
Abstract
Beta-adrenergic receptors (β-ARs) play a role in angiogenic processes that characterize neovascularization-associated retinal diseases, but the role of β3-ARs has not been disclosed yet. We used ex vivo retinal explants to investigate the role of β3-ARs in regulating vascular endothelial growth factor (VEGF) release associated with hypoxia. Whether nitric oxide (NO) mediates β3-AR regulation of VEGF release was also investigated. β3-AR activation was obtained using BRL 37344, whereas SR59230A, L-748,337, or specific siRNAs were used to block β3-ARs. Pharmacological approaches were used to interfere with the NO pathway. Western blot was used to determine β-AR levels. Enzyme-linked immunosorbent assay was used to measure VEGF release. NO production was assessed by a colorimetric assay. We found that hypoxia upregulates β3-ARs. In addition, we observed that β3-AR activation with BRL 37344 increases VEGF release in response to hypoxia. Either β3-AR blocker or β3-AR silencing downregulates drastically hypoxic levels of VEGF. With experiments using NO synthase (NOS) blockade with L-NAME, NOS activation with fluvastatin or NO supplementation with SNAP, we demonstrated that β3-ARs and VEGF are functionally coupled via the NO pathway. In summary, the data presented here support the assumption that β3-ARs are involved in the regulation of angiogenic responses to hypoxia through the NO signalling, a key pathway in hypoxic/ischemic diseases. Although extrapolation of these data to the human situation is difficult, these findings may help to explore the possible role of β3-ARs in vascularization-associated disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Angiogenesis is a multistep process, controlled by opposing regulatory factors, which plays a crucial role in several ocular diseases (Sapieha et al. 2010). In the retina, hypoxia is considered to be one of the key factors to trigger angiogenesis by inducing vascular endothelial growth factor (VEGF; Arden and Sivaprasad 2011). In this respect, anti-VEGF therapy has become a mainstay of managing vascularization-associated diseases in the retina, although some limitations including recurrence of neovascularization and a number of side effects may be taken into account (Tremolada et al. 2012). In this respect, new pharmacological strategies to counteract retinal diseases characterized by angiogenesis are urgently needed.
Recently, the beta-adrenergic system has been shown to interfere with angiogenesis-dependent diseases (Pérez-Sayáns et al. 2010; Storch and Hoeger 2010). For instance, in C57BL/6J mice with oxygen-induced retinopathy (OIR), a well-established model of retinopathy of prematurity (Chen and Smith 2007), we have demonstrated that β-AR blockade with propranolol downregulates VEGF and reduces proangiogenic effects of hypoxia (Ristori et al. 2011). We have also found that β2-AR blockade with ICI 118,551 or β2-AR desensitization after prolonged isoproterenol administration reduces VEGF upregulation and retinal neovascularization in response to hypoxia (Dal Monte et al. 2012; Martini et al. 2011). β3-AR blockade does not seem to affect angiogenic response to hypoxia in the OIR model (Martini et al. 2011); however, several findings are indicative of a role of β3-ARs in retinal angiogenesis. Indeed, β3-ARs are localized to blood vessels in rodent retinas (Chen et al. 2012; Mori et al. 2010; Ristori et al. 2011), they are upregulated by hypoxia in OIR mice (Chen et al. 2012; Ristori et al. 2011), and β3-AR activation induces markers of angiogenesis in human retinal and choroidal endothelial cells (Steinle et al. 2005; 2003).
A possible role of β3-ARs in retinal angiogenesis may involve nitric oxide (NO). Although no data demonstrating NO as a downstream effector of β3-AR are available in the retina, increasing evidence indicates that β3-ARs promote NO synthesis in different experimental models. For instance, β3-AR agonism activates NO signaling cascade in mouse cardiac myocytes (Cawley et al. 2011) and in rat aortic rings (Ignarro et al. 2002) and stimulates lipolysis by inducing NO production in rat adipocytes (Hodis et al. 2011). There is also accumulating evidence that NO is an important modulator of the expression of endogenous angiogenic factors such as VEGF (Ziche and Morbidelli 2009). In this respect, NO is known to be implicated in pathogenic retinal angiogenesis (Wilkinson-Berka 2004), and inhibiting NO production results in decreased VEGF levels and attenuated retinal neovascularization (He et al. 2007; Zhang et al. 2009).
In the present study, we used ex vivo mouse retinal explants to investigate whether (1) β3-ARs regulate VEGF release in response to hypoxia and (2) NO is involved in mediating this regulation. Although the ex vivo model does not exactly recapitulate in vivo homeostasis, it does permit direct retinal manipulation, allows greater control of retinal environment, and provides better efficiency of compounds (Curatola et al. 2005; Kaempf et al. 2008). In addition, mouse retinal explants are a well-established model to study angiogenesis-related events (DeNiro et al. 2009; Martini et al. 2011; Mei et al. 2012).
Materials and methods
Materials
Dulbecco’s modified Eagle medium (DMEM) and fetal calf serum (FCS) were obtained from Lonza (Basel, Switzerland). Millicell-CM culture inserts (0.4 mm × 30 mm) and the enhanced chemiluminescence reagent were from Millipore (Billerica, MA, USA). The β3-AR agonist BRL 37344 ((±)-(R*,R*)-[4-[2-[[2-(3-Chlorophenyl)-2-hydroxyethyl]amino]propyl]phenoxy]acetic acid sodium hydrate), the β3-AR antagonist SR59230A (3-(2-ethylphenoxy)-1-[[(1S)-1,2,3,4-tetrahydronaphth-1-yl]amino]-(2S)-2-propanol oxalate salt), the nitric oxide synthase (NOS) inhibitor l-NAME (Nω-nitro-l-arginine methyl ester hydrochloride), the NOS activator fluvastatin ((±)-(3R*,5S*,6E)-7-[3-(4-fluorophenyl)-1-(1-methyethyl)-1H-indol-2-yl]-3,5-dihydroxy-6-heptenoic acid sodium salt hydrate), the NO donor SNAP (S-nitroso-N-acetyl-dl-penicillamine), the primary monoclonal antibody directed to β-actin and the rabbit anti-mouse horseradish peroxidase (HRP)-labeled secondary antibody were from Sigma-Aldrich (St. Louis, MO, USA). The β3-AR antagonist L-748,337 was purchased from Tocris Bioscience (Bristol, UK). The protease inhibitor cocktail Complete was obtained from Roche Applied Science (Indianapolis, IN, USA). Polyvinylidene difluoride (PVDF) membrane was obtained from GE Healthcare (Piscataway, NJ). The primary mouse monoclonal antibody directed to mitogen-activated protein kinase 1 (MAPK1), primary rabbit polyclonal antibodies directed to β1- or β2-ARs, primary goat polyclonal antibody directed to β3-ARs, the mouse anti-rabbit HRP-labeled secondary antibody, and the rabbit anti-goat HRP-labeled secondary antibody were from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). The enzyme-linked immunosorbent assay (ELISA) kit for the detection of VEGF was from R&D Systems (Minneapolis, MN, USA). The RNeasy Mini Kit, the QuantiTect Reverse Transcription Kit, pre-designed Flexitube siRNAs directed to β3-ARs, a non-silencing siRNA, a positive control siRNA directed to mitogen-activated protein kinase 1 and the HiPerfect transfection reagent were purchased from Qiagen (Valencia, CA, USA). The iQ Sybr Green Supermix was obtained from Bio-Rad Laboratories (Hercules, CA, USA). The colorimetric assay kit for the detection of NO production was from Enzo Life Sciences (Plymouth Meeting, PA, USA).
Animals
Adult C57BL/6J mice (Charles River Laboratories Italia, Calco, Italy) at 6–8 weeks after birth were used. Procedures involving animals were carried out in agreement with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and in compliance with the Italian guidelines for animal care (DL 116/92) and the European Communities Council Directive (86/609/EEC). Procedures were approved by the Ethical Committee in Animal Experiments of the University of Pisa. All efforts were made to reduce both animal suffering and the number of animals used. Experiments were performed on a total of 141 mice of both sexes. In all experiments, mice were anesthetized with halothane (4 %) and killed by cervical dislocation, and the eyes were enucleated.
Retinal explants
Retinal explants were cultured according to Mei et al. (2012). Briefly, enucleated eyes were immediately transferred to DMEM containing 10 % FCS plus Fungizone (1.25 μg/mL). The choroid, sclera, cornea, and iris were removed from the globe under a dissecting microscope. The remaining retina, vitreous, and lens were incubated in a dish with fresh DMEM for 30–60 min at 37 °C. Then, retinal pigment epithelium was gently teased away from the retina using fine forceps. The isolated retina was transferred onto a culture insert with enough fresh DMEM to barely cover the explant. The retina was cut at 3, 6, 9, and 12 o’clock to flatten the whole mount and placed photoreceptor-side down onto the center of the insert. The medium was removed from the insert leaving only a moist film covering the tissue. The edges of the retina were flattened by light brushing. Inserts were placed in six-well tissue culture plates containing 1 mL of DMEM with 10 % FCS plus Fungizone (1.25 μg/mL) without or with drugs or siRNAs. Retinal explants were cultured in either normoxia (20 % O2, 5 % CO2, 37 °C) or hypoxia (1 % O2, 5 % CO2, 37 °C) in line with a previous study (Mei et al. 2012). Hypoxic conditions were achieved using an incubator (MiniGalaxy A; RS Biotech, Irvine, Scotland, UK) flushed with 5 % (v/v) CO2 and N2 to reduce O2 level. The O2 concentration was monitored and maintained at 1 ± 0.1 %. Retinal explants were incubated in hypoxia for 24 h (Mei et al. 2012). Drugs were added to the medium at the onset of hypoxia. In the silencing experiments, siRNAs were added to the medium 24 h before the onset of hypoxia (Mei et al. 2012).
β3-AR agonists and antagonists
BRL 37344 is an established β3-AR agonist (Vrydag and Michel 2007) which has been previously used in mice (Aragón et al. 2011). BRL 37344 agonistic effects vary in different species. For instance, its EC50 toward the lipolytic response in white adipocytes is in the nanomolar range in rat, while it is in the micromolar range in human (Carpéné et al. 1999). In addition, BRL 37344 has been found to be effective at 10 μM in the human isolated internal anal sphincter model (Ballester et al. 2010) and in human retinal endothelial cells (Steinle et al. 2003). Based on these data, we used BRL 37344 at 1, 3, and 10 μM.
SR59230A is the most widely used β3-AR antagonist (Vrydag and Michel 2007), and it has been previously used in mice (Martini et al. 2011). Its IC50 toward the lipolytic response in rat white adipocytes is in the micromolar range, with maximal inhibitory effects at 10 μM (Carpéné et al. 1999). Similar values for IC50 and maximal inhibitory effects have been reported in the human detrusor strip model (Kanie et al. 2012). Based on these data, we used SR59230A at 1, 3, and 10 μM.
L-748,337 is one of the very few antagonists with high selectivity for β3-ARs (Wuest et al. 2009). Its IC50 toward cAMP production in CHO cells transfected with β3-ARs is in the nanomolar range, with maximal inhibitory effects in the micromolar range (Candelore et al. 1999). In addition, in the murine isolated urinary bladder, L-748,337 at 10 μM has been reported to inhibit the effect of fenoterol, a β2-AR agonist which also displays an agonistic activity at β3-ARs (Palea et al. 2012). Based on these data, we used L-748,337 at 1, 3, and 10 μM.
BRL 37344 was dissolved in phosphate buffer (PB), while SR59230A and L-748,337 were dissolved in DMSO (final concentration 0.1 %). Control experiments were performed with 0.1 % DMSO in PB (vehicle).
Pharmacology of the NO pathway
NOS inhibition was obtained using 1 mM l-NAME as previously reported (Nakagawa et al. 2006). NOS activation was induced using 1 μM fluvastatin, a concentration known to activate both endothelial and inducible NOS (Chen et al. 2000; Meda et al. 2010). To increase NO production, we also used a different approach bypassing NOS activation with NO supplementation. To this aim, we used 2 mM SNAP, a stable analogous of endogenous S-nitrosothiols which acts as NO donor, as previously reported (West et al. 2006). l-NAME and fluvastatin were dissolved in PB, while SNAP was dissolved in DMSO (final concentration 0.1 %).
Western blot analysis
Western blot analysis was performed using antibodies directed against MAPK1, β1-, β2-, β3-ARs, and β-actin in line with Dal Monte et al. (2012). Retinas were homogenized in 10 mM Tris–HCl, pH 7.6, containing 5 mM EDTA, 3 mM EGTA, 250 mM sucrose, and protease inhibitor cocktail, and centrifuged at 22,000×g for 30 min at 4 °C. The supernatant containing cytosolic proteins was used to detect MAPK1 (not shown). The pellet was resuspended in 20 mM HEPES, pH 7.4 containing 150 mM NaCl, 5 mM EDTA, 3 mM EGTA, 4 mg/mL N-dodecyl-β-maltoside and protease inhibitor cocktail and centrifuged at 22,000×g for 30 min at 4 °C. The supernatant containing membrane proteins were used to detect β1-, β2-, and β3-ARs. Protein concentration was determined using a fluorometer (Qubit; Invitrogen). Aliquots of each sample containing equal amounts of protein (40 μg) were subjected to SDS-PAGE. The gels were transblotted onto PVDF membrane. PVDF membranes were blocked in 5 % skim milk for 1 h at room temperature and then incubated overnight at 4 °C with a primary mouse monoclonal antibody directed to MAPK1 (1:100 dilution), primary rabbit polyclonal antibodies directed to β1- (1:200 dilution) or β2-ARs (1:200 dilution) or a primary goat polyclonal antibody directed to β3-ARs (1:200 dilution). The same PVDF membrane was reblotted with a primary mouse monoclonal antibody directed to β-actin (1:2,500 dilution) as loading control. Finally, PVDF membranes were incubated for 1 h at room temperature with rabbit anti-mouse (1:25,000), mouse anti-rabbit (1:1,000), or rabbit anti-goat (1:5,000) HRP-labeled secondary antibodies, as appropriate, and developed with the enhanced chemiluminescence reagent. Images were acquired (Chemidoc XRS+; Bio-Rad Laboratories). The OD of the bands was evaluated (Image Lab 3.0 software; Bio-Rad Laboratories). The data were normalized to the level of β-actin as loading control. For each experimental condition, six samples from six different mice were used. Each sample refers to the protein extracted from one retina. All experiments were run in duplicate. After statistics, data were averaged and plotted in the same graph.
Enzyme-linked immunosorbent assay
VEGF release was measured in the culture medium using a commercially available ELISA kit. VEGF release was measured spectrophotometrically (Microplate Reader 680 XR; Bio-Rad Laboratories) at 450 nm (correction wavelength set at 570 nm). For each experimental condition, six samples from six different mice were used. Each sample refers to the culture medium of one retina. Data were collected as picograms VEGF per milliliter culture medium. All experiments were run in duplicate. After statistical analysis, data were averaged and plotted in the same graph.
siRNA transfection
In preliminary experiments, four different pre-designed siRNAs targeting β3-AR gene, each at three different concentrations (5, 15, and 50 nM), were used to identify the siRNA which gave the maximum reduction in the expression of β3-AR at the lowest concentration (Online Resource 1). A non-silencing siRNA was used as negative control together with mock control. siRNA transfection was performed using the transfection reagent. For each experimental condition, six samples from six different mice, each containing one retina, were used. In order to evaluate transfection efficiency, Western blot was used to measure MAPK1 expression after transfection with MAPK1-positive control siRNA. After MAPK1 silencing, MAPK1 expression was reduced by ∼85 % as compared with untransfected, mock-transfected, or non-silencing-transfected explants (not shown). The success of transfection with β3-AR-siRNAs was demonstrated by Quantitative real-time RT-PCR (qPCR) (Online Resource 1, panel a). The silencing efficiency was confirmed by Western blot (Online Resource 1, panel b). After these preliminary experiments, we used β3-AR-siRNA4 at 50 nM. After transfection with β3-AR-siRNA4 or non-silencing siRNA, retinal explants were cultured for 24 h in normoxia followed by 24 h hypoxia.
RNA isolation and cDNA preparation
From each retina, total RNA was extracted (RNeasy Mini Kit; Qiagen), purified, resuspended in RNase-free water and quantified using a fluorometer (Qubit; Invitrogen, Milan, Italy). First-strand cDNA was generated from 1 μg of total RNA (QuantiTect Reverse Transcription Kit; Qiagen).
Quantitative real-time RT-PCR
qPCR was performed using published primers for β3-ARs (Ristori et al. 2011). qPCR was performed using SYBR Green on a detection system (MiniOpticon Two-Color; Bio-Rad Laboratories). Polymerase chain reaction (PCR) reactions included 1 μL of cDNA, 300 nM of each primer, 7.5 μL of mastermix (iQ Sybr Green Supermix; Bio-Rad Laboratories), and RNase-free water to a final volume of 15 μL. The target gene was run concurrently with the ribosomal protein L13A gene (Rpl13a), which encodes a ribosomal protein that is a component of the 60S subunit (Mazumder et al. 2003). Rpl13a is known to be a stable housekeeping gene in OIR mice and mouse retinal explants (Dal Monte et al. 2009, 2010; Martini et al. 2011; Mei et al. 2012; Ristori et al. 2011). Negative control PCR reactions without any template cDNA were performed. The qPCR was performed with hot-start denaturation step at 95 °C for 3 min, followed by 40 cycles at 95 °C for 10 s, and 58 °C for 20 s. The fluorescence was read during the reaction, allowing a continuous monitoring of the amount of PCR product. Primers were initially used to generate a standard curve over a large dynamic range of cDNA quantity, which allows calculation of the amplification efficiency for the primer pair (not shown). Amplification efficiency was close to 100 % (Opticon Monitor 3 software; Bio-Rad Laboratories). After amplification, first derivative melting curve analysis was used to confirm that the signal corresponded to a unique amplicon (not shown). PCR products were analyzed by nucleic acid staining on a gel stain (GelStar) containing 3 % agarose to verify the correct product size (not shown). After performing qPCR, the results were analyzed using the comparative cycle threshold method (Livak and Schmittgen 2001). Changes in mRNA expression were relative to the hypoxic levels after normalization to Rpl13a. For each experimental condition, six samples from six different mice were used. Each sample refers to the mRNA extracted from one retina. All reactions were run in triplicate. After statistical analysis, data were averaged and plotted in the same graph.
Colorimetric assay
Total amount of NO (micrometer) was assessed by the Griess reaction using a colorimetric assay kit that detects nitrite, a stable reaction product of NO (Kaur et al. 2009). Measurements were performed according to the manufacturer’s instructions on the supernatants containing the cytosolic proteins used in Western blot. OD was measured spectrophotometrically (Microplate Reader 680 XR; Bio-Rad Laboratories) at 540 nm. For each experimental condition, six samples from six different mice were used. Each sample refers to the protein extracted from one retina. All experiments were run in duplicate. After statistics, data were averaged in the same graph.
Statistics
Statistical significance was evaluated using unpaired Student's t test or one-way analysis of variance (ANOVA) followed by Newman–Keuls multiple-comparison post-test as appropriate. The results were expressed as means ± SEM of the indicated n values. Prism 4 (GraphPad Software, Inc., La Jolla, CA, USA) was used to analyze data. Differences with P < 0.05 were considered statistically significant.
Results
Effects of β3-AR agonism/antagonism or β3-AR silencing
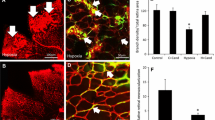
In a preliminary set of experiments, we confirmed that, similar to what was found in the retina of the OIR model (Ristori et al. 2011), β3-ARs were upregulated by hypoxia, whereas β1- or β2-ARs were unaffected (Fig. 1).

Hypoxia regulation of β3-ARs. The expression of β1- (a), β2- (b), and β3-ARs (c) was evaluated by Western blot and densitometric analysis in normoxic and hypoxic retinal explants. Protein expression was relative to the loading control β-actin. Each column represents the mean ± SEM of data from six independent samples, each containing one retina. *P < 0.001 versus normoxic (unpaired Student's t test)
β3-AR agonists/antagonists were then used in order to investigate whether β3-ARs play a role in regulating VEGF release in response to hypoxia. As shown in Fig. 2, VEGF release was increased by ∼4-fold by hypoxia (**P < 0.001 versus normoxic). No effects were observed after vehicle treatment (not shown). β3-AR agonists/antagonists dose-dependently affected hypoxia-induced VEGF release. BRL 37344 did not affect VEGF release at 1 μM, while significantly increased VEGF release at 3 or 10 μM (∼26 %, § P < 0.05 and ∼46 %, §§ P < 0.001 versus hypoxic, respectively). SR59230A did not influence VEGF release at 1 or 3 μM, while it significantly reduced VEGF release at 10 μM (∼54 %, §§ P < 0.001 versus hypoxic). L-748,337 significantly reduced VEGF release by ∼28 % at 1 μM (§ P < 0.05 versus hypoxic), ∼51 % at 3 μM (§§ P < 0.001 versus hypoxic), and ∼74 % at 10 μM (§§ P < 0.001 versus hypoxic), respectively. After SR59230A at 10 μM, VEGF release was ∼140 % higher than in normoxia (*P < 0.05 versus normoxic), whereas after L-748,337 at 10 μM, VEGF release did not differ significantly from that measured in normoxia.

VEGF release after BRL 37344, SR59230A, or L-748,337. VEGF release was evaluated by ELISA in normoxic and in hypoxic retinal explants either untreated or treated with BRL 37344, SR59230A, or L-748,337. All drugs were used at 1, 3, or 10 μM. Each column represents the mean ± SEM of data from six independent samples, each containing one retina. *P < 0.05 and **P < 0.001 versus normoxic; § P < 0.05 and §§ P < 0.001 versus hypoxic (ANOVA followed by Newman–Keuls multiple-comparison post-test)
Specificity of β3-AR pharmacological activation/blockade was confirmed by experiments with β3-AR silencing. As shown in Fig. 3a, after β3-AR-siRNA4 at 50 nM, VEGF release was significantly lower than in hypoxia (∼52 %, § P < 0.001 versus hypoxic) but ∼135 % higher than in normoxia (*P < 0.05 versus normoxic). In addition, the effects of BRL 37344, SR59230A, or L-748,337 (10 μM) on VEGF release were prevented by β3-AR silencing (Fig. 3b). Effects of BRL 37344, SR59230A, or L-748,337 on VEGF release were not influenced by non-silencing siRNA (not shown).

VEGF release after β3-AR silencing and effects of BRL 37344, SR59230A, or L-748,337 on VEGF release after β3-AR silencing. a VEGF release was evaluated by ELISA in normoxic and hypoxic retinal explants either untreated or treated with non-silencing siRNA or 50 nM β3-AR-siRNA4. b VEGF release was evaluated by ELISA in hypoxic retinal explants treated with β3-AR agonists/antagonists at 10 μM either in the absence or in the presence of 50 nM β3-AR-siRNA4. Each column represents the mean ± SEM of data from six independent samples, each containing one retina. *P < 0.05 and **P < 0.001 versus normoxic; § P < 0.001 versus hypoxic (ANOVA followed by Newman–Keuls multiple-comparison post-test)
Role of NO signalling in the β3-AR-induced regulation of VEGF release
On the basis of evidence supporting NO as a downstream effector of β3-ARs (Hodis et al. 2011; Ignarro et al. 2002; Massion et al. 2004) and of the reported regulation of VEGF expression by NO (Ziche and Morbidelli 2009), we investigated whether NO signaling participates in the β3-AR-induced regulation of VEGF release.
We first determined whether NO production in response to hypoxia may be influenced by β3-AR activation, blockade, or silencing. No effects were observed after vehicle treatment or non-silencing siRNA (not shown). As shown in Fig. 4, hypoxia significantly increased NO levels (205 %, ***P < 0.001 versus normoxic). BRL 37344 at 10 μM increased hypoxic levels of NO by ∼25 % (§ P < 0.05 versus hypoxic), while SR59230A and L-748,337 at 10 μM or β3-AR-siRNA4 at 50 nM significantly reduced hypoxic levels of NO (∼31 %, §§ P < 0.01, ∼45 % and ∼53 %, §§§ P < 0.001 versus hypoxic, respectively).

NO production after BRL 37344, SR59230A, L-748,337, or β3-AR-siRNA4. NO production was evaluated by a colorimetric assay in normoxic and hypoxic retinal explants either untreated or treated with β3-AR agonists/antagonists at 10 μM or 50 nM β3-AR-siRNA4. Each column represents the mean ± SEM of data from six independent samples, each containing one retina. *P < 0.05, **P < 0.01, and ***P < 0.001 versus normoxic; § P < 0.05, §§ P < 0.01, and §§§ P < 0.001 versus hypoxic (ANOVA followed by Newman–Keuls multiple-comparison post-test)
In a second set of experiments, we interfered with NO signaling in order to establish whether β3-AR-induced regulation of VEGF release in response to hypoxia occurs through NO. To this aim, we used 1 mM L-NAME in combination with BRL 37344 at 10 μM in order to determine whether NOS inhibition prevents VEGF upregulation in response to β3-AR agonism. Fluvastatin at 1 μM or SNAP at 2 mM was also used in combination with SR59230 and L-748,337 at 10 μM or β3-AR-siRNA4 at 50 nM in order to determine whether NOS activation or NO supplementation prevents VEGF downregulation in response to β3-AR antagonism/silencing. As shown in Fig. 5, L-NAME prevented BRL 37344-induced increase of VEGF. In addition, effects of SR59230, L-748,337, or β3-AR-siRNA4 were abolished by fluvastatin or SNAP. In these experimental conditions, VEGF levels did not differ from those measured after BRL 37344 alone.

Effects of NOS inhibition after BRL 37344 and effects of NOS activation or NO supplementation after SR59230A, L-748,337, or β3-AR-siRNA4 on VEGF release. VEGF release was evaluated by ELISA in hypoxic retinal explants treated with 10 μM BRL 37344 either in the absence or in the presence of 1 mM L-NAME or with 10 μM SR59230A, 10 μM L-748,337, or 50 nM β3-AR-siRNA4 either in the absence or in the presence of 1 μM fluvastatin or 2 mM SNAP. Each column represents the mean ± SEM of data from six independent samples, each containing one retina. § P < 0.001 versus hypoxic (ANOVA followed by Newman–Keuls multiple-comparison post-test)
Discussion
In this study, we report a novel role for β3-ARs as endogenous regulators of angiogenesis-associated events in the retina. The fact that VEGF release is modulated by β3-ARs indicates that these receptors may participate in maintaining VEGF homeostasis and that β3-AR blockade may be effective in counteracting the damage produced by a proangiogenic environment. The additional finding that β3-AR effects on VEGF release may be regulated by NOS activation, NOS inhibition, or NO supplementation demonstrates that NO is one of the functional links between β3-ARs and VEGF in hypoxic retinas.
β3-AR role in regulating VEGF release
Recently, the β3-AR has emerged as a potential target for the treatment of ischemic disorders because of its upregulation in response to hypoxia and its resistance to desensitization (Dessy and Balligand 2010).
As shown by present results, β3-ARs regulate VEGF release in hypoxic retinas. In fact, BRL 37344 increases VEGF release suggesting that activation of β3-ARs may constitute an important part of the retinal response to hypoxia. In line with our present results, BRL 37344 induces VEGF expression in mouse adipocytes (Fredriksson et al. 2000) and causes proliferation of human retinal endothelial cells (Steinle et al. 2003) indicating β3-ARs as a possible regulatory switch in retinal angiogenesis. Some issues have been reported in the use of BRL 37344. For instance, studies in the human heart support the idea that BRL 37344 may have an intrinsic activity at β1- or β2-ARs (Pott et al. 2003). In addition, BRL 37344 may have effects unrelated to β-ARs that may occur at similar concentrations as those used for the activation of β3-ARs (Kubota et al. 2002; Leblais et al. 2005). On the other hand, our finding that β3-AR knockdown with selective siRNAs prevents the BRL 37344-mediated VEGF upregulation in hypoxic conditions supports the selectivity of BRL 37344 in our experimental setting and excludes that BRL 37344 can exert an off-target effect.
The fact that β3-AR antagonists prevent VEGF upregulation in response to hypoxia indicates that blocking β3-AR signaling may affect angiogenesis-related events. In this respect, SR59230A, a widely used β3-AR antagonist, may lead in some cases to misleading conclusions. For instance, SR59230A may also act as partial agonist at β3-ARs (Vrydag and Michel 2007) or it may not suppress the receptor constitutive activity, which is typical of β3-ARs (Perrone and Scilimati 2011). On the other hand, the fact that L-748,337, a β3-AR antagonist belonging to a different chemical class than SR59230A, prevents VEGF upregulation supports the selectivity of β3-AR blockade and demonstrates that interfering with β3-AR signaling is effective in attenuating retinal angiogenesis. The additional finding that β3-AR silencing reproduces the effects of β3-AR antagonism on VEGF release also supports the selectivity of β3-AR blockers. It is noteworthy that L-748,337 has potency greater than SR59230A in decreasing VEGF. This is in line with the finding that L-748,337 prevents vasodilation of rat retinal arterioles with a potency of one order of magnitude higher than SR59230A (Mori et al. 2010).
The fact that β3-ARs participate in VEGF regulation is consistent with the finding that β3-ARs are localized to retinal blood vessels and are upregulated by hypoxia (Chen et al. 2012; Ristori et al. 2011) indicating the possibility that β3-ARs may modulate VEGF release by endothelial cells. There is evidence that, in the retina, endothelial cells express and release VEGF (Tugues et al. 2011), although a major source of retinal VEGF is represented by Müller cells, particularly under hypoxic–ischemic conditions (Kaur et al. 2008). Thus, β3-AR localization to endothelial cells may not necessarily be linked to the regulation of VEGF release. In this respect, we have previously demonstrated that β2-ARs localized to Müller cells play an important role in regulating retinal VEGF in OIR (Martini et al. 2011).
The observed potent effect of β3-AR blockade on VEGF release in retinal explants would suggest an equally potent action of β3-AR blockade against pathologic angiogenesis. However, it has been observed recently that SR59230A does not affect either VEGF release or retinal neovascularization in the OIR model (Martini et al. 2011). This can be explained by assuming that, in OIR, systemic administration of SR59230A does not provide the retina with a sufficient drug concentration to interfere with VEGF release. As shown by our results, SR 59230A concentrations lower than 10 μM do not affect VEGF release in retinal explants.
Another possibility is that different post-receptor mechanisms may be activated in retinal explants and OIR. For instance, β3-ARs may hetero-oligomerize with β2-ARs, generating a β-adrenergic signaling unit with functional properties distinct from those of β2- and β3-ARs (Breit et al. 2004). Since the formation of hetero-oligomers between membrane receptors is influenced by oxidative stress caused by hypoxia–ischemia (van der Wijk et al. 2005; Li et al. 2012), we cannot exclude that differences in oxygen tension in the two models may be responsible of the different β3-AR function in retinal explants and OIR. In fact, in retinal explants, hypoxia corresponds to 1 % oxygen, whereas in OIR hypoxia corresponds to atmospheric oxygen tension when mice are returned to normoxia after hyperoxia.
β3-AR coupling to NO signalling
As shown by the present results, β3-ARs regulate NO production. This result is consistent with previous experimental evidence demonstrating that β3-ARs are coupled to NO pathway in the ischemic heart (Dessy and Balligand 2010), in human coronary microarteries (Dessy et al. 2004), in rat thoracic aorta (Trochu et al. 1999), and in rat retinal vessels (Mori et al. 2010).
Our results also show that modulating the NO pathway affects the β3-AR-induced VEGF release indicating that β3-AR-induced regulation of VEGF response to hypoxia is mediated, at least in part, by NO signaling. In this respect, complex but not completely elucidated relationships between NO and VEGF have been reported. In fact, NO may act upstream of VEGF, regulating its expression (Dulak and Józkowicz 2003). For instance, inhibition of the inducible isoform of NOS reduces VEGF levels in OIR (He et al. 2007; Zhang et al. 2009). Conversely, VEGF may act as an upstream regulator of NO (Wilkinson-Berka 2004). For instance, a VEGF trap reduces the diabetes-related NO increase in the rat retina (Joussen et al. 2002). This complexity may reflect overlapping and complementary functions between NO and VEGF pathways, suggesting their relationships as a nodal point in the control of angiogenesis-related retinal diseases.
Our evidence that β3-ARs regulate VEGF release through NO suggests possible pathways by which β3-AR agonists/antagonists may modulate VEGF. In fact, we may hypothesize that β3-ARs, expressed by VEGF-containing endothelial cells, modulate VEGF release acting through NO both in the same endothelial cells and, since NO is a diffusible molecule (Bryan et al. 2009), on surrounding cells, including Müller cells (Fig. 6 ).

A schematic diagram of the possible pathway by which β3-ARs regulate VEGF release through NO. β3-ARs expressed by endothelial cells modulate the production of NO (1) which, in turn, regulate VEGF release by the same endothelial cells (2, 3) or, since NO is a diffusible molecule (4, 5) by surrounding cells, including Müller cells (6, 7)
Conclusions and clinical implications
Overall, the present results suggest that VEGF accumulation, a key factor in many hypoxic–ischemic vision-threatening retinal diseases, depends at least in part on β3-AR activity, suggesting β3-AR blockers as attractive therapeutic tools. Although extrapolation of these data to the human situation is difficult, these results may help to explore the possible role of β3-ARs in vascularization-associated disorders. In this respect, the fact that β3-ARs are upregulated by hypoxia and that they influence retinal VEGF through the involvement of NO pathway suggests that blocking this signaling pathway may be effective in reducing VEGF-stimulated retinal angiogenesis.
References
Aragón JP, Condit ME, Bhushan S, Predmore BL, Patel SS, Grinsfelder DB, Gundewar S, Jha S, Calvert JW, Barouch LA, Lavu M, Wright HM, Lefer DJ (2011) Beta3-adrenoreceptor stimulation ameliorates myocardial ischemia-reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J Am Coll Cardiol 58:2683–2691
Arden GB, Sivaprasad S (2011) Hypoxia and oxidative stress in the causation of diabetic retinopathy. Curr Diabetes Rev 7:291–304
Ballester C, Sarriá B, García-Granero E, Mata M, Milara J, Morcillo EJ, Lledó S, Cortijo J (2010) Relaxation by beta 3-adrenoceptor agonists of the isolated human internal anal sphincter. Life Sci 86:358–364
Breit A, Lagacé M, Bouvier M (2004) Hetero-oligomerization between beta2- and beta3-adrenergic receptors generates a beta-adrenergic signaling unit with distinct functional properties. J Biol Chem 279:28756–28765
Bryan NS, Bian K, Murad F (2009) Discovery of the nitric oxide signaling pathway and targets for drug development. Front Biosci 14:1–18
Candelore MR, Deng L, Tota L, Guan XM, Amend A, Liu Y, Newbold R, Cascieri MA, Weber AE (1999) Potent and selective human beta(3)-adrenergic receptor antagonists. J Pharmacol Exp Ther 290:649–655
Carpéné C, Galitzky J, Fontana E, Atgié C, Lafontan M, Berlan M (1999) Selective activation of beta3-adrenoceptors by octopamine: comparative studies in mammalian fat cells. Naunyn Schmiedebergs Arch Pharmacol 359:310–321
Cawley SM, Kolodziej S, Ichinose F, Brouckaert P, Buys ES, Bloch KD (2011) sGC{alpha}1 mediates the negative inotropic effects of NO in cardiac myocytes independent of changes in calcium handling. Am J Physiol Heart Circ Physiol 301:H157–H163
Chen J, Smith LE (2007) Retinopathy of prematurity. Angiogenesis 10:133–140
Chen H, Ikeda U, Shimpo M, Ikeda M, Minota S, Shimada K (2000) Fluvastatin upregulates inducible nitric oxide synthase expression in cytokine-stimulated vascular smooth muscle cells. Hypertension 36:923–928
Chen J, Joyal JS, Hatton CJ, Juan AM, Pei DT, Hurst CG, Xu D, Stahl A, Hellstrom A, Smith LE (2012) Propranolol inhibition of β-adrenergic receptor does not suppress pathologic neovascularization in oxygen-induced retinopathy. Invest Ophthalmol Vis Sci 53:2968–2977
Curatola AM, Moscatelli D, Norris A, Hendricks-Munoz K (2005) Retinal blood vessels develop in response to local VEGF-A signals in the absence of blood flow. Exp Eye Res 81:147–158
Dal Monte M, Ristori C, Cammalleri M, Bagnoli P (2009) Effects of somatostatin analogues on retinal angiogenesis in a mouse model of oxygen-induced retinopathy: involvement of the somatostatin receptor subtype 2. Invest Ophthalmol Vis Sci 50:3596–3606
Dal Monte M, Ristori C, Videau C, Loudes C, Martini D, Casini G, Epelbaum J, Bagnoli P (2010) Expression, localization, and functional coupling of the somatostatin receptor subtype 2 in a mouse model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci 51:1848–1856
Dal Monte M, Martini D, Latina V, Pavan B, Filippi L, Bagnoli P (2012) Beta-adrenoreceptor agonism influences retinal responses to hypoxia in a model of retinopathy of prematurity. Invest Ophthalmol Vis Sci 53:2181–2192
DeNiro M, Alsmadi O, Al-Mohanna F (2009) Modulating the hypoxia-inducible factor signaling pathway as a therapeutic modality to regulate retinal angiogenesis. Exp Eye Res 89:700–717
Dessy C, Balligand JL (2010) Beta3-adrenergic receptors in cardiac and vascular tissues emerging concepts and therapeutic perspectives. Adv Pharmacol 59:1351–1363
Dessy C, Moniotte S, Ghisdal P, Havaux X, Noirhomme P, Balligand JL (2004) Endothelial beta3-adrenoceptors mediate vasorelaxation of human coronary microarteries through nitric oxide and endothelium-dependent hyperpolarization. Circulation 110:948–954
Dulak J, Józkowicz A (2003) Regulation of vascular endothelial growth factor synthesis by nitric oxide: facts and controversies. Antioxid Redox Signal 5:123–132
Fredriksson JM, Lindquist JM, Bronnikov GE, Nedergaard J (2000) Norepinephrine induces vascular endothelial growth factor gene expression in brown adipocytes through a beta-adrenoreceptor/cAMP/protein kinase A pathway involving Src but independently of Erk1/2. J Biol Chem 275:13802–13811
He T, Xing YQ, Zhao XH, Ai M (2007) Interaction between iNOS and COX-2 in hypoxia-induced retinal neovascularization in mice. Arch Med Res 38:807–815
Hodis J, Vaclavíková R, Farghali H (2011) Beta-3 agonist-induced lipolysis and nitric oxide production: relationship to PPARgamma agonist/antagonist and AMP kinase modulation. Gen Physiol Biophys 30:90–99
Ignarro LJ, Sisodia M, Trinh K, Bedrood S, Wu G, Wei LH, Buga GM (2002) Nebivolol inhibits vascular smooth muscle cell proliferation by mechanisms involving nitric oxide but not cyclic GMP. Nitric Oxide 7:83–90
Joussen AM, Poulaki V, Qin W, Kirchhof B, Mitsiades N, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP (2002) Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol 160:501–509
Kaempf S, Walter P, Salz AK, Thumann G (2008) Novel organotypic culture model of adult mammalian neurosensory retina in co-culture with retinal pigment epithelium. J Neurosci Methods 173:47–58
Kanie S, Otsuka A, Yoshikawa S, Morimoto T, Hareyama N, Okazaki S, Kobayashi R, Hasebe K, Nakao K, Hayashi R, Mochizuki H, Matsumoto R, Ozono S (2012) Pharmacological effect of TRK-380, a novel selective human β3-adrenoceptor agonist, on mammalian detrusor strips. Urology 79:744, e1-7
Kaur C, Foulds WS, Ling EA (2008) Blood-retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog Retin Eye Res 27:622–647
Kaur C, Sivakumar V, Foulds WS, Luu CD, Ling EA (2009) Cellular and vascular changes in the retina of neonatal rats after an acute exposure to hypoxia. Invest Ophthalmol Vis Sci 50:5364–5374
Kubota Y, Nakahara T, Yunoki M, Mitani A, Maruko T, Sakamoto K, Ishii K (2002) Inhibitory mechanism of BRL37344 on muscarinic receptor-mediated contractions of the rat urinary bladder smooth muscle. Naunyn Schmiedebergs Arch Pharmacol 366:198–203
Leblais V, Pourageaud F, Ivorra MD, Marthan R, Muller B (2005) Comparison of the alpha-adrenoceptor-mediated effects of beta3-adrenoceptor ligands in rat pulmonary artery. Naunyn Schmiedebergs Arch Pharmacol 371:535–539
Li SY, Fu ZJ, Lo ACY (2012) Hypoxia-induced oxidative stress in ischemic retinopathy. Oxid Med Cell Longev 2012:426769
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408
Martini D, Dal Monte M, Ristori C, Cupisti E, Mei S, Fiorini P, Filippi L, Bagnoli P (2011) Antiangiogenic effects of β2 -adrenergic receptor blockade in a mouse model of oxygen-induced retinopathy. J Neurochem 119:1317–1329
Massion PB, Dessy C, Desjardins F, Pelat M, Havaux X, Belge C, Moulin P, Guiot Y, Feron O, Janssens S, Balligand JL (2004) Cardiomyocyte-restricted overexpression of endothelial nitric oxide synthase (NOS3) attenuates beta-adrenergic stimulation and reinforces vagal inhibition of cardiac contraction. Circulation 110:2666–2672
Mazumder B, Sampath P, Seshadri V, Maitra RK, DiCorleto PE, Fox PL (2003) Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell 115:187–198
Meda C, Plank C, Mykhaylyk O, Schmidt K, Mayer B (2010) Effects of statins on nitric oxide/cGMP signaling in human umbilical vein endothelial cells. Pharmacol Rep 62:100–112
Mei S, Cammalleri M, Azara D, Casini G, Bagnoli P, Dal Monte M (2012) Mechanisms underlying somatostatin receptor 2 down-regulation of vascular endothelial growth factor expression in response to hypoxia in mouse retinal explants. J Pathol 226:519–533
Mori A, Miwa T, Sakamoto K, Nakahara T, Ishii K (2010) Pharmacological evidence for the presence of functional beta(3)-adrenoceptors in rat retinal blood vessels. Naunyn Schmiedebergs Arch Pharmacol 382:119–126
Nakagawa T, Sato W, Sautin YY, Glushakova O, Croker B, Atkinson MA, Tisher CC, Johnson RJ (2006) Uncoupling of vascular endothelial growth factor with nitric oxide as a mechanism for diabetic vasculopathy. J Am Soc Nephrol 17:736–745
Palea S, Rekik M, Rouget C, Camparo P, Botto H, Rischmann P, Lluel P, Westfall TD (2012) Fenoterol functionally activates the β3-adrenoceptor in human urinary bladder, comparison with rat and mouse: implications for drug discovery. Eur J Pharmacol 690:202–206
Pérez-Sayáns M, Somoza-Martín JM, Barros-Angueira F, Diz PG, Gándara Rey JM, García-García A (2010) Beta-adrenergic receptors in cancer: therapeutic implications. Oncol Res 19:45–54
Perrone MG, Scilimati A (2011) β(3)-Adrenoceptor ligand development history through patent review. Expert Opin Ther Pat 21:505–536
Pott C, Brixius K, Bundkirchen A, Bölck B, Bloch W, Steinritz D, Mehlhorn U, Schwinger RH (2003) The preferential beta3-adrenoceptor agonist BRL 37344 increases force via beta1-/beta2-adrenoceptors and induces endothelial nitric oxide synthase via beta3-adrenoceptors in human atrial myocardium. Br J Pharmacol 138:521–529
Ristori C, Filippi L, Dal Monte M, Martini D, Cammalleri M, Fortunato P, la Marca G, Fiorini P, Bagnoli P (2011) Role of the adrenergic system in a mouse model of oxygen-induced retinopathy: antiangiogenic effects of beta-adrenoreceptor blockade. Invest Ophthalmol Vis Sci 52:155–170
Sapieha P, Hamel D, Shao Z, Rivera JC, Zaniolo K, Joyal JS, Chemtob S (2010) Proliferative retinopathies: angiogenesis that blinds. Int J Biochem Cell Biol 42:5–12
Steinle JJ, Booz GW, Meininger CJ, Day JN, Granger HJ (2003) Beta 3-adrenergic receptors regulate retinal endothelial cell migration and proliferation. J Biol Chem 278:20681–20686
Steinle JJ, Zamora DO, Rosenbaum JT, Granger HJ (2005) Beta 3-adrenergic receptors mediate choroidal endothelial cell invasion, proliferation, and cell elongation. Exp Eye Res 80:83–91
Storch CH, Hoeger PH (2010) Propranolol for infantile haemangiomas: insights into the molecular mechanisms of action. Br J Dermatol 163:269–274
Tremolada G, Del Turco C, Lattanzio R, Maestroni S, Maestroni A, Bandello F, Zerbini G (2012) The role of angiogenesis in the development of proliferative diabetic retinopathy: impact of intravitreal anti-VEGF treatment. Exp Diabetes Res 2012:728325
Trochu JN, Leblais V, Rautureau Y, Bévérelli F, Le Marec H, Berdeaux A, Gauthier C (1999) Beta 3-adrenoceptor stimulation induces vasorelaxation mediated essentially by endothelium-derived nitric oxide in rat thoracic aorta. Br J Pharmacol 128:69–76
Tugues S, Koch S, Gualandi L, Li X, Claesson-Welsh L (2011) Vascular endothelial growth factors and receptors: anti-angiogenic therapy in the treatment of cancer. Mol Aspects Med 32:88–111
van der Wijk T, Blanchetot C, den Hertog J (2005) Regulation of receptor protein-tyrosine phosphatase dimerization. Methods 35:73–79
Vrydag W, Michel MC (2007) Tools to study beta3-adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol 374:385–398
West MB, Hill BG, Xuan YT, Bhatnagar A (2006) Protein glutathiolation by nitric oxide: an intracellular mechanism regulating redox protein modification. FASEB J 20:1715–1717
Wilkinson-Berka JL (2004) Vasoactive factors and diabetic retinopathy: vascular endothelial growth factor, cycoloxygenase-2 and nitric oxide. Curr Pharm Des 10:3331–3348
Wuest M, Eichhorn B, Grimm MO, Wirth MP, Ravens U, Kaumann AJ (2009) Catecholamines relax detrusor through beta 2-adrenoceptors in mouse and beta 3-adrenoceptors in man. J Pharmacol Exp Ther 328:213–222
Zhang Q, Zhang J, Guan Y, Zhang S, Zhu C, Xu GT, Wang L (2009) Suppression of retinal neovascularization by the iNOS inhibitor aminoguanidine in mice of oxygen-induced retinopathy. Graefes Arch Clin Exp Ophthalmol 247:919–927
Ziche M, Morbidelli L (2009) Molecular regulation of tumour angiogenesis by nitric oxide. Eur Cytokine Netw 20:164–170
Acknowledgments
This work was supported by the Meyer Foundation, “A. Meyer” University Children’s Hospital (PB) and the Ente Cassa di Risparmio of Florence (LF). The authors thank Prof. Giovanni Casini for critical reading of the manuscript and Dr. Angelo Gazzano and Gino Bertolini for assistance with the mouse colonies.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
β3-AR mRNA and protein after siRNA transfection in hypoxic retinal explants (PDF 764 kb)
Rights and permissions
About this article
Cite this article
Dal Monte, M., Filippi, L. & Bagnoli, P. Beta3-adrenergic receptors modulate vascular endothelial growth factor release in response to hypoxia through the nitric oxide pathway in mouse retinal explants. Naunyn-Schmiedeberg's Arch Pharmacol 386, 269–278 (2013). https://doi.org/10.1007/s00210-012-0828-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-012-0828-x