
Abstract
The aim of this work was to study the mechanism of cross-modulation between cannabinoid and opioid systems for analgesia during acute and chronic exposure. Acute coadministration of ineffectual subanalgesic doses of the synthetic cannabinoid CP-55,940 (0.2 mg/kg i.p.) and morphine (2.5 mg/kg i.p.) resulted in significant antinociception. In chronic studies, a low dose of CP-55,940 (0.2 mg/kg, i.p.) that per se did not induce analgesia in naive animals produced a significant degree of antinociception in rats made tolerant to morphine, whereas in rats made tolerant to CP-55,940, morphine challenge did not produce any analgesic response. To identify the mechanism of these asymmetric interactions during chronic treatment, we investigated the functional activity of cannabinoid and μ opioid receptors and their effects on the cyclic AMP (cAMP) cascade. Autoradiographic-binding studies indicated a slight but significant reduction in cannabinoid receptor levels in the hippocampus and cerebellum of morphine-tolerant rats, whereas CP-55,940-stimulated [35S]GTPγS binding showed a significant decrease in receptor/G protein coupling in the limbic area. In CP-55,940 exposed rats, μ opioid receptor binding was significantly raised in the lateral thalamus and periaqueductal gray (PAG), with an increase in DAMGO-stimulated [35S]GTPγS binding in the nucleus accumbens. Finally, we tested the cAMP system's responsiveness to the cannabinoid and opioid in the striatum and dorsal mesencephalon. In vivo chronic morphine did not affect CP-55,940's ability to inhibit forskolin-stimulated cAMP production in vitro and actually induced sensitization in striatal membranes. In contrast, in vivo chronic CP-55,940 desensitized DAMGO's efficacy in inhibiting forskolin-stimulated cAMP production in vitro. The alterations to the cAMP system seem to mirror the behavioral responses, indicating that the two systems may interact at the postreceptor level. This might open up new therapeutic opportunities for relief of chronic pain through cannabinoid–opioid coadministration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cannabinoids and opioids share similar pharmacological effects, including analgesia, sedation, hypothermia, inhibition of motor activity, hypotension and sedation (Manzanares et al. 1999; Massi et al. 2001). Both produce analgesia through a G protein-coupled mechanism that blocks the release of pain-propagating neurotransmitters in the brain and spinal cord (Cichewicz 2004). Opioids such as morphine are commonly prescribed for chronic and persistent pain, but the analgesic benefits/disadvantages of a cannabinoids and opioids combined have not been fully explored. Prolonged clinical use of morphine is often limited because it causes tolerance after repeated administration. To overcome tolerance to the analgesic effects, higher doses are necessary but are often accompanied by untoward effects such as constipation, nausea, and respiratory depression (Ellison 1993).
The analgesic potential of cannabinoids has not been well explored in humans, aside from anecdotal reports. Clinical data support the use of cannabinoids for peripheral inflammatory pain (Sawynok 2003), but question their utility in cancer-related pain on account of the adverse effects (Campbell et al. 2001). However, chronic pain patients frequently use cannabis (Ware et al. 2003). The cannabinoids' adverse psychological effects (including psychomotor and cognitive impairment, anxiety and panic attack, acute psychosis, and paranoia) may limit their use (Ashton 1999; Tramer et al. 2001). New safe, effective agonists at the cannabinoid receptor whose therapeutic and psychotropic effects are dissociated could be useful to reduce side effects.
In several models of acute pain, low doses of cannabinoids coadministered with opioid receptor agonists gave greater antinociceptive effects than either drug alone (Welch and Stevens 1992; Welch et al. 1995; Smith et al. 1998, Cichewicz et al. 1999; Massi et al. 2001; Cichewicz and McCarthy 2003). There is also synergy in the opposite direction because morphine can enhance the antinociception induced by tetrahydrocannabinol (THC; Reche et al. 1996). Interactions between these two drugs would be important in the prevention of chronic pain. After chronic treatment, some authors detected cross-tolerance for analgesia between cannabinoids and opioids (Thorat and Bhargava 1994; Rowen et al. 1998; Massi et al. 2001), whereas Rubino et al. (1997) reported potentiation of the antinociceptive effects of cannabinoids in morphine-dependent rats. Sandra Welch's group has now found that cotreatment of mice with a low dose of THC prevents morphine tolerance, suggesting these drugs might be employed in combination therapy to maintain morphine's long-term efficacy (Cichewicz and Welch 2003).
The interactions between opioids and cannabinoids might be explained by links in the mechanisms of action. Several hypotheses have been formulated including the release and/or synthesis of endogenous opioid peptides by cannabinoids, alterations to the endocannabinoid system after exposure to opioids, an interaction at their signal-transduction mechanisms or direct interaction at the receptor (see review Manzanares et al. 1999; Varvel et al. 2004). Indeed, opioid and cannabinoid receptors are codistributed in areas of the dorsal horn of the spinal cord (Welch and Stevens 1992; Hohmann et al. 1999; Salio et al. 2001) and in parts of the brain controlling nociceptive responses, such as periaqueductal gray (PAG), raphe nuclei, and central–medial thalamic nuclei (Mansour et al. 1988; Herkenham et al. 1991; Mailleux and Vanderhaeghen 1992). Studies at the cellular level have assessed the effects of chronic opioid and cannabinoid treatment on CB1 and μ receptor levels and efficiency. However, the findings are contradictory and region dependent, with increases (Rubino et al. 1997; Gonzalez et al. 2002), decreases (Gonzalez et al. 2002; Viganò et al. 2003; Gonzalez et al. 2003), or no change in CB1 receptor function after chronic morphine (Romero et al. 1998). Differences in the species and the method employed to render animals tolerant to morphine may well explain these contradictions. Thorat and Bhargava (1994) reported that μ opioid receptors were unaffected in the brain and spinal cord of THC-tolerant mice, whereas Corchero et al. (2004) found that repeated doses of THC raised μ opioid receptor density in most brain regions, including the caudate putamen, amygdala, hindbrain and thalamic regions, hypothalamus, and hippocampus, suggesting that cannabinoids induce time-related differences in responsiveness to opioids. Our group (Viganò et al. 2003) and others (Gonzalez et al. 2003) also reported that prolonged activation of opioid receptors altered endocannabinoid levels, suggesting the cannabinoid system was involved in morphine tolerance.
The present study investigated the cross modulation between cannabinoid and opioid systems in parallel with behavioral and biochemical studies. In rats we characterized the effect of (1) acute inactive doses of the synthetic cannabinoid compound CP-55,940 with morphine on analgesic effect; (2) acute CP-55,940 in morphine-tolerant rats and acute morphine in rats tolerant to CP-55,940 analgesia. Finally, to identify the molecular mechanism, we checked the effects of repeated doses of morphine and CP-55,940 on the CB1 and μ opioid receptor, respectively, (receptor density and receptor G protein/coupling) and their action on the cyclic AMP (cAMP) cascade.
Materials and methods
Animals
Male Sprague–Dawley rats (Charles River, Calco, Italy) weighing 125–150 g at the beginning of the experiment were housed three per cage in a controlled environment, at constant temperature and humidity, on a 12-h light/dark cycle, with free access to food and water. The experimental protocols were approved as required by Italian Governmental Decree no. 94/2000-A. All animal procedures met the guidelines of the European Community directives regulating animal research. The number of animals used and their suffering were minimized.
Drugs
Morphine hydrochloride was obtained from Salars (Como, Italy), dissolved in saline and CP-55,940 from Tocris (Bristol, UK), dissolved in ethanol, cremophor, and saline (1:1:18).
Acute drug combinations
Rats received CP-55,940 intraperitoneally (i.p.), 0.2 mg/kg or its vehicle, and morphine subcutaneously (s.c.), 2.5 mg/kg or saline. The drugs were injected 30 min apart, alternating the order. These doses were chosen on the basis of our previous results (Massi et al. 2001). Analgesia was assessed by the tail flick test 30, 60, 90, 120 min after the second drug.
Chronic interaction studies
Chronic morphine
Animals received morphine hydrochloride twice a day (between 9:00 and 10:00 a.m. and 5:00 and 6:00 p.m.), 5 mg/kg s.c. for 4.5 days. Control rats received the same treatment with saline. This regimen induced clear development of tolerance, as previously demonstrated (Massi et al. 2001). On day 5, the animals treated with morphine were divided into two groups: one received the morning injection of morphine, but the second was again split into two parts and given CP-55,940 at two different doses (0.2 or 0.4 mg/kg i.p.). Control animals were divided into two groups and given vehicle or acute CP-55,940 (0.2, 0.4 mg/kg i.p.). Therefore, this schedule consists of four treatments: chronic morphine + morphine (chronic morphine), chronic morphine + CP-55,940 0.2 or 0.4, chronic saline + CP-55,940 (acute CP-55,940 0.2 or 0.4), and chronic saline + CP-55,940 vehicle (control). On days 1 and 5, the analgesic effect was evaluated by the tail-flick test to check for tolerance.
Chronic CP-55,940
Rats received CP-55,940 twice a day (between 9:00 and 10:00 a.m. and 5:00 and 6:00 p.m.), 0.4 mg/kg i.p. for 6.5 days. Controls received the vehicle at the same times. Full tolerance developed to the analgesic effect of CP-55,940 (Rubino et al. 1998). On day 7, the animals given CP-55,940 were divided into two groups: one received the morning injection of CP-55,940, the second was given a single dose of morphine (5 mg/kg s.c.). Control animals were divided into two groups, given saline or acute morphine (5 mg/kg s.c.). Therefore, this schedule consists of four treatments: chronic CP-55,940 + CP-55,940 (chronic CP-55,940), chronic CP-55,940 + morphine 5, chronic vehicle + morphine (acute morphine 5), and chronic vehicle + saline (control). On days 1, 3, and 7, the analgesic effect was evaluated by the tail-flick test to check for tolerance.
Tail flick test
The analgesic effect was evaluated using the tail flick test according to D'Amour and Smith (1941) with a cut-off of 15 s to prevent tissue damage. Pain thresholds were evaluated 30, 60, 90, and 120 min after the last dose and expressed as total area under the time-response curve (AUC) over the 120 min.
Autoradiographic-binding studies
Rats were decapitated 1 h after the last morphine done (day 5) or CP-55,940 (day 7). Brains were rapidly removed and frozen in liquid nitrogen and stored at −80°C until processing. Twenty-micron coronal and sagittal sections were cut on a cryostat and thaw-mounted on gelatin-coated slides. The sections were briefly dried at 30°C and stored at −80°C until they were processed for autoradiographic-binding studies.
[3H]DAMGO receptor autoradiographic binding
Slides from rats chronically treated with cannabinoid were incubated for 1 h at room temperature with 4.5 nM [3H][d-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin ([3H]-DAMGO) (Tocris) for the μ opioid receptor in binding buffer (50 mM Tris–HCl, pH 7.4, 10 mM bacitracin). Adjacent cerebral sections were incubated in parallel with 2.5 mM naloxone to assess nonspecific binding. Sections were washed three times for 5 min at 4°C in 50 mM Tris–HCl, pH 7.4, then dipped briefly in distilled water, and dried under a cool air stream. Autoradiograms were generated by exposing the dried sections to Hyperfilm-3H tritium-sensitive film (Amersham Pharmacia Biotech, Milan, Italy) for 30–40 days.
[3H]CP-55,940 receptor autoradiographic binding
Slides from rats chronically exposed to morphine were processed for [3H]CP-55,940 receptor binding. Slides were brought to room temperature, then incubated for 2.5 h at 37°C with 10 nm[3H]CP-55,940 (Perkin Elmer Life Sciences, Milan, Italy) in binding buffer (50 mM Tris–HCl, pH 7.4, 5% BSA). Adjacent cerebral sections were incubated in parallel with 10 μM CP-55,940 to assess nonspecific binding. Sections were washed for 1 h at 4°C in 50 mM Tris–HCl, pH 7.4, 1% BSA buffer and again for 3 h in the same conditions. They were then dipped in 50 mM Tris–HCl buffer (pH 7.4, 5 min) to remove excess BSA, dipped briefly in distilled water, and dried under a cool air stream. Autoradiograms were generated by exposing the dried sections for 7 days to Hyperfilm-3H (Amersham Pharmacia Biotech).
DAMGO- and CP-55,940-stimulated [35S]GTPγS binding in autoradiography
This was determined as described by Sim et al. (1996), with slight modifications. Briefly, slides were incubated in assay buffer (50 mM Tris–HCl, 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, 10 mU/ml adenosine deaminase, 0.1% BSA, pH 7.4) at 25°C for 10 min then in 3 mM GDP in assay buffer at 25°C for 15 min. They were then transferred to assay buffer containing 3 mM GDP and 0.04 nM [35S]GTPγS with (stimulated) or without (basal) 3 μM DAMGO or 5 μM CP-55,940 and incubated at 25°C for 2 h. Slides were rinsed twice in 50 mM cold Tris buffer and once in deionized water, dried, and exposed to βmax film (Amersham Pharmacia Biotech) for 48 h.
Image analysis
The intensity of the autoradiographic films was assessed by measuring the gray levels with an image analysis system consisting of a scanner connected to a PC running Microsoft Windows. The images were analyzed using Image-Pro Plus 5.0 (MediaCybernetics, Silver Spring, USA). Each area of both sides of the brain was traced with the mouse cursor using the Paxinos and Watson (1986) atlas as reference, and light transmittance was determined as the gray level. The gray level of densitometric measurements calculated after subtraction of the film background density was established within the linear range, determined using tritium standards (3H Microscales, Amersham Pharmacia Biotech) for receptor-binding studies and 35S standards prepared in the laboratory for [35S]GTPγS-binding studies.
cAMP level
Brains from rats chronically exposed to CP-55,940 and morphine were quickly removed, and the striatum and dorsal mesencephalon were obtained within a few minutes by gross dissection on ice, according to the Paxinos and Watson (1986) atlas. Tissues were homogenized in ice-cold lysis buffer containing 0.25 M sucrose, 50 mM Tris–HCl, pH 7.5, 5 mM EGTA, 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 0.1 mM dithiothreitol, 0.1% Triton X-100, 10 mg/ml leupeptin, and 10 mg/ml aprotinin. The tissues were then centrifuged at 1,000×g for 5 min (4°C), and the supernatant was centrifuged at 35,000×g for 10 min (4°C). The pellet was resuspended in buffer solution (50 mM Tris–HCl, 5 mM EDTA, pH 7.5), and protein content was assayed as described by Bradford (1976).
For cAMP levels, striatal and mesencephalic membranes (40 μg protein/100 μl) were preincubated with DAMGO (5 μM) or CP-55,940 (3, 10, 30 μM) or their vehicle at 37°C for 15 min. The cell preparation was then stimulated with 0.1 μM forskolin (except basal) and a reaction mixture containing 13 mM Tris–HCl pH 7.5, 0.2 mM ATP, 10 mM GTP, 32 mM MgSO4, 0.16 mM 3-isobutyl-1-methylxanthine, 160 mM NaCl, 8 mM phosphocreatine, 20 U/150 ml creatine phosphokinase, and incubated for 10 min at 37°C. The reaction was stopped by heating the samples for 5 min at 90°C. After 5-min centrifugation at 12,000×g at 4°C, cAMP in the supernatant was measured using a TRK 432 radioreceptor assay kit (Amersham Pharmacia Biotech).
Statistical analysis
Differences in behavioral scores between groups at each time were assessed by one-way ANOVA followed by Tukey's test. Biochemical data were analyzed using Student's t test or one-way ANOVA, followed by Tukey's test.
Results
Behavioral studies
Acute combinations
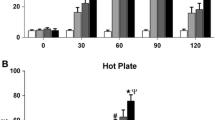
Figure 1 shows the result of the tail-flick test in rats given subanalgesic doses of the synthetic cannabinoid compound CP-55,940 and morphine (2.5 and 0.2 mg/kg, respectively). Neither drug alone induced significant analgesia, but the two together, in either order, produced significant analgesic effect, as indicated by the AUC, which was triple than that of control animals.

Effect of an acute dose of morphine (2.5 mg/kg s.c.) with CP-55,940 (0.2 mg/kg i.p.) on analgesia expressed as area under the analgesic curve (AUC). Data are mean±SEM of at least five animals. *p<0.001 vs control, °p<0.001 vs morphine alone, #p<0.001 vs CP-55,940 alone (Tukey's test)
Chronic interaction studies
To evaluate a possible long-term interaction between the drugs, we checked the effect of acute CP-55,940 or morphine in rats made tolerant, respectively, to morphine or CP-55,940.
Acute CP-55,940 in morphine-tolerant rats
As expected, rats given morphine, 5 mg/kg, twice a day for 4.5 days, developed full tolerance to its analgesic effect (Fig. 2). On day 5, the animals were given two different doses of CP-55,940 (0.2 or 0.4 mg/kg). The low dose, per se, did not induce analgesia in naive animals, but produced a significant degree of analgesia in morphine-tolerant rats, suggesting they had become sensitized to CP-55,940-induced analgesia. The higher dose of CP-55,940 (0.4 mg/kg) had similar analgesic effects in naive and morphine-tolerant rats (Fig. 2).

Analgesic response induced by different doses of CP-55,940 (0.2 or 0.4 mg/kg i.p.) in rats made tolerant to morphine (5 mg/kg s.c., twice a day for 4.5 days). Data are expressed as area under the analgesic curve (AUC) and are the mean±SEM of at least five animals. *$p<0.001, *$*$p<0.0001 vs control, °p<0.001 vs acute morphine, *p<0.05, **p<0.001 vs chronic morphine, #p<0.001 vs acute CP-55,940 (Tukey's test)
Acute morphine in CP-55,940-tolerant rats
To determine whether the interaction between the two systems could be bidirectional, we tested the effect of an acute dose of morphine in rats tolerant to CP-55,940. Rats treated with CP-55,940, 0.4 mg/kg, twice a day for 6.5 days, developed full tolerance to its analgesic effect (Fig. 3). An acute dose of morphine (5 mg/kg) on day 7 did not have any significant analgesic effect, indicating cross-tolerance between the two drugs.

Analgesic response induced by morphine (5 mg/kg s.c.) in rats made tolerant to CP-55,940 (0.4 mg/kg i.p., twice a day for 6.5 days). Data are expressed as area under the analgesic curve (AUC) and are the mean±SEM of at least five animals. *p<0.001 vs control, °p<0.01 vs acute morphine, #p<0.01 vs acute CP-55,940 (Tukey's test)
Autoradiographic-binding studies
To investigate the molecular mechanism of this asymmetric interaction, we checked the functional activity of the cannabinoid and μ opioid receptors after chronic morphine and CP-55,940.
Effect of chronic morphine on cannabinoid receptor function
Morphine-tolerant and saline-injected rats had similar levels of cannabinoid receptor binding in most brain structures (basal ganglia, molecular layer of the cerebellum, cerebral cortex, several limbic nuclei, and central gray substance (Table 1). Only in the cerebellum and hippocampus there was a slight (15%) but significant down-regulation of the CB1 cannabinoid receptors.
We also assayed CP-55,940-stimulated [35S]GTPγS binding in brain regions from rats tolerant to morphine analgesia. Basal [35S]GTPγS binding was not affected by chronic morphine in any of these areas (data not shown). Prolonged exposure to morphine significantly lowered the net-binding value only in the nucleus accumbens (40%) (Table 1).
Effect of chronic CP-55,940 on μ opioid receptor function
Figure 4a shows the densitometric analysis of μ opioid receptor binding on brain coronal sections of rats given CP-55,940 or vehicle. Seven days' exposure to CP-55,940 induced a slight but significant up-regulation of opioid receptors in the lateral thalamus (15% increase vs vehicle) and PAG (40% increase vs vehicle), without any changes in the other regions. We also assayed agonist-stimulated [35S]GTPγS binding with DAMGO as the agonist. Chronic CP-55,940 did not affect basal [35S]GTPγS binding in any of the cerebral areas (data not shown), but longer exposure significantly raised the net-binding value in the nucleus accumbens (25%), without affecting the other brain regions (Fig. 4b).

Effect of CP-55,940 chronic treatment (0.4 mg/kg i.p., twice a day for 6.5 days) on opioid receptor binding (a) and on net DAMGO-stimulated [35S]GTPγS binding determined by subtracting basal [35S]GTPγS binding (b) in different brain regions. Gray levels obtained with densitometric analysis were transformed into fmol/mg tissue using [3H]standards. Bars indicate the mean±SEM of at least five animals. *p<0.05 vs vehicle (Student's t test). CPu Caudate putamen, NAc nucleus accumbens, ctx cortex, TC central thalamus, TL lateral thalamus, TV ventral thalamus, hippo hippocampus, amyg anterior amygdala, PAG periaqueductal gray, coll colliculus, nigra substantia nigra
cAMP level
To see whether the cAMP pathway was the second messenger involved in the asymmetric opioid–cannabinoid interaction after chronic treatment, we tested the ability of DAMGO and CP-55,940 to inhibit forskolin-stimulated cAMP production in vitro in membranes of the striatum and dorsal mesencephalon (including the PAG) from rats made tolerant to CP-55,940 or morphine. These areas were chosen as they are involved in the functional control of antinociception and contain both CB1 and μ opioid receptors (Mansour et al. 1988; Lichtman et al. 1996; Rodriguez et al. 2001).
Cerebral areas from rats after chronic morphine or CP-55,940 were exposed in vitro to DAMGO (5–10 μM) or CP-55,940 (3, 10, 30 μM). Apart from the chronic treatment in vivo (morphine or CP-55,940), basal cAMP levels in striatal and mesencephalic membranes were the same in treated and control animals, and forskolin raised them similarly in both groups (data not shown).
The ability of DAMGO (5 μM) to inhibit forskolin-stimulated cAMP production in vitro was reduced in membranes from rats tolerant to morphine (30% in the striatum and 64% in the dorsal mesencephalon), indicating desensitization to this effect (Fig. 5). However, when CP-55,940 at the three concentrations was added to membranes from chronic morphine rats, the responses differed, depending on the area considered. In the striatum CP-55,940's ability to inhibit forskolin-stimulated cAMP production tended to increase, the rise reaching significance at 10 μM, whereas in the mesencephalic membranes, CP-55,940's inhibitory effect was much the same independently of in vivo treatment (morphine or saline) (Fig. 5).

The effect of DAMGO (5 μM) and CP-55,940 (3, 10, 30 μM) in vitro on forskolin-stimulated cAMP accumulation on striatal (a) and dorsal mesencephalic membranes (b) of rats chronically treated in vivo with morphine (5 mg/kg s.c., twice a day for 4.5 days). Data are expressed as percentages of inhibition vs forskolin (FSK). Bars indicate the mean±SEM of at least six animals. *p<0.05 vs vehicle (Student's t test)
These data show that CP-55,940's ability to inhibit cAMP production is not only maintained in morphine-tolerant rats, but there might even be sensitization (striatum, CP-55,940 10 μM).
Figure 6 shows the effect of chronic CP-55,940 in vivo on the ability of CP-55,940 or DAMGO to inhibit forskolin-stimulated cAMP production in vitro. Chronic CP-55,940 caused a weak desensitization of CP-55,940's (10 μM) capacity to inhibit forskolin-stimulated cAMP production in both cerebral areas examined (Fig. 6). However, when the membranes from rats chronically treated with CP-55,940 were exposed in vitro to different DAMGO concentrations (5–10 μM), the opioid agonist's inhibitory capacity decreased, mainly in the striatum, suggesting cross desensitization between the drugs.

The effect of CP-55,940 (10 μM) and DAMGO (5, 10 μM) in vitro on forskolin-stimulated cAMP accumulation on striatal (a) and dorsal mesencephalic membranes (b) of rats chronically treated in vivo with CP-55,940 (0.4 mg/kg i.p., twice a day for 6.5 days). Data are expressed as percentages of inhibition vs forskolin (FSK). Bars indicate the mean±SEM of at least six animals. *p<0.05 vs vehicle (Student t test)
Discussion
Opioid and cannabinoid agonists share several pharmacological actions that might offer new therapeutic potential, particularly as analgesics. We set out to characterize the functional interaction between cannabinoid and opioid agonists on analgesia after acute or repeated doses.
In acute studies, coadministration of morphine and CP-55,940 led to enhancement of their analgesic effect, as already reported for THC and morphine (Welch et al. 1995; Smith et al. 1998; Cichewicz and McCarthy 2003; Cichewicz 2004). Subanalgesic doses of CP-55,940 and morphine given together had a slightly greater analgesic effect than that predicted by simple addition of their separate effects, suggesting that the combination might offer a means of enhancing the pain-relieving effect of morphine without the side effects of either drug.
More studies have examined the interaction between morphine and THC than morphine and the synthetic cannabinoid compound CP-55,940, but the result of interaction could be different depending on the cannabinoid used. In vivo CP-55,940 did not interact with morphine in the spinal cord (Welch and Stevens 1992); pretreatment of mice with CP-55,940 did not enhance the antinociceptive effect of morphine intrathecally, whereas pretreatment with THC reduces the ED50 for morphine tenfold. However, the synthetic compound, like THC, shifted the morphine dose-response curve nearly ten times after intracerebroventricular injection (Welch et al. 1995) and increased morphine antinociception by about 45% when given i.p. (Massi et al. 2001). Thus, the antinociceptive potency of morphine might also depend on the administration route and whether CP-55,940 or THC pretreatment is evaluated, suggesting a possible involvement of multiple cannabinoid receptor subtypes at spinal or supra-spinal levels (Pugh et al. 1997).
The second important point concerns the asymmetric interaction between the two drugs given chronically. A subanalgesic dose of CP-55,940 induced significant analgesia in rats tolerant to morphine, and the maximal CP-55,940 dose (0.4 mg/kg) was still fully active. Thus, the potency of the subanalgesic CP-55,940 dose was greatly increased in morphine-tolerant rats, suggesting the development of sensitization, whereas the fully effective dose maintained its action, indicating that cross-tolerance did not develop. Similarly, tolerance to morphine was prevented in animals receiving daily cotreatment with an oral non-analgesic dose of THC or challenged with a single injection of THC at the end of chronic exposure to morphine (Cichewicz and Welch 2003). In an earlier study, we too found that the antinociceptive effect of THC was potentiated in morphine-tolerant rats (Rubino et al. 1997), indicating hypersensitivity to THC analgesia. In addition, there was no cross-tolerance between morphine and THC in a rat model of neuropathic pain resulting from chronic constriction nerve injury (Mao et al. 2000). In contrast, another group observed cross-tolerance between the analgesic effects of THC and morphine (Thorat and Bhargava 1994). Differences in species, strains, and methods for inducing tolerance to morphine or cannabinoid agonists used might account for these differences.
One clinical implication of our result is that cannabinoids and opioids, in combination but also as replacement therapy, may be more effective analgesics for a longer time than morphine alone.
Surprisingly, an acute dose of morphine given to rats made tolerant to CP-55,940 led to cross-tolerance to the former's analgesic action. Thus, in chronic treatment, the interaction between cannabinoid and opioids does not appear to be two-directional but depends on the drug used in chronic exposure, with sensitization to cannabinoid in morphine-tolerant rats and cross-tolerance to morphine in CP-55,940-tolerant animals. Previous reports have already described the cross-tolerance between morphine or k-receptor agonists and THC (Thorat and Bhargava 1994; Smith et al. 1994; Rowen et al. 1998; Welch 1997). These behavioral data confirm that there are different levels of long-term interaction for drug-induced analgesia and that the phenomenon is asymmetric and differs with the experimental set-up.
The final goal of this study was to correlate the asymmetric interaction observed at the behavioral level with molecular mechanisms. Several hypotheses have tried to explain the interaction between cannabinoid and opioid systems including interaction at the level of the receptor and signal transduction system and the release of opioid peptides by cannabinoids or endocannabinoids by opioids (see review Manzanares et al. 1999; Varvel et al. 2004).
Here we evaluated the effects of prolonged exposure to morphine and CP-55,940 on the status of cannabinoid and μ opioid receptors in different brain regions to see whether some alteration might explain the behavioral sensitization/cross-tolerance. Chronic morphine slightly reduced the cannabinoid receptor level in the cerebellum and hippocampus, whereas agonist-stimulated [35S]GTPγS binding showed a strong decrease in receptor/G protein coupling in the limbic area. Thus, CB1 receptor function is slightly reduced in density in two areas and significantly reduced in G protein coupling in the limbic area. These data conflict with the behavioral results showing sensitization to cannabinoid analgesia in morphine-tolerant rats, suggesting that the CB1 receptor machinery might be more and not less efficient. In a previous study (Rubino et al. 1997), we reported an increase in CB1 receptor density in rats made tolerant to morphine through pellet implantation that fitted well with the increased analgesic response to THC in the tail flick test. The morphine schedule used in the present study was milder than pellet implantation, so the alteration in CB1 receptor function might possibly be a final event following other cellular adaptations that trigger the altered response to cannabinoid after chronic opioid exposure. Thus, the alterations we observed do not seem strictly related to morphine treatment, but could be nonspecific effects not related to the behavioral phenomenon, as supported by the fact that, except for the limbic area, the cerebral regions where there were alterations are not strictly involved in analgesia. In the literature too, we could not find a concordant picture of how chronic opioid affects CB1 receptor density/coupling; depending on the animals used—mice or rats—and the protocol of administration, some authors reported no significant changes in CB1 receptors in mice chronically treated with morphine (Thorat and Bhargava 1994; Romero et al. 1998), but others reported an increase in the caudate putamen and limbic structures (Gonzalez et al. 2002). Some studies do not try to correlate the biochemical effect with a pharmacological effect, so we can only assume that the discordant findings presumably reflect different species (rats or mice), strains, and protocols (mild or strong) to induce morphine tolerance that might influence the cannabinoid system differently.
Only a few studies have examined changes in opioid receptor function in animals tolerant to cannabinoids. In CP-55,940-tolerant rats, our data indicated a slight up-regulation of the μ opioid receptor in the lateral thalamus and PAG accompanied by an increase in net binding in the nucleus accumbens. These findings are in line with a study by Corchero et al. (2004) where there was a time-dependent increase in μ opioid receptor density in the forebrain and hindbrain region and in the thalamic nuclei of rat brain after repeated exposure to THC. The μ opioid receptor increased most on day 1, 3 s after THC administration, persisted until day 7, and returned to baseline at the end of the experiment (day 14), except in the thalamic nuclei where alterations were still detectable. The authors did not report the pharmacological consequences of this interaction and concluded that the brain regions altered are part of a well-established circuit that mediates the descending pain-suppressive actions of opioids and cannabinoids (Walker et al. 1999a; Meng et al. 1998), confirming the neuroanatomical evidence of common opiate/cannabinoid neuronal substrates for analgesia. We observed only a very small alteration in μ receptor binding, and once again, it is very difficult to correlate that with the presence of cross-tolerance. As previously suggested, therefore, the alteration does not seem to be the molecular basis of cross-tolerance to morphine in cannabinoid-tolerant rats.
The same conclusion holds for the increase in μ opioid receptor agonist-stimulated [35S]GTPγS binding in the nucleus accumbens (with a tendency in the caudate putamen) in the brain of CP-55,940-tolerant animals. Again, our data agree with those of Corchero et al. (1999) who described a μ opioid receptor-activated G protein in the caudate putamen of animals treated with THC for 7 days, suggesting that opioids and cannabinoids may interact at the postreceptor level only in specific areas depending on the treatment protocol (the length of treatment) or on the agonist selected to induce tolerance (natural or synthetic cannabinoid). In any case, the alterations in receptor efficiency appear to be anatomically discrete and relatively small, and their possible functional significance depended on the brain region where they occur. The areas involved are closely related to the mechanisms of cannabinoid/opioid analgesia (thalamic structures and PAG, Walker et al. 1999a,b; Meng et al. 1998) or drug addiction (limbic structures, nucleus accumbens).
One of the earliest and most important signaling events initiated by ligand binding to cannabinoid and opioid receptors is the inhibition of adenylate cyclase that leads to a decrease in the production and accumulation of intracellular cAMP. Our last step was to determine whether cAMP pathways provide the molecular link for explaining the opioid–cannabinoid functional interaction seen at the behavioral step. In view of the many mechanistic similarities between opioid and cannabinoid agonists, the possibility of concomitant regulation of a common intracellular pathway is not surprising (Pugh et al. 1994; Shapira et al. 1998, 2003).
We used two brain areas, the caudate putamen and dorsal mesencephalon, because the latter is involved in the functional control of antinociception, and both contain cannabinoid and opioid receptors. Results differ depending on the experimental set-up but in line with the analgesic response: in vivo chronic treatment with morphine sensitized/maintained CP-55940's ability in vitro to inhibit forskolin-stimulated cAMP production in both brain areas. A significant sensitized effect in the striatum is observed only with CP-55,940 10 μM, the relevant increase (100%) with CP-55,940 3 μM not reached the statistical significance due to high variability in the vehicle group, whereas the lack of significance with CP-55,940 30 μM is probably due to the reaching of the maximal physiological cAMP inhibition. On the other hand, in vivo chronic treatment with CP-55,940 desensitizes the in vitro inhibitory efficacy of DAMGO. These results provide evidence that the cAMP level is regulated concomitantly by the drugs. This interaction was asymmetric and coincided with the asymmetric cross-interaction seen at the behavioral level. When we observed cross-tolerance (chronic CP-55,940 + morphine) besides the increase in μ receptor binding in the PAG, we found cross-desensitization on the cAMP pathway, whereas when we observed potentiation (chronic morphine + CP-55,940), we found sensitization in the cAMP pathway. Several other brain areas need to be analyzed to confirm this, but for the first time, we can suggest that not only in vitro (Pugh et al. 1994; Shapira et al. 1998, 2003) but also in vivo cannabinoid and opioid chronic exposure could end up in an interaction at postreceptor level whose nature might differ depending on the chronic drug exposure. The nature of this interaction needs to be clarified in other brain regions and for other opioid receptors, but it appears to offer promise for improving the therapeutic approach to chronic pain relief.
References
Ashton CH (1999) Adverse effects of cannabis and cannabinoids. Br J Anaesth 83(4):637–649
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Campbell FA, Tramer MR, Carroll D, Reynolds DJ, Moore RA, McQuay HJ (2001) Are cannabinoids an effective and safe treatment option in the management of pain? A qualitative systematic review. BMJ 323:13–16
Cichewicz DL (2004) Synergistic interactions between cannabinoid and opioid analgesics. Life Sci 74(11):1317–1324
Cichewicz DL, McCarthy EA (2003) Antinociceptive synergy between delta(9)-tetrahydrocannabinol and opioids after oral administration. J Pharmacol Exp Ther 304(3):1010–1015
Cichewicz DL, Welch SP (2003) Modulation of oral morphine antinociceptive tolerance and naloxone-precipitated withdrawal signs by oral Delta 9-tetrahydrocannabinol. J Pharmacol Exp Ther 305(3):812–817
Cichewicz DL, Martin ZL, Smith FL, Welch SP (1999) Enhancement mu opioid antinociception by oral delta9-tetrahydrocannabinol: dose-response analysis and receptor identification. J Pharmacol Exp Ther 289(2):859–867
Corchero J, Romero J, Berrendero F, Fernandez-Ruiz J, Ramos JA, Fuentes JA, Manzanares J (1999) Time-dependent differences of repeated administration with Delta9-tetrahydrocannabinol in proenkephalin and cannabinoid receptor gene expression and G-protein activation by mu-opioid and CB1-cannabinoid receptors in the caudate-putamen. Brain Res Mol Brain Res 67(1):148–157
Corchero J, Oliva JM, Garcia-Lecumberri C, Martin S, Ambrosio E, Manzanares J (2004) Repeated administration with Delta9-tetrahydrocannabinol regulates mu-opioid receptor density in the rat brain. J Psychopharmacol 18(1):54–58
D'Amour FE, Smith DL (1941) A method for determining loss of pain sensation. J Pharmacol Exp Ther 72:74–86
Ellison NM (1993) Opioid analgesics for cancer pain: toxicities and their treatments. In: Patt RB (ed) Cancer pain. JB Lippincott, USA
Gonzalez S, Fernandez-Ruiz JJ, Sparpaglione V, Parolaro D, Ramos JA (2002) Chronic exposure to morphine, cocaine or ethanol in rats produced different effects in brain cannabinoid CB1 receptor binding and mRNA levels. Drug Alcohol Depend 66:77–84
Gonzalez S, Schmid PC, Fernandez-Ruiz J, Krebsbach R, Schmid HH, Ramos JA (2003) Region-dependent changes in endocannabinoid transmission in the brain of morphine-dependent rats. Addict Biol 8(2):159–166
Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC (1991) Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11(2):563–583
Hohmann AG, Briley EM, Herkenham M (1999) Pre- and postsynaptic distribution of cannabinoid and mu opioid receptors in rat spinal cord. Brain Res 822(1–2):17–25
Lichtman AH, Cook SA, Martin BR (1996) Investigation of brain sites mediating cannabinoid-induced antinociception in rats: evidence supporting periaqueductal gray involvement. J Pharmacol Exp Ther 276(2):585–593
Mailleux P, Vanderhaeghen JJ (1992) Distribution of neuronal cannabinoid receptor in the adult rat brain: a comparative receptor binding radioautography and in situ hybridization histochemistry. Neuroscience 48(3):655–668
Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ (1988) Anatomy of CNS opioid receptors. Trends Neurosci 11(7):308–314
Manzanares J, Corchero J, Romero J, Fernandez-Ruiz JJ, Ramos A, Fuentes JA (1999) Pharmacological and biochemical interactions between opioids and cannabinoids. Trends Pharmacol Sci 20:287–294
Mao J, Price DD, Lu J, Keniston L, Mayer DJ (2000) Two distinctive antinociceptive systems in rats with pathological pain. Neurosci Lett 280(1):13–16
Massi P, Vaccani A, Romorini S, Parolaro D (2001) Comparative characterization in the rat of the interaction between cannabinoids and opiates for their immunosuppressive and analgesic effects. J Neuroimmunol 117:116–124
Meng ID, Manning BH, Martin WJ, Fields HL (1998) An analgesia circuit activated by cannabinoids. Nature 395:381–383
Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates. Academic, New York
Pugh G Jr, Welch SP, Bass PP (1994) Modulation of free intracellular calcium and cAMP by morphine and cannabinoids, alone and in combination in mouse brain and spinal cord synaptosomes. Pharmacol Biochem Behav 49:1093–1100
Pugh G Jr, Mason DJ Jr, Combs V, Welch SP (1997) Involvement of dynorphin B in the antinociceptive effects of the cannabinoid CP55,940 in the spinal cord. J Pharmacol Exp Ther 281(2):730–737
Reche I, Fuentes JA, Ruiz-Gayo M (1996) Potentiation of delta 9-tetrahydrocannabinol-induced analgesia by morphine in mice: involvement of mu- and kappa-opioid receptors. Eur J Pharmacol 318(1):11–16
Rodriguez JJ, Mackie K, Pickel VM (2001) Ultrastructural localization of the CB1 cannabinoid receptor in mu-opioid receptor patches of the rat caudate putamen nucleus. J Neurosci 21(3):823–833
Romero J, Fernandez-Ruiz JJ, Vela G, Ruiz-Gayo M, Fuentes JA, Ramos JA (1998) Autoradiographic analysis of cannabinoid receptor binding and cannabinoid agonist-stimulated [35S]GTPgS binding in morphine-dependent mice. Drug Alcohol Depend 50:241–249
Rowen DW, Embrey JP, Moore CH, Welch SP (1998) Antisense oligodeoxynucleotides to Kappa1 receptor enhance D9-THC-induced antinociceptive tolerance. Pharmacol Biochem Behav 59:399–404
Rubino T, Tizzoni L, Viganò D, Massi P, Parolaro D (1997) Modulation of rat brain cannabinoid receptors after chronic morphine treatment. NeuroReport 8:3219–3223
Rubino T, Patrini G, Massi P, Fuzio D, Viganò D, Giagnoni G, Parolaro D (1998) Cannabinoid-precipitated withdrawal: a time-course study of the behavioral aspect and its correlation with cannabinoid receptors and G protein expression. J Pharmacol Exp Ther 285(2):813–819
Salio C, Fischer J, Franzoni MF, Mackie K, Kaneko T, Conrath M (2001) CB1-cannabinoid and mu-opioid receptor co-localization on postsynaptic target in the rat dorsal horn. NeuroReport 12(17):3689–3692
Sawynok J (2003) Topical and peripherally acting analgesics. Pharmacol Rev 55(1):1–20
Shapira M, Gafni M, Sarne Y (1998) Independence of, and interactions between, cannabinoid and opioid signal transduction pathways in N18TG2 cells. Brain Res 806(1):26–35
Shapira M, Gafni M, Sarne Y (2003) Long-term interactions between opioid and cannabinoid agonists at the cellular level: cross-desensitization and downregulation. Brain Res 960:190–200
Sim LJ, Selley DE, Xiao R, Childers SR (1996) Differences in G-protein activation by mu- and delta-opioid, and cannabinoid, receptors in rat striatum. Eur J Pharmacol 307(1):97–105
Smith PB, Welch SP, Martin BR (1994) Interactions between delta 9-tetrahydrocannabinol and kappa opioids in mice. J Pharmacol Exp Ther 268(3):1381–1387
Smith FL, Cichewicz D, Martin ZL, Welch SP (1998) The enhancement of morphine antinociception in mice by delta9-tetrahydrocannabinol. Pharmacol Biochem Behav 60(2):559–566
Thorat SN, Bhargava HN (1994) Evidence for a bidirectional cross-tolerance between morphine and D9-tetrahydrocannabinol in mice. Eur J Pharmacol 260:5–13
Tramer MR, Carroll D, Campbell FA, Reynolds DJ, Moore RA, McQuay HJ (2001) Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ 323:16–21
Varvel SA, Cichewicz DL, Lichtman AH (2004) Interactions between cannabinoids and opioids. In: Wenger T (ed) Recent advances in pharmacology and physiology of cannabinoids. Research Signpost Trivandrum, Kerala, India, pp 157–182
Viganò D, Grazia Cascio M, Rubino T, Fezza F, Vaccani A, Di Marzo V, Parolaro D (2003) Chronic morphine modulates the contents of the endocannabinoid, 2-arachidonoyl glycerol, in rat brain. Neuropsychopharmacology 28:1160–1167
Walker JM, Hohmann AG, Martin WJ, Strangman NM, Huang SM, Tsou K (1999a) The neurobiology of cannabinoid analgesia. Life Sci 65:665–673
Walker JM, Huang SM, Strangman NM, Tsou K, Sanudo-Pena MC (1999b) Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci U S A 96:12198–12203
Ware MA, Doyle CR, Woods R, Lynch ME, Clark AJ (2003) Cannabis use for chronic non-cancer pain: results of a prospective survey. Pain 102:211–216
Welch SP (1997) Characterization of anandamide-induced tolerance: comparison to delta 9-THC-induced interactions with dynorphinergic systems. Drug Alcohol Depend 45:39–45
Welch S, Stevens DL (1992) Antinociceptive activity of intrathecally administered cannabinoids alone, and in combination with morphine, in mice. J Pharmacol Exp Ther 262(1):10–18
Welch SP, Thomas C, Patrick GS (1995) Modulation of cannabinoid-induced antinociception after intracerebroventricular versus intrathecal administration to mice: possible mechanisms for interaction with morphine. J Pharmacol Exp Ther 272:310–321
Acknowledgements
This research was supported by funds from the Italian Ministry of University and Research (PRIN 2002, FIRB 2003) and funds from University of Insubria (FAR 2003).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Viganò, D., Rubino, T., Vaccani, A. et al. Molecular mechanisms involved in the asymmetric interaction between cannabinoid and opioid systems. Psychopharmacology 182, 527–536 (2005). https://doi.org/10.1007/s00213-005-0114-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-005-0114-4