
Abstract
Several lines of evidence implicate NMDA receptor dysfunction in the cognitive deficits of schizophrenia, suggesting that pharmacological manipulation of the NMDA receptor may be a feasible therapeutic strategy for treatment of these symptoms. Although direct manipulation of regulatory sites on the NMDA receptor is the most obvious approach for pharmacological intervention, targeting the G-protein coupled metabotropic glutamate (mGlu) receptors may be a more practical strategy for long-term regulation of abnormal glutamate neurotransmission. Heterogeneous distribution, both at structural and synaptic levels, of at least eight subtypes of mGlu receptors suggests that selective pharmacological manipulation of these receptors may modulate glutamatergic neurotransmission in a regionally and functionally distinct manner. Two promising targets for improving cognitive functions are mGlu5 or mGluR2/3 receptors, which can modulate the NMDA receptor-mediated signal transduction by pre- or postsynaptic mechanisms. Preclinical studies indicate that activation of these subtypes of mGlu receptors may be an effective strategy for reversing cognitive deficits resulting form reduced NMDA receptor mediated neurotransmission.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As described in recent reviews (Tamminga 1998; Coyle et al. 2002; Harrison and Owen 2003; Konradi and Heckers 2003; Krystal et al. 2003; Moghaddam 2003), genetic linkage studies as well as postmortem and psychopharmacological findings strongly implicate glutamate neurotransmission in the pathophysiology of schizophrenia. In particular, reduced activation of the NMDA receptor subtype may play a major role in cognitive deficits of schizophrenia, because blocking active NMDA channels in healthy volunteers impairs cognitive functioning in a manner that is similar to the cognitive deficits of schizophrenia (Krystal et al. 1994; Adler et al. 1999). These findings suggest that targeting the NMDA receptor may be a feasible strategy for treating the cognitive symptoms of schizophrenia. In addition to these findings, and regardless of the validity of the notion that NMDA dysfunction is involved in schizophrenia, an elementary line of reasoning implicates glutamate-mediated neurotransmission as a primary therapeutic target for cognitive symptoms of schizophrenia. Specifically, abnormal cortical functioning is the most consistent finding in schizophrenia (Weinberger et al. 1986; Benes et al. 1992; Goldman-Rakic 1994; Andreasen et al. 1997; Gur et al. 2000). Although there may be considerable debate about which cortical area is most pertinent to the expression of which symptoms, there is little debate that disruption in cortical processing of information is involved in the expression of the cognitive deficits of schizophrenia. All cortical efferents and cortico-cortical connections are glutamatergic. Therefore, regardless of the nature and the etiology of a cortical dysfunction in schizophrenia, glutamatergic neurons are the pathway by which information abnormally processed in the cortex becomes expressed as aberrant behavior. This suggests that even if the primary pathology in schizophrenia did not involve the glutamate synapse, modulation of glutamate-mediated signal transduction might still provide a mechanism for ameliorating aberrant information transfer within and from cortex.
Options for pharmacotherapeutic approaches that target glutamate receptors
The idea that glutamate functions as a neurotransmitter was introduced in the 1960s (Curtis et al. 1960; Crawford and Curtis 1964). Although the majority of glutamate found in the brain is involved in intermediary metabolism and other non-neuronal functions, the neuronal pool of glutamate is the most prevalent of all neurotransmitter pools. Glutamate receptors mediate nearly half of synaptic transmission throughout the central nervous system (Hollmann and Heinemann 1994). Glutamate neurotransmission is involved in nearly all aspects of brain function, and direct or circumstantial evidence has implicated a role for abnormal glutamate neurotransmission in the etiology or pathophysiology of most neurological and psychiatric disorders. Therefore, there has been a great deal of interest in developing therapeutic strategies that can influence the function of glutamate receptors. However, because of the ubiquitous nature of the glutamate synapse, drugs that interfere with glutamate receptor function were expected to impact glutamatergic function throughout the central nervous system, resulting in an overt disruption in basic brain function. The discovery of a distinct class of glutamate receptors, called metabotropic glutamate (mGlu) receptors, however, has changed the classical view of glutamate receptors and has brought forth the opportunity of developing drugs that modify glutamate neurotransmission in a functionally selective manner (Conn and Pin 1997; Schoepp 2001; Schoepp and Conn 2002). Before the discovery of mGlu receptors, it was thought that glutamate exerts its physiological action through receptors that act directly as ion channels. These “ionotropic” receptors were classified into three broad subtypes according to their preferential agonists as the NMDA, kainate, and AMPA receptors (Hollmann and Heinemann 1994; Huntley et al. 1994). Binding of glutamate to these subclasses of receptors stimulates Ca2+ and Na+ entry into neurons through channels formed either by the receptor itself (as is the case with the NMDA receptor subtype) or by opening voltage-sensitive ion channels that are on the cell membrane. The ionotropic glutamate receptors are expressed by nearly all subtypes of neurons, and mediate fast excitatory neurotransmission throughout the brain. Thus, direct pharmacological manipulation of this group of receptors may produce a global disruption in brain function and produce profound side effects ranging from disruption of movement to impairment of attention and memory. Our understanding of glutamate-mediated neurotransmission was changed profoundly after the discovery of a novel receptor with high affinity for glutamate which, in contrast to fast-acting ionotropic receptors, activate second messenger systems through coupling with G-proteins (Sladeczek et al. 1985; Nicoletti et al. 1986; Sugiyama et al. 1987). The initial reports about these so called metabotropic receptors were followed by an explosion of findings about many other structurally and functionally related receptors (Conn and Pin 1997). Interestingly, the advances in molecular methodologies presented the field with a novel chronology of discoveries: in contrast to nearly all the other neurotransmitter receptors in the brain whereby specific ligands and transduction mechanisms were discovered long before the application of cloning technology allowed for identification of their structure, the amino acid sequence and structure of the metabotropic glutamate receptors were characterized before we knew anything about their functional characteristics or had identified ligands that targeted these receptors.
At least eight metabotropic glutamate receptors, termed mGlu1–8, have been cloned. These eight receptors have been classified into three groups (termed groups I–III) primarily based on sequence identity and transduction mechanisms (Table 1; for recent reviews, see De Blasi et al. 2001; Schoepp 2001). Several characteristics of metabotropic glutamate receptors that distinguish them from ionotropic receptors also make them important pharmacotherapeutic targets. First, unlike the ionotropic glutamate receptors that mediate fast synaptic neurotransmission, activation of metabotropic glutamate receptors modulates neuronal activity in a manner similar to neuromodulators such as dopamine and serotonin, which have been effective targets of psychoactive drugs for treatment of most psychiatric disorders (Conn and Pin 1997; Schoepp 2001). Second, the distribution and function of these receptors is highly diverse and heterogeneous. Different subclasses of mGlu receptors are localized differently at both regional and cellular levels. For example, mGlu2 and mGlu3 are found in high density in the cerebral cortex and limbic regions (Phillips et al. 2000; Tamaru et al. 2001), whereas the mGlu6 receptor is found almost exclusively in the retina (Vardi et al. 2000). In addition, different classes of metabotropic glutamate receptors are found on different neuronal and non-neuronal elements such postsynaptic or presynaptic membranes or glial cells. Thus, unlike ligands, that target the ionotropic glutamate receptors and produce non-discriminate excitation or inhibition of fast synaptic neurotransmission throughout the nervous system, heterogeneous distribution of various subtypes of metabotropic glutamate receptors with distinct functional and anatomical properties may allow for modification of glutamate neurotransmission in a functionally selective manner.
Targeting metabotropic glutamate receptors to treat the cognitive symptoms of schizophrenia
As mentioned in the Introduction, several lines of evidence make a convincing case that the glutamatergic system may be disrupted in schizophrenia. First, antagonists of the NMDA subtype of glutamate receptors impair cognitive functioning in healthy volunteers in a manner that is very similar to the cognitive deficits observed in patients with schizophrenia (Krystal et al. 1994; Adler et al. 1998, 1999; Newcomer et al. 1999; Krystal et al. 2000). Second, most of the genes that are associated with increased vulnerability to develop schizophrenia express proteins that can, directly or indirectly, affect excitatory neurotransmission (Harrison and Owen 2003; Moghaddam 2003). Third, an accumulating body of postmortem data has found changes in schizophrenic brains that may have resulted from abnormal neurotransmission in the glutamate synapse (Harrison 1999; McCullumsmith and Meador-Woodruff 2002; Clinton et al. 2003). Finally, postmortem, imaging, and psychological lines of investigation consistently point to cortical regions as the primary site of dysfunction in schizophrenia (Goldman-Rakic 1994; Lewis and Anderson 1995; Weinberger et al. 2001), and the only neurons that can transmit abnormally processed information out of the cortex are glutamate-containing neurons. Thus, targeting glutamate neurotransmission is a plausible strategy for treating the cognitive symptoms of schizophrenia.
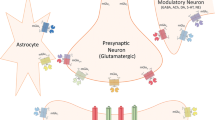
In general, the cognitive deficit-inducing properties of NMDA antagonists in healthy volunteers suggest that the primary glutamatergic abnormality in schizophrenia may be reduced NMDA function. Therapeutic strategies so far have focused mostly on enhancing NMDA receptor function through mechanisms that produce subtle positive modulation of this receptor. In particular, activation of the glycine/d-serine modulatory site on the NMDA receptor by increasing oral intake of d-serine or exogenous ligands such as d-cycloserine has shown promise in clinical trials (Coyle et al. 2002; Javitt 2002). Modulation of NMDA receptors may also be achieved by targeting metabotropic glutamate receptors (Marino and Conn 2002). Specifically, preclinical studies suggest that two subtypes of mGluRs have the potential of ameliorating cognitive deficits resulting from NMDA receptor dysfunction. These include mGlu5 receptors, which can directly modulate the function of NMDA channel, and the mGlu2/3 receptors, which regulate the release of glutamate (Fig. 1).

Activation of two types of metabotropic glutamate receptors may attenuate the effects of NMDA receptor function and have utility for treating the cognitive deficits of schizophrenia. These include the mGlu2/3 receptors, which can regulate the release of glutamate and mGlu5 which modulates the activity of NMDA receptors
Activation of mGlu5 receptors leads to potentiation of NMDA-evoked responses in cortical and other neural tissues (Doherty et al. 1997; Pisani et al. 1997; Awad et al. 2000; Attucci et al. 2001; Mannaioni et al. 2001). The selective mGlu5 receptor antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) blocks NMDA-induced membrane depolarization in striatal and cortical neurons (Pisani et al. 1997; Attucci et al. 2001). These cellular studies suggest that manipulation of the mGlu5 receptors may influence behavioral effects of NMDA antagonists in vivo. In fact, several recent studies suggest that there is a synergistic interaction between NMDA and mGlu5 receptors at a behavioral level. Specifically, mGlu5 receptor antagonists enhance the effects of NMDA receptor antagonists on hyperlocomotion and disruption of prepulse inhibition (Henry et al. 2002; Kinney et al. 2002, 2003), and working memory (Homayoun et al. 2004). These findings suggest that activation of mGlu5 receptors may be an effective therapeutic strategy for ameliorating NMDA receptor deficiency. Unfortunately, because of the rapid rate of mGlu5 receptor desensitization, mGlu5 receptor agonists are not considered effective therapeutic targets. However, recent studies have identified allosteric sites on the mGlu5 receptor that positively modulate the function of mGlu5 receptors (Knoflach et al. 2001; O’Brien et al. 2003). Preclinical studies so far suggest that these ligands may have therapeutic potential (Marino et al. 2003). Thus, this class of allosteric modulators is expected to reduce the cognitive effects of NMDA antagonists and possibly be of potential of therapeutic value for treating the cognitive symptoms of schizophrenia.
Based on extensive preclinical and recent, albeit limited, clinical data, ligands that activate mGlu2 and/or mGlu3 receptors may also have therapeutic value for treating cognitive deficits of schizophrenia. These receptors are primarily distributed in forebrain regions such as cerebral cortex, caudate putamen, nucleus accumbens, amygdala, and hippocampus (Phillips et al. 2000; Tamaru et al. 2001) that have been implicated in most psychiatric and neurological disorders. They are localized in various combinations of presynaptic, postsynaptic, extrasynaptic, and glial localization. Several studies indicate that activation of this group of receptors “normalizes” glutamate release presumably via presynaptic receptors localized on excitatory terminals (Battaglia et al. 1997; Cartmell and Schoepp 2000). Preclinical studies suggest that agonists of mGlu2/3 receptors diminish the behavioral, including cognitive impairing, effects of NMDA antagonists (Moghaddam and Adams 1998). The mechanism for this normalization of behavior appears to be as follows. One of the secondary effects of systemic exposure to NMDA antagonists, that may be critical to the behavioral effects of these drugs and to the state of glutamate dysfunction in schizophrenia, is an increase in the efflux of glutamate in prefrontal cortex (Moghaddam et al. 1997). The activation of cortical glutamate release by NMDA antagonists probably results from disinhibition of GABAergic input to cortical glutamate neurons (Greene 2001), and would be expected to lead to over-activation of glutamate neurotransmission at non-NMDA receptors and disruption of cognitive functioning (Moghaddam et al. 1997; Lorrain et al. 2003). Blocking this secondary effect by reducing the enhanced release of glutamate in the frontal cortex may provide a feasible strategy to selectively target mechanisms that influence cognitive functioning. The best example of this pharmacological approach, which has been tested in preclinical and clinical models of schizophrenia, involves LY354740 an agonist of mGlu2/3 receptors, which, as alluded to above, autoregulate the release of glutamate. In rodents, agonists of mGlu2/3 receptors normalize the increase in glutamate overflow after NMDA antagonist treatment (Moghaddam and Adams 1998; Lorrain et al. 2003). More importantly, these drugs reduce many of the behavioral abnormalities produced by NMDA receptor antagonist treatment, including stereotypy (Moghaddam and Adams 1998; Cartmell et al. 1999) and working memory deficits (Moghaddam and Adams 1998). Positive allosteric modulators of mGlu2 receptors, which may be more feasible for long-term treatment than direct agonists, have recently been discovered (Johnson et al. 2003). Nonetheless, direct mGlu2/3 agonists have been successfully used in clinical trials for treatment for anxiety (Schoepp et al. 2003), and limited clinical trials with LY354740 in healthy volunteers treated with subanesthetic doses of the NMDA antagonist ketamine indicate that this strategy reverses some of the cognitive impairments induced by ketamine (Krystal et al. 2003). Considering the accumulated evidence suggesting that NMDA receptor dysfunction may be critical to etiology and pathophysiology of schizophrenia (Coyle et al. 2002; Harrison and Owen 2003; Moghaddam 2003), these basic and clinical findings would suggest that potentiating mGlu2/3 receptor function may be an effective treatment strategy for the cognitive symptoms of schizophrenia. Assessment of the effect of this class of drugs on the cognitive functioning in schizophrenia is hopefully not far in the future.
References
Adler CM, Goldberg TE, Malhotra AK, Pickar D, Breier A (1998) Effects of ketamine on thought disorder, working memory, and semantic memory in healthy volunteers. Biol Psychiatry 43:811–816
Adler CM, Malhotra AK, Elman I, Goldberg T, Egan M, Pickar D, Breier A (1999) Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am J Psychiatry 156:1646–1649
Andreasen NC, O’Leary DS, Flaum M, Nopoulos P, Watkins GL, Boles Ponto LL, Hichwa RD (1997) Hypofrontality in schizophrenia: distributed dysfunctional circuits in neuroleptic-naive patients. Lancet 349:1730–1734
Attucci S, Albani-Torregrossa S, Moroni F, Pellegrini-Giampietro DE (2001) Metabotropic glutamate receptors stimulate phospholipase D via different pathways in the adult and neonate rat hippocampus. Neurochem Res 26:1151–1155
Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ (2000) Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci 20:7871–7879
Battaglia G, Monn JA, Schoepp DD (1997) In vivo inhibition of veratridine-evoked release of striatal excitatory amino acids by the group II metabotropic glutamate receptor agonist LY354740 in rats. Neurosci Lett 229:161–164
Benes FM, Vincent SL, Alsterberg G, Bird ED, SanGiovanni JP (1992) Increased GABAA receptor binding in superficial layers of cingulate cortex in schizophrenics. J Neurosci 12:924–929
Cartmell J, Schoepp DD (2000) Regulation of neurotransmitter release by metabotropic glutamate receptors. Journal of Neurochemistry 75:889–907
Cartmell J, Monn JA, Schoepp DD (1999) The metabotropic glutamate 2/3 receptor agonists LY354740 and LY379268 selectively attenuate phencyclidine versus d-amphetamine motor behaviors in rats. J Pharmacol Exp Ther 291:161–170
Clinton SM, Haroutunian V, Davis KL, Meador-Woodruff JH (2003) Altered transcript expression of NMDA receptor-associated postsynaptic proteins in the thalamus of subjects with schizophrenia. Am J Psychiatry 160:1100–1109
Conn JP, Pin J-P (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37:205–237
Coyle JT, Tsai G, Goff DC (2002) Ionotropic glutamate receptors as therapeutic targets in schizophrenia. Curr Drug Target CNS Neurol Disord 1:183–189
Crawford J, Curtis D (1964) The excitation and depression of mammalian cortical neurons by amino acids. Br J Pharmacol 23:323–329
Curtis D, Phillis J, Watkins J (1960) The chemical excitation of spinal neurons by certain acidic amino acids. J Physiol 150:656–682
De Blasi A, Conn PJ, Pin J, Nicoletti F (2001) Molecular determinants of metabotropic glutamate receptor signaling. Trends Pharmacol Sci 22:114–120
Doherty AJ, Palmer MJ, Henley JM, Collingridge GL, Jane DE (1997) (RS)-2-chloro-5-hydroxyphenylglycine (CHPG) activates mGlu5, but no mGlu1, receptors expressed in CHO cells and potentiates NMDA responses in the hippocampus. Neuropharmacology 36:265–267
Goldman-Rakic P (1994) Cerebral cortical mechanisms in schizophrenia. Neuropsychopharmacology 10:22S–27S
Greene R (2001) Circuit analysis of NMDAR hypofunction in the hippocampus, in vitro, and psychosis of schizophrenia. Hippocampus 11:569–577
Gur RE, Cowell PE, Latshaw A, Turetsky BI, Grossman RI, Arnold SE, Bilker WB, Gur RC (2000) Reduced dorsal and orbital prefrontal gray matter volumes in schizophrenia. Arch Gen Psychiatry 57:761–768
Harrison PJ (1999) The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain 122:593–624
Harrison PJ, Owen MJ (2003) Genes for schizophrenia? Recent findings and their pathophysiological implications [comment]. Lancet 361:417–419
Henry SA, Lehmann-Masten V, Gasparini F, Geyer MA, Markou A (2002) The mGluR5 antagonist MPEP, but not the mGluR2/3 agonist LY314582, augments PCP effects on prepulse inhibition and locomotor activity. Neuropharmacology 43:1199–1209
Hollmann M, Heinemann S (1994) Cloned glutamate receptors. Annu Rev Neurosci 17:31–108
Homayoun H, Stefani MR, Adams BW, Tamagan GD, Moghaddam B (2004) Functional interaction between NMDA and mGlu5 receptors: effects on working memory, instrumental learning, motor behaviors, and dopamine release. Neuropsychopharmacology (in press)
Huntley G, Vickers J, Morrison J (1994) Cellular and synaptic localization of NMDA and non-NMDA receptor subunits in neocortex: organizational features related to cortical circuitry, function and disease. Trends Neurosci 17:536–543
Javitt DC (2002) Glycine modulators in schizophrenia. Curr Opin Invest Drugs 3:1067–1072
Johnson MP, Baez M, Jagdmann GE, Jr., Britton TC, Large TH, Callagaro DO, Tizzano JP, Monn JA, Schoepp DD (2003) Discovery of allosteric potentiators for the metabotropic glutamate 2 receptor: synthesis and subtype selectivity of N-(4-(2-methoxyphenoxy)phenyl)-N-(2,2,2- trifluoroethylsulfonyl)pyrid-3-ylmethylamine. J Med Chem 46:3189–3192
Kinney G, Wittmann M, Bristow L, Campbell U, Conn P (2002) Behavioral consequences of mGluR5 and NMDA receptor antagonist interaction: implications for schizophrenia. Neuropharmacology 43:292
Kinney G, Burno M, Campbell U, Hernandez L, Rodriguez D, Bristow L, Conn P (2003) Metabotropic glutamate subtype 5 receptors modulate locomotor activity and sensorimotor gating in rodents. J Pharmacol Exp Ther 306:116–123
Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, Wichmann J, Kemp JA (2001) Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site.[erratum appears in Proc Natl Acad Sci USA 2001 Dec 18;98(26):15393]. Proc Natl Acad Sci USA 98:13402–13407
Konradi C, Heckers S (2003) Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther 97:153–179
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers Jr M, Charney DS (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51:199–214
Krystal JH, Bennett A, Abi-Saab D, Belger A, Karper LP, D’Souza DC, Lipschitz D, Abi-Dargham A, Charney DS (2000) Dissociation of ketamine effects on rule acquisition and rule implementation: possible relevance to NMDA receptor contributions to executive cognitive functions. Biol Psychiatry 47:137–143
Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R (2003) NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology 169:215–233
Lewis DA, Anderson SA (1995) The functional architecture of the prefrontal cortex and schizophrenia. Psychol Med 25:887–894
Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA (2003) Effects of ketamine and N-methyl-d-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience 117:697–706
Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ (2001) Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci 21:5925–5934
Marino MJ, Conn PJ (2002) Direct and indirect modulation of the N-methyl d-aspartate receptor. Curr Drug Target CNS Neurol Disord 1:1–16
Marino MJ, Williams DL Jr, O’Brien JA, Valenti O, McDonald TP, Clements MK, Wang R, DiLella AG, Hess JF, Kinney GG, Conn PJ (2003) Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc Natl Acad Sci USA 100:13668–13673
McCullumsmith RE, Meador-Woodruff JH (2002) Striatal excitatory amino acid transporter transcript expression in schizophrenia, bipolar disorder, and major depressive disorder. Neuropsychopharmacology 26:368–375
Moghaddam B (2003) Bringing order to the glutamate chaos in schizophrenia. Neuron 40:881–884
Moghaddam B, Adams B (1998) Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 281:1349–1352
Moghaddam B, Adams B, Verma A, Daly D (1997) Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17:2921–2927
Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, Craft S, Olney JW (1999) Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology 20:106–118
Nicoletti F, Meek JL, Iadarola MJ, Chuang DM, Roth BL, Costa E (1986) Coupling of inositol phospholipid metabolism with excitatory amino acid recognition sites in rat hippocampus. J Neurochem 46:40–46
O’Brien JA, Lemaire W, Chen TB, Chang RS, Jacobson MA, Ha SN, Lindsley CW, Schaffhauser HJ, Sur C, Pettibone DJ, Conn PJ, Williams DL Jr (2003) A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol Pharmacol 64:731–740
Phillips T, Rees S, Augood S, Waldvogel H, Faull R, Svendsen C, Emson P (2000) Localization of metabotropic glutamate receptor type 2 in the human brain. Neuroscience 95:1139–1156
Pisani A, Calabresi P, Centonze D, Bernardi G (1997) Enhancement of NMDA responses by group I metabotropic glutamate receptor activation in striatal neurones. Br J Pharmacol 120:1007–1014
Schoepp DD (2001) Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther 299:12–20
Schoepp DD, Conn PJ (2002) Metabotropic glutamate receptors. Pharmacol Biochem Behav 74:255–256
Schoepp DD, Wright RA, Levine LR, Gaydos B, Potter WZ (2003) LY354740, an mGlu2/3 receptor agonist as a novel approach to treat anxiety/stress. Stress 6:189–197
Sladeczek F, Pin JP, Recasens M, Bockaert J, Weiss S (1985) Glutamate stimulates inositol phosphate formation in striatal neurones. Nature 317:717–719
Sugiyama H, Ito I, Hirono C (1987) A new type of glutamate receptor linked to inositol phospholipid metabolism. Nature 325:531–533
Tamaru Y, Nomura S, Mizuno N, Shigemoto R (2001) Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience 106:481–503
Tamminga CA (1998) Schizophrenia and glutamatergic transmission. Crit Rev Neurobiol 12:21–36
Vardi N, Duvoisin R, Wu G, Sterling P (2000) Localization of mGluR6 to dendrites of ON bipolar cells in primate retina. J Comp Neurol 423:402–412
Weinberger D, Berman K, Zec R (1986) Physiological dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow (rCBF) evidence. Arch Gen Psychiatry 43:114–125
Weinberger DR, Egan MF, Bertolino A, Callicott JH, Mattay VS, Lipska BK, Berman KF, Goldberg TE (2001) Prefrontal neurons and the genetics of schizophrenia. Biol Psychiatry 50:825–844
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moghaddam, B. Targeting metabotropic glutamate receptors for treatment of the cognitive symptoms of schizophrenia. Psychopharmacology 174, 39–44 (2004). https://doi.org/10.1007/s00213-004-1792-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-004-1792-z