, 57]. The anionic one is very competitive to (higher in energy by only 0.001 eV) the previously regarded global minimum structure in terms of our DFT calculations. In addition, by using dispersion-corrected ωB97XD functional and diffusive basis set, a more convincing ranking of these structures (by energy) was obtained.
For neutral (NaCl)3(H2O) n clusters, the water-doped cuboid and the structures derived from adding water molecules on the Na–Cl edges of the cuboid are energetically favored. We found that the most stable structure of (NaCl)3(H2O)2 is a water-doped cuboid (2A in Fig. 5). This finding expands the class of dopant sodium chloride clusters: This study shows not only the metals [58] and hydroxide [59], but also water molecules can be the proper candidate for the doped NaCl clusters (crystals). The cuboid is so stable that the low-energy structures of (NaCl)3(H2O) n (n = 3–6) are those that all maintain the cuboid. The fused face-sharing cuboid structure appears when n is up to 6 though its energy is slightly higher than the water-doped cuboid structure. Finally, the derivative structures from the fused face-sharing cuboid become dominant for the low-energy structures of (NaCl)3(H2O)7. The lowest energy structures change from n = 1 to 2 and 6 to 7 are well reflected on the two maxima, which can be seen from the plot of the water binding energy (WBE) versus the cluster size (Fig. 6). Through this study, the pattern of water binding on (NaCl)3 is established. From the structure evolution of the cuboid (Fig. 5), we can clearly find that the water molecule preferentially binds to the Na–Cl edge if the NaCl ion pair has larger partial charges than others. The fused face-sharing cuboid (6D) is the first structure in which the salt is fully covered by one layer of water molecules. The seventh water molecule again binds to the Na–Cl edge instead of other water molecules in the low-energy structures of (NaCl)3(H2O)7. Thus, water molecules will preferentially bind to the Na–Cl edges than interact with other water molecules.
Finally, we want to note that the anionic clusters are more distorted and have higher configuration diversity than the corresponding neutral ones. For example, for the water-free (NaCl)3 cluster, only three local minima were found for neutral form with relatively low energies, but for anionic clusters, seven low-energy stable structures were identified. For (NaCl) −3 (H2O) n (n = 1–6) clusters, although the majority of structures are similar to the corresponding neutral ones, the Na+ and Cl− ions are much more hydrated in the former.
References
Collins KD, Neilson GW, Enderby JE (2007) Biophys Chem 128:95
Tobias DJ, Hemminger JC (2008) Science 319:1197
Gao YQ (2011) J Phys Chem B 115:12466
Gao YQ (2012) J Phys Chem B 116:9934
Yang LJ, Fan YB, Gao YQ (2011) J Phys Chem B 115:2456
Oum KW, Lakin MJ, DeHaan DO et al (1998) Science 279:74
Knipping EM, Lakin MJ, Foster KL et al (2000) Science 288:301
Finlayson-Pitts BJ (2003) Chem Rev 103:4801
Ohtaki H, Radnai T (1993) Chem Rev 93:1157
Dedonder-Lardeux C, Gre´goire G, Jouvet C et al (2000) Chem Rev 100:4023
Blandamer MJ, Fox MF (1970) Chem Rev 70:59
Ohtaki H, Fukushima N (1992) J Solu Chem 21:23
Ault BS (1978) J Am Chem Soc 100:2426
Mizoguchi A, Ohshima Y, Endo Y (2003) J Am Chem Soc 125:1716
Mizoguchi A, Ohshima Y, Endo Y (2011) J Chem Phys 135:064307
Partanen L, Mikkelä M-H, Huttula M et al (2013) J Chem Phys 138:044301
Belch AC, Berkowitz M, McCammon JA (1986) J Am Chem Soc 108:1755
Karim OA, McCammon JA (1986) J Am Chem Soc 108:1762
Ohtaki H, Fukushima N (1988) Pure Appl Chem 61:179
Ohtaki H, Fukushima N, Hayakawat E et al (1988) Pure Appl Chem 61:1321
Makov G, Nitzan A (1992) J Phys Chem 96:2965
Woon DE, Thom H, Dunning J (1995) J Am Chem Soc 117:1090
Petersen CP, Gordon MS (1999) J Phys Chem A 103:4162
Jungwirth P (1999) J Phys Chem A 104:145
Yamabe S, Kouno H, Matsumura K (2000) J Phys Chem B 104:10242
Godinho SSMC, do Couto PC, Cabral BJC (2004) Chem Phys Lett 399:200
Olleta AC, Lee HM, Kim KS (2006) J Chem Phys 124:024321
Asada T, Nishimoto K (1995) Chem Phys Lett 232:518
Laria D, Fernández-Prini R (1995) J Chem Phys 102:7664
Smith DE, Dang LX (1994) J Chem Phys 100:3757
Siu C-K, Fox-Beyer BS, Beyer MK et al (2006) Chem Eur J 12:6382
Joung IS, Luchko T, Case DA (2013) J Chem Phys 138:044103
Ghosh MK, Re S, Feig M et al (2013) J Phys Chem B 117:289
Sunil KK, Jordan KD (1987) J Chem Phys 91:1710
Anusiewicz I, Skurski P (2003) J Phys Chem A 107:2356
Malliavin M-J, Coudray C (1997) J Chem Phys 106:2323
Ayuela A, López JM, Alonso JA et al (1993) Z Phys D: At, Mol Clusters 26:213
Hooton IE, Jacobs PWM (1988) Can J Chem 66:830
Tsyro S, Aas W, Soares J et al (2011) Atmos Chem Phys 11:10367
Ming Y, Russell LM (2001) J Geophys Res D: Atmos 106:28
Wick CD (2009) J Phys Chem C 113:2497
Li RZ, Liu CW, Gao YQ et al (2013) J Am Chem Soc 135:5190
Gao YQ (2008) J Chem Phys 128:134111
Liu C-W, Wang F, Yang L et al (2014) J Phys Chem B 118:743
Hou G-L, Liu C-W, Gao YQ et al Unpublished Results
Xu H-G, Zhang Z-G, Feng Y et al (2010) Chem Phys Lett 487:204
Yang L, Shao Q, Gao YQ (2008) J Phys Chem B 113:803
Case DA, Darden TA, Cheatham ITE et al (2006) Amber 9. University of California, San Francisco
Joung IS, Cheatham TE (2008) J Phys Chem B 112:9020
Berendsen HJC, Grigera JR, Straatsma TP (1987) J Chem Phys 91:6269
Frisch MJ, Trucks GW, Schlegel HB et al (2009) Gaussian 09 Revision A.02. Gaussian Inc, Wallingford
Chai J, Head-Gordon M (2008) Phys Chem Chem Phys 10:6615
Grimme S (2011) Wiley Interdiscip Rev Comput Mol Sci 1:211
Turi L (1999) J Chem Phys 110:10364
Yang YA, Conover CWS, Bloomfield LA (1989) Chem Phys Lett 158:279
Pramanik A, Kalagi RP, Barge VJ et al (2005) Theor Chem Acc 114:129
Phillips NG, Conover CWS, Bloomfield LA (1991) J Chem Phys 94:4980
Watterich A (1978) Phys Status Solidi B 88:K51
Barnett RN, Landman U (1996) J Phys Chem 100:13950
Acknowledgments
Y.Q.G. acknowledges the NSFC (91027044, 21233002, and 21125311) and National Key Basic Research Foundation of China (2012CB917304) for financial support. W.-J.Z. acknowledges the Natural Science Foundation of China (Grant No. 21273246) and the Knowledge Innovation Program of the Chinese Academy of Sciences (Grant No. KJCX2-EW-H01) for finanical support. The calculations were performed on the supercomputers of the Shanghai Supercomputer Center (SSC).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dedicated to Professor Guosen Yan and published as part of the special collection of articles celebrating his 85th birthday.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, CW., Hou, GL., Zheng, WJ. et al. Adsorption of water molecules on sodium chloride trimer. Theor Chem Acc 133, 1550 (2014). https://doi.org/10.1007/s00214-014-1550-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-014-1550-1