
Abstract
Although genome-wide association studies (GWASs) have identified over 100 colorectal cancer (CRC) risk loci, an understanding of causal genes or risk variants and their biological functions in these loci remain unclear. Recently, genomic loci 10q26.12 with lead SNP rs1665650 was identified as an essential CRC risk loci of Asian populations. However, the functional mechanism of this region has not been fully clarified. Here, we applied an RNA interfering-based on-chip approach to screen for the genes essential for cell proliferation in the CRC risk loci 10q26.12. Notably, HSPA12A had the most significant effect among the identified genes and functioned as a crucial oncogene facilitating cell proliferation. Moreover, we conducted an integrative fine-mapping analysis to identify putative casual variants and further explored their association with CRC risk in a large-scale Chinese population consisting of 4054 cases and 4054 controls and also independently validated in 5208 cases and 20,832 controls from the UK biobank cohort. We identified a risk SNP rs7093835 in the intron of HSPA12A that was significantly associated with an increased risk of CRC (OR 1.23, 95% CI 1.08–1.41, P = 1.92 × 10–3). Mechanistically, the risk variant could facilitate an enhancer–promoter interaction mediated by the transcriptional factor (TF) GRHL1 and ultimately upregulate HSPA12A expression, which provides functional evidence to support our population findings. Collectively, our study reveals the important role of HSPA12A in CRC development and illustrates a novel enhancer–promoter interaction module between HSPA12A and its regulatory elements rs7093835, providing new insights into the etiology of CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer is the third most common cancer with the second highest mortality rate worldwide (Sung et al. 2021). The burden of colorectal cancer incidence and mortality is also rapidly rising in China with the continuous improvement of living conditions and diagnosis techniques (Chen et al. 2016). With the ascending tendency of CRC, it has grown up an urgent need to investigate the pathogenesis for cancer prevention and diagnosis. Convincing evidence is noted that CRC carcinogenesis and progression are affected by heredity and environment, in which genetic factors play an indispensable role (Gong et al. 2018; Song et al. 2015).
Genome-wide association studies have become an effective research strategy to reveal genetic susceptibility factors for human cancers and successfully identified over 100 CRC risk loci so far (Huyghe et al. 2019; Lu et al. 2019; Zhang et al. 2014). Nevertheless, most of GWAS-identified single-nucleotide polymorphisms (SNPs) are located in non-coding regions and at large distances from nearby annotated genes (Kopp and Mendell 2018). It remains a huge challenge to explore the potential functional mechanism of genetic variants and their target genes on cancer development, which are critical for translating GWAS findings into clinical application (Gong et al. 2016; Wang et al. 2013). Therefore, first, identifying the causal genes in these loci, which are different from traditional fine-mapping analysis, appears to be a feasible way to elucidate targets for prevention and therapy. Extensive research has shown that functional genomic screen using high-throughput RNA interference (RNAi) is regarded as an effective method for the recognition of massive key cancer-related genes (Fulco et al. 2016; McDonald et al. 2017; Meyers et al. 2017; Zhao et al. 2017). This approach may also be a helpful tool for systematic identification of causal genes in GWAS loci.
In light of our previous comprehensive researches focusing on the CRC risk locus 12q13.12, we elucidated the mechanism of ATF1 and its regulatory elements dbSNP: rs61926301 and dbSNP: rs7959129 in the development of CRC (Tian et al. 2019a). However, there are also many genomic loci identified by GWASs whose exact functional mechanisms have not been fully elucidated. Previous studies pointed out that genomic region 10q26.12 with lead SNP rs1665650 was significantly relevant to CRC risk in east Asian population, but little is known about the underlying functional mechanism for this signal (Jia et al. 2013). In this study, we integrated an RNA interfering-based on-chip approach, a large-scale population study and multiple biological experiments to reveal the potential role of genes located in 10q26.12 loci during CRC progression and investigate the underlying regulatory mechanisms. Our results indicated that HSPA12A was verified to be an important oncogene in the CRC risk loci 10q26.12 promoting CRC cell proliferation. We also found that a risk SNP rs7093835, resided in the intron of HSPA12A, facilitated an enhancer–promoter interaction mediated by GRHL1 and upregulated HSPA12A expression, thus significantly predisposing to CRC risk (OR 1.23, 95%CI 1.08–1.41, P = 1.92 × 10–3). These findings might deepen our understanding of the CRC etiology and provide a new insight into precision prevention for CRC.
Materials and methods
A functional genomic screen with an RNA interfering-based on-chip assay in 10q26.12 GWAS loci
We focused on the 10q26.12 loci which is significantly associated with CRC risk in the Asian population. To select candidate genes in this region for functional screening, we performed fine mapping by extending 1 Mb upstream and downstream of the tag SNP rs1665650. In total, we select 11 protein-coding genes in this loci for a proliferation measurement of CRC cells by a large-scale RNAi interrogation, excluding microRNAs, non-coding RNAs, and pseudogenes. The screening threshold value is P < 0.05 and an n-fold change > 1.1 or < 0.9. Details can be viewed in our previous study (Tian et al. 2019a).
Integrative expression quantitative trait locus (eQTL) analysis and genotype imputation
SNPs in LD (r2 > 0.2, MAF > 0.05) with tag SNP rs1665650 were downloaded from the HaploReg database. HSPA12A mRNA expression and individual genotypes were downloaded from the TCGA (The Cancer Genome Atlas) database. To improve the ability of eQTL analysis, we imputed the variants for all CRC samples from TCGA with IMPUTE2, using 1000 Genomes Phase 3 as the reference panel (Ardlie et al. 2015; Howie et al. 2009). Then, integrative eQTL analysis evaluating the association between these SNPs and HSPA12A mRNA expression was conducted by adjusting for the effect of population structures (principal components) and clinical parameters (age, sex, and tumor stage) on gene expression. The details of principal components analysis and the imputation can be viewed in our previous study (Tian et al. 2019b).
Next, we performed a functional annotation for eQTLs with multiple bioinformatic tools, including HaploReg, RegulomeDB, and CistromeDB, and this annotation integrated multiple histone modification ChIP-seq peaks, TF ChIP-seq peaks, and DNase hypersensitive site data. Finally, we selected functional variants with the highest potential in the LD block (r2 ≥ 0.8) for further population and experimental validation.
Cell lines and culture
HCT116 and SW480 cells were obtained from the China Center for Type Culture Collection (Wuhan, China). Cells were cultured in Dulbecco's modified eagle's medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% antibiotics (100 U/mL penicillin and 0.1 mg/mL streptomycin) at 37 °C in a humidified atmosphere containing 5% CO2. Both cell lines used in this study were authenticated by short tandem repeat (STR) profiling (Applied Biosystems, USA) and tested for the absence of mycoplasma contamination (MycoAlert, USA); the latest date of test was October 1, 2021.
Construction of plasmids and RNA interference
A total of 1000-bp DNA fragments containing SNP rs7093835 C or T allele were commercially synthesized and subcloned into the pGL3-promoter vector (Promega, USA). The full-length cDNA of HSPA12A was subcloned into the pcDNA3.1 (+) vector (Invitrogen). All plasmids were commercially synthesized by Genewiz Biological Technology (Shuzhou, China).
For RNA interference, the siRNA oligonucleotides targeting GRHL1 and the non-targeting siRNA control were purchased from RiboBio (Guangzhou, China) and transfected using Lipofectamine RNAiMAX (Invitrogen, USA). The siRNA sequences are shown in Table S1 and the knockdown effect was determined by qRT-PCR (Fig. 4H).
Quantitative reverse transcription PCR (qRT-PCR)
Total RNA of cells and patients' tissues were extracted with TRIzol reagent (Thermo Fisher Scientific, USA). Reverse transcription was performed using the SuperScript®III First-Strand Synthesis System (Invitrogen, USA) and quantitative PCR was performed with Power SYBRTM Green PCR Master Mix (Applied Biosystems, USA). Target gene expression was normalized to that of GAPDH. All specific primers used for qPCR are listed in Table S1.
154 pairs of CRC tumor and normal tissues and blood samples were collected from Tongji Hospital of Huazhong University of Science and Technology. The expression levels of two candidate genes, HSPA12A and GRHL1, were measured using qRT-PCR assays in our own CRC discovery set. The P values were calculated by a two-sided Student’s t test. Written informed consent was obtained from each subject and the study was conducted under the approval of participating hospitals.
Dual-luciferase reporter assay
We co-transfected constructed luciferase vector containing either the rs7093835[C] or rs7093835[T] allele and the pRL-SV40 Renilla luciferase plasmid (Promega, USA) into cells. The luciferase reporter assays were performed using the Dual-Luciferase Reporter Kit (Promega, USA) as the manufacturer’s recommendations. The luciferase activities were normalized by the luminescence value of Renilla luciferase to that of firefly luciferase.
Chromatin immunoprecipitation qPCR (ChIP-qPCR)
ChIP assays were performed by ChIP assay kit (Cat# 10086, Millipore, USA) according to the manufacturer’s instructions. Genomic DNA was extracted from the fixed-chromatin cells and sheared by sonication. Next, an antibody against GRHL1 (Novus Biologicals, NBP1-81321) and a nonspecific rabbit IgG ((Santa Cruz, as control) were, respectively, incubated with cross-linked protein/DNA overnight for immunoprecipitation using protein A/G magnetic beads; the purification and collection of DNA fragments using the Dr. GenTLE Precipitation Carrier Kit (Takara, Japan). The purified DNA library was analyzed by qPCR. The primers used in ChIP-qPCR are shown in Table S1.
Cell proliferation and colony-formation assays
In cell proliferation assays, cells were seeded and transfected in 24-well plates (5 × 104 cells per well). The cells were harvested by trypsin digestion after 24 h and then seeded in 96-well plates (2500 cells in 100 μL of cell suspension per well). Cell viability was measured using CCK-8 assays (Dojindo) in four time points (24, 48, 72, and 96 h).
In colony-formation assays, cells were seeded in 6-well cell culture plates (2000 cells per well). After 10 days, the cells were washed with cold PBS twice, fixed with 3.7% formaldehyde, and were stained with crystal violet. The colony number in each well was counted.
SNP genotyping for our own CRC samples
Genomic DNA was extracted from peripheral blood samples using the Relax Gene Blood DNA System Kit (Tiangen, China) according to the protocol. SNPs were genotyped using the TaqMan SNP Genotyping system in both stages. Quality control was implemented as previously described (Tian et al. 2020).
Study subjects in association analyses between the candidate eQTL and CRC risk
To evaluate the association between the candidate eQTL and CRC risk, we first conducted a two-stage case–control study in our own samples. The characteristics of the study subjects are described in Table S2. The first stage contained 1000 CRC patients and 1000 cancer-free controls recruited from the cancer hospital of Chinese Academy of Medical Sciences in Beijing, China. The second stage consisting of 3054 cases and 3054 cancer-free controls were recruited from Tongji Hospital of Huazhong University of Science and Technology (HUST), Wuhan, China. All controls were cancer-free individuals selected from a community nutritional survey in the same region during the same period when patients were recruited. Peripheral blood samples and demographic characteristics, including age, gender, smoking status, and drinking status, were obtained from the medical records and interviews. Written informed consent was obtained from each subject and the study was conducted under the approval of the Chinese Academy of Medical Sciences Cancer Institute and the Institutional Review Board of Tongji hospital of HUST.
Furthermore, we also screened participants in the UK Biobank (UKBB) cohort to strengthen the finding in our own samples (Bycroft et al. 2018). CRC cases were defined as subjects with primary invasive CRC diagnosed (1020–1023), or CRC deaths according to ICD9 (1530–1534, 1536–1541) or ICD10 (C180, C182-C189, C19, C20) codes. For each case, we selected 4 eligible controls from subjects without invasive CRC by nearest neighbor matching in R package MatchIt, with enrollment age, race/ethnicity, and sex as matching criteria. Finally, 5,208 CRC cases and 20,832 matched controls were included. Demographic characteristics are demonstrated in Table S3.
Statistical analysis
Differences in demographic characteristics between cases and controls were assessed by two-sided Student's t test or Pearson’s χ2 test. For the association analysis, unconditional multivariate logistic regression was employed to estimate odds ratios (ORs) and 95% confidence intervals (CIs) for the association between the candidate eQTL and the CRC risk, with adjustments for gender, age group, smoking status, and drinking status. Additive genetic model was applied to assess the genetic susceptibility of eQTLs to CRC. For functional assays, figure legends denoted the statistical details of experiments, including the statistical tests used, the numbers of replicates, and the data presentation type in relevant figures. All statistical analyses were performed by R (3.30) or SAS (9.4) and P < 0.05 was considered as statistical significance.
Results
HSPA12A was identified as a potential oncogene in CRC based on an RNA-interference functional genomic screen
As shown in Fig. 1, we systematically integrated an RNAi-based on-chip approach and bioinformatics analysis to screen for vital casual genes in CRC GWAS loci 10q26.12. Among 11 protein-coding genes, HSPA12A was identified to have the most significantly effect on cell proliferation in both HCT116 and SW480 cell lines (Tian et al. 2019a) (Fig. 2A, B). The potential role of HSPA12A in cell proliferation was also verified in the CRC SW948 cell line from the genome-wide CRISPR-Cas9-based loss-of-function screening data (Fig. 2C and Fig. S1). Additionally, we also compared the HSPA12A mRNA expression in tumor and adjacent normal tissues using data from TGCA and GEO datasets (Fig. 2D–F and Fig. S2A). We observed that HSPA12A was significantly overexpressed in tumor tissues than those in normal tissues, in consistent with these results in our own samples (Fig. 2G). Data from the Cancer Cell Line Encyclopedia (CCLE) also indicated that HSPA12A was highly expressed in CRC cell lines, which ranks ahead of 1158 human cancer cell lines (Fig. S2B). Furthermore, we overexpressed HSPA12A in CRC cell lines to investigate the function of HSPA12A on cell malignant phenotypes. The results demonstrated that upregulation of HSPA12A could significantly increase cell proliferation rates and colony-formation abilities of CRC cells, compared with control vector (Fig. 2H–K). Collectively, these findings provide multiple lines of evidence, supporting that HSPA12A might act as a potential oncogene promoting CRC progression.

Flowchart of a comprehensive strategy to identify the causal gene and its regulatory variants in CRC risk loci. First, a high-throughput RNA-interference functional screen was conducted to identify the genes essential for proliferation in the CRC risk loci 10q26.12. Next, by integrating a fine-mapping analysis and large-scale population studies, we systematically screened functional risk variants associated with CRC risk. Furthermore, multipronged biological experiments were conducted to elucidate the critical role of HSPA12A and a novel carcinogenic mechanism between HSPA12A and its regulatory variants in CRC development

Functional genomic screening reveals that HSPA12A is an oncogene in CRC. A, B Functional genomic screening based on high-throughput RNAi interrogation was used to identify genes important for cell proliferation in the CRC risk loci 10q26.12 in HCT116 and SW480 cells. C The data of genome-wide CRISPR/Cas9-based loss-of-function screen showed that HSPA12A was essential for cell growth in the CRC cell line. Negative scores imply cell growth inhibition or death following gene knockout and higher absolute scores indicated an elevated dependency of cell viability on given genes. D–G HSPA12A was overexpressed in tumor tissues than adjacent normal tissues from TCGA (D), Gaedcke (GEO: GSE20842) (E), Gaspar (GEO: GSE9689) (F), and our own CRC tissues (G). Data were presented as the median (minimum to maximum). All P values were calculated by a two-sided Student's t test in TCGA and Gaspar tissues, whereas were calculated by a paired two-sided Student's t test in Gaedcke and our own CRC tissues. H, I The effect of HSPA12A overexpression on the proliferation rates in HCT116 (H) and SW480 (I) cells. Data were shown as the mean ± SEM from three experiments, each with six replicates. All *P < 0.05 and **P < 0.005 values were derived from comparison with controls by a two-sided Student's t test. J, K The effect of HSPA12A overexpression on colony-formation abilities in HCT116 (J) and SW480 cells (K). The results presented colony-formation ability relative to control cells (set to 100%). Data were shown as the mean ± SD from three experiments, each with three replicates. All ****P < 0.00005 values were calculated by a two-sided Student’s t test
Fine mapping identifies a putative functional variants affecting HSPA12A expression
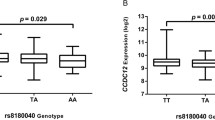
rs1665650 was identified as the tag SNP in 10q26.12 region based on east Asian population and causal variants in this region have not been fully clarified. Therefore, we comprehensively conducted an eQTL analysis between all SNPs in LD (r2 ≥ 0.2) with the tag SNP rs1665650 and HSPA12A mRNA expression. The result indicated that an LD block (r2 ≥ 0.6) showed significant eQTLs with HSPA12A expression (Fig. 3A, B). Subsequently, we performed functional annotation for SNPs in this LD block using Haploreg database, ANNOVAR, CistromeDB, RegulomeDB, and 3DGenome Browser database. Notably, rs7093835 showed the highest potential to be functional in this LD block and was selected as the candidate causal variant. We found that rs7093835, resided in HSPA12A intron, was significantly enriched in active histone modification peaks (H3K4me1 and H3K27ac) and open chromatin accessibility (ATAC-seq peaks and DNase-seq) (Fig. 3C). Moreover, this variant has a statistically significant eQTL with HSPA12A expression level in GTEx transverse colon samples (Fig. 3D), which was consistent with the result from our CRC samples (Fig. 3E).

The variant rs7093835 is identified by integrative eQTL analysis and associated with the expression of HSPA12A. A Regional plots of eQTL results and recombination rates of rs1665650 LD SNPs (r2 ≥ 0.2) and HSPA12A expression. The eQTL P values [− log10 (P value)] of the SNPs (y axis) are presented according to their chromosomal positions (x axis). The genetic recombination rates (cM/Mb) estimated using the 1000 Genomes June 2014 ASN samples are illustrated with a blue line. The SNPs within the interested region are annotated. The r2 values of these SNPs with the tag SNP rs1665650 are represented by different colors. B LD block plot (r2 ≥ 0.6) showing the r2 values of SNPs having a significant eQTL with HSPA12A expression. The r2 values between variants were based on the 1000 Genomes June 2014 ASN samples. The most potentially functional variant in the LD block (r2 ≥ 0.6) is labeled in red, for further population and experimental validation. C Epigenetic annotation for the region surrounding SNP rs7093835 in CRC cell lines. Data including ATAC-seq peaks, DNase peaks, TF (GRHL1) peaks, and multiple histone (H3k4me1 and H3k27ac) modification peaks were obtained from the ENCODE database. D eQTL results of rs7093835 genotypes with HSPA12A expression in the GTEx database. Data were shown as the mean ± SD. ***P value < 0.0005 was calculated by linear regression analysis. E eQTL results of rs7093835 genotypes with HSPA12A expression from our CRC patients. Data were presented as the median (minimum to maximum), and ***P value < 0.0005 was calculated by linear regression analysis. F, G Relative reporter gene activity of the constructs containing the SNP rs7093835[T]/[C] allele in CRC HCT116 and SW480 cell lines. All ****P < 0.00005 values were calculated by a two-sided Student’s t test
To further determine the effect of rs7093835 on target gene expression, we performed dual-luciferase reporter assays in two CRC cell lines and found that the construct containing the rs7093835[T] allele exhibited higher enhancer activity than that containing the rs7093835[C] allele (Fig. 3F, G). Overall, these results demonstrated that rs7093835 might exert an allele-specific enhancer activity, thus affecting HSPA12A expression.
Effect of rs7093835 on transcriptional activity was mediated by GRHL1
Having demonstrated that rs7093835 was capable of influencing HSPA12A mRNA expression level, we next sought to elucidate the underlying regulatory mechanisms. Considering that the allele-specific activity of SNPs in regulatory regions might be due to the different binding affinity of TFs, we first conducted TF motif analysis using multiple databases, including Cistrome and JASPAR, and identified GRHL1 as a candidate factor that might specifically bind to the rs7093835[T] allele (Fig. 4A). Furthermore, ChIP-seq data from the ENCODE database also provided supporting evidence that GRHL1 maps within the region surrounding SNP rs7093835 in CRC LoVo cells (Fig. 3C). GRHL1, as a member of Grainyhead-like family of transcription factors, plays an essential role in cancer development (Frisch et al. 2017). GRHL1 was significantly overexpressed in tumor tissues compared to their normal tissues from multiple datasets, such as TCGA, Hong, and Skrzypczak (Fig. 4B–D), which was in line with the result in our own samples (Fig. 4E). Moreover, positive correlations between the expression of GRHL1 and HSPA12A expression were observed in TCGA CRC samples (Fig. 4F), GTEx colon tissues (Fig. S3A), and our own CRC cohort (Fig. 4G and Fig. S3B). Intriguingly, higher correlations were observed in carriers with the rs7093835[T] allele (Fig. S3), indicating that the regulatory effect of GRHL1 on HSPA12A expression might occur in an allele-specific manner.

Effect of rs7093835 on transcriptional activity was mediated by GRHL1. A The rs7093835[T] allele resides within a GRHL1-binding motif. B–E GRHL1 is significantly overexpressed in tumor tissues compared with normal tissues from multiple independent database, including TCGA (B), Hong (GEO: GSE9348) (C), Skrzypczak (GSE20916) (D), and our own CRC tissues (E). Data were shown as the median (minimum to maximum). All P values were calculated by a two-sided Student’s t test in TCGA, Hong, and Skrzypczak tissues, whereas they were calculated by a paired two-sided Student’s t test in our own CRC tissues. F, G The correlations between GRHL1 expression with HSPA12A expression were measured in TCGA CRC tissues (F) and our own CRC patients (G). All P values and r values were calculated by Pearson’s correlation analyses. H Relative expression levels of GRHL1 in HCT116 and SW480 cell lines transfected with siRNA. Data shown were representative of three independent experiments, each with three technical replicates. Data were presented as the mean ± SD and ****P < 0.00005 values were calculated by a two-sided Student’s t test. I, J Effect of GRHL1 knockdown on the luciferase activity of combined constructs containing rs7093835 in HCT116 (I) and SW480 (J) cells. All experiments were performed in triplicate and each with three technical replicates. Data were shown as the mean ± SD and **P < 0.005 and ****P < 0.00005 were calculated by a two-sided Student’s t test. (K) ChIP-qPCR results show that GRHL1 binds rs7093835[T] allele in allele-specific manner in CRC cells carrying different rs7093835 genotypes (HCT116[CT] and SW480[CC]). Data were presented as the median (minimum to maximum) from three repeated experiments, each with three replicates. ****P < 0.00005 were calculated by a two-sided Student’s t test
To experimentally validate whether the effect of rs7093835 C > T was mediated by the predicted TF GRHL1, we performed reporter gene assays with the knockdown of GRHL1 by siRNAs in HCT116 and SW480 cell lines. The knockdown efficiencies in the two cells lines after 36 h reached approximately 73% (Fig. 4H). As the GRHL1 expression was knocked down by specific siRNAs, the luciferase activity of plasmids containing rs7093835[T] allele reduced to the similar level of the C allele in two cell lines (Fig. 4I, J). Additionally, we further validated the binding of GRHL1 to the region surrounding SNP rs7093835 by ChIP-qPCR assays in cell lines with different rs7093835 genotypes (SW480[CC] and HCT116[CT]). As illustrated in Fig. 4K, a stronger GRHL1 binding is enriched in this region and the binding is more statistically significant in HCT116 cells carrying rs7093835[T] allele than in SW480 cells lacking this allele. Intriguingly, these binding signals are significantly attenuated when GRHL1 is knocked down. Collectively, these findings revealed that rs7093835 modulates GRHL1 binding to affect regulatory element activity and then upregulate HSPA12A expression.
The variant rs7093835 is associated with the risk of CRC
To robustly demonstrate the association between the functional variant rs7093835 and CRC risk, we performed a two-stage case–control study, consisting of 4054 cases and 4054 controls in our own CRC samples. The demographic characteristics of the study subjects are detailed in Table S2. As shown in Table 1, rs7093835 conferred a significant genetic predisposition to CRC in both stages after adjusting for gender, age group, smoking status, and drinking status. Moreover, we combined the results from the two stages, and observed that the rs7093835[T] allele was still associated with an increased risk of CRC with an OR of 1.23 (95% CI 1.08–1.41, P = 1.92 × 10–3 in the additive model).
Following this, we further adopted CRC genotype data from the UK biobank cohort, which includes 5208 CRC patients and 20,832 controls to strengthen the findings in our own samples. The demographic characteristics are listed in Table S3. Results are summarized in Table S4 and the candidate eQTL rs7093835 was also validated to be associated with CRC risk in UKBB CRC samples (OR 1.08, 95% CI 1.03–1.13, P = 2.69 × 10–3 in the additive model).
Discussion
Although genome-wide association studies have identified thousands of loci associated with human traits and diseases, the underlying mechanisms of risk variants or genes in GWAS-identified loci have not been fully elucidated (Jia et al. 2013; Wang et al. 2016; Zeng et al. 2016). Here, by utilizing an RNAi functional genomic screen, our study substantiated that HSPA12A, located in GWAS loci 10q26.12, might function as an important oncogene and facilitate CRC cell proliferation. Moreover, we performed an integrative fine-mapping analysis to explore the potential functional variant and further examined its association with CRC risk in a large-scale Chinese population consisting of 4054 cases and 4054 controls and also independently validated in 5208 cases and 20,832 controls using GWAS-chip data from the UKBB cohort. We identified a risk SNP rs7093835 located within an intron of HSPA12A that contributed to an increased risk of CRC (OR 1.23, 95% CI 1.08–1.41, P = 1.92 × 10–3). Mechanistically, the risk variant could facilitate an enhancer–promoter interaction mediated by GRHL1, thus upregulating HSPA12A mRNA expression (Fig. 5).

Graphical representation of the regulation and function of HSPA12A in CRC. Compared with the SNP rs7093835[C] allele, the CRC risk SNP rs7093835[T] allele enhances the binding of transcription factor GRHL1 to the promoter of HSPA12A, facilitating an enhancer–promoter interaction that upregulates HSPA12A expression. Subsequently, the overexpression of HSPA12A promotes CRC cell proliferation, thus leads to an increasing risk of CRC
Heat shock protein A12A (HSPA12A) as a novel member of the HSP70 family is widely involved in multiple cancer development through several different mechanisms, such as regulation of angiogenesis, proliferation, and migration (Wu et al. 2017). Recent evidence indicated that HSPA12A could promote CRC migration and invasion by stabilizing N‐cadherin mRNA and inducing EMT progression (Pan et al. 2022). Based on the published studies discussed above, the role of HSPA12A in CRC was further examined in our study. Similarly, we observed that HSPA12A is overexpressed in CRC tumor tissues compared with normal tissues in multiple independent databases as well as in our own CRC samples and HSPA12A overexpression could substantially promote CRC cell proliferation. Taken together, these findings provide multiple lines of evidence, supporting that HSPA12A might act as a potential oncogene promoting CRC progression.
In view of the critical role of HSPA12A in CRC, greater efforts are needed to investigate the precise regulatory mechanisms of HSPA12A-enhanced tumor activity. Extensive research has shown that the allele-specific binding of transcription factor to the regulatory elements such as enhancers or promoters is a general model of gene expression modulation (Weintraub et al. 2017). Enhancer–promoter interaction, one of major transcriptional regulation mechanisms, has been proven to be vital in cancer proliferation and invasion (Zhang et al. 2013). In this study, by integrating multiple bioinformatical tools and experimental validations, we identified that SNP rs7093835 could facilitate an enhancer–promoter interaction mediated by GRHL1, which ultimately upregulated the HSPA12A expression and thus conferred susceptibility to CRC. This finding was also consistent with the eQTL results, revealing that HSPA12A expression was gradually elevated as the number of risk alleles increased. GRHL1 has been recently characterized as an essential pioneer transcription factor that could access DNA-binding sites within typically inaccessible chromatin to drive the direct target gene transcription, which is closely related with cell growth and differentiation of tumors (Gasperoni et al. 2022; He et al. 2021). Similarly, we found that GRHL1 was overexpressed in tumors compared with normal tissues derived from various databases and predicted to bind to the rs7093835. Moreover, the luciferase activity of plasmids containing rs7093835[T] allele was significantly reduced by the knockdown of GRHL1. The genotype-specific ChIP-qPCR results also showed that the binding of GRHL1 to the region containing rs7093835 is more statistically significant in HCT116 cells carrying rs7093835[T] than in SW480 cells lacking this allele. Taken together, these results elucidated that the risk SNP rs7093835, as an allele-specific enhancer, facilitated enhancer–promoter interactions mediated by GRHL1 to modulate HSPA12A expression, which provided functional evidence to support our population findings.
However, some limitations are still present in our study. First, we only chose the candidate genes in GWAS-identified loci of Asian population, and those genes and genetic variants in other genomic regions may also have potential effects on CRC susceptibility. Second, non-coding RNAs, which have been suggested to play important roles in cancers, are not involved in this study and the extensive work will be carried out in future. Thirdly, more experiments in vitro and in vivo need to be done to clarify the mechanism of the interaction in gene regulation regulated by non-coding variants.
In conclusion, by integrating an RNAi-based functional interrogation, large-scale epidemiological studies, and multipronged biological experiments, we demonstrated that HSPA12A might function as an essential oncogene in GWAS loci 10q26.12 and further provided a regulatory circuit in CRC development by which a functional variant rs7093835 facilitated enhancer–promoter interaction mediated by GRHL1 to promote the expression of HSPA12A, ultimately contributing an increased CRC risk. Overall, our findings not only deepen the understanding of the genetic susceptibility of CRC, but also shed light on the biological basis for cancer etiology.
Data availability
All accession codes or web links for publicly available datasets are described in the paper. Data are available from corresponding author on reasonable request.
References
Ardlie KG, Deluca DS, Segrè AV et al (2015) Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science (new York, NY) 348(6235):648–660. https://doi.org/10.1126/science.1262110
Bycroft C, Freeman C, Petkova D et al (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature 562(7726):203–209. https://doi.org/10.1038/s41586-018-0579-z
Chen W, Zheng R, Baade PD et al (2016) Cancer statistics in China, 2015. CA Cancer J Clin 66(2):115–132. https://doi.org/10.3322/caac.21338
Frisch SM, Farris JC, Pifer PM (2017) Roles of grainyhead-like transcription factors in cancer. Oncogene 36(44):6067–6073. https://doi.org/10.1038/onc.2017.178
Fulco CP, Munschauer M, Anyoha R et al (2016) Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science (new York, NY) 354(6313):769–773. https://doi.org/10.1126/science.aag2445
Gasperoni JG, Fuller JN, Darido C, Wilanowski T, Dworkin S (2022) Grainyhead-like (Grhl) target genes in development and cancer. Int J Mol Sci 23(5):2735. https://doi.org/10.3390/ijms23052735
Gong J, Tian J, Lou J et al (2016) A functional polymorphism in lnc-LAMC2-1:1 confers risk of colorectal cancer by affecting miRNA binding. Carcinogenesis 37(5):443–451. https://doi.org/10.1093/carcin/bgw024
Gong J, Tian J, Lou J et al (2018) A polymorphic MYC response element in KBTBD11 influences colorectal cancer risk, especially in interaction with an MYC-regulated SNP rs6983267. Ann Oncol 29(3):632–639. https://doi.org/10.1093/annonc/mdx789
He Y, Gan M, Wang Y et al (2021) EGFR-ERK induced activation of GRHL1 promotes cell cycle progression by up-regulating cell cycle related genes in lung cancer. Cell Death Dis 12(5):430. https://doi.org/10.1038/s41419-021-03721-9
Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5(6):e1000529. https://doi.org/10.1371/journal.pgen.1000529
Huyghe JR, Bien SA, Harrison TA et al (2019) Discovery of common and rare genetic risk variants for colorectal cancer. Nat Genet 51(1):76–87. https://doi.org/10.1038/s41588-018-0286-6
Jia WH, Zhang B, Matsuo K et al (2013) Genome-wide association analyses in East Asians identify new susceptibility loci for colorectal cancer. Nat Genet 45(2):191–196. https://doi.org/10.1038/ng.2505
Kopp F, Mendell JT (2018) Functional classification and experimental dissection of long noncoding RNAs. Cell 172(3):393–407. https://doi.org/10.1016/j.cell.2018.01.011
Lu Y, Kweon SS, Tanikawa C et al (2019) Large-scale genome-wide association study of east Asians identifies loci associated with risk for colorectal cancer. Gastroenterology 156(5):1455–1466. https://doi.org/10.1053/j.gastro.2018.11.066
McDonald ER 3rd, de Weck A, Schlabach MR et al (2017) Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale. Deep RNAi Screen Cell 170(3):577-592.e10. https://doi.org/10.1016/j.cell.2017.07.005
Meyers RM, Bryan JG, McFarland JM et al (2017) Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49(12):1779–1784. https://doi.org/10.1038/ng.3984
Pan YJ, Huo FC, Kang MJ, Liu BW, Wu MD, Pei DS (2022) Alternative splicing of HSPA12A pre-RNA by SRSF11 contributes to metastasis potential of colorectal cancer. Clin Transl Med 12(11):e1113. https://doi.org/10.1002/ctm2.1113
Song M, Garrett WS, Chan AT (2015) Nutrients, foods and colorectal cancer prevention. Gastroenterology 148(6):1244–60.e16. https://doi.org/10.1053/j.gastro.2014.12.035
Sung H, Ferlay J, Siegel RL et al (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249. https://doi.org/10.3322/caac.21660
Tian J, Chang J, Gong J et al (2019a) Systematic functional interrogation of genes in GWAS loci identified ATF1 as a key driver in colorectal cancer modulated by a promoter-enhancer interaction. Am J Hum Genet 105(1):29–47. https://doi.org/10.1016/j.ajhg.2019.05.004
Tian J, Wang Z, Mei S et al (2019b) CancerSplicingQTL: a database for genome-wide identification of splicing QTLs in human cancer. Nucleic Acids Res 47(D1):D909-d916. https://doi.org/10.1093/nar/gky954
Tian J, Lou J, Cai Y et al (2020) Risk SNP-mediated enhancer–promoter interaction drives colorectal cancer through both FADS2 and AP002754.2. Cancer Res 80(9):1804–1818. https://doi.org/10.1158/0008-5472.CAN-19-2389
Wang H, Haiman CA, Burnett T et al (2013) Fine-mapping of genome-wide association study-identified risk loci for colorectal cancer in African Americans. Hum Mol Genet 22(24):5048–5055. https://doi.org/10.1093/hmg/ddt337
Wang M, Gu D, Du M et al (2016) Common genetic variation in ETV6 is associated with colorectal cancer susceptibility. Nat Commun 7:11478. https://doi.org/10.1038/ncomms11478
Weintraub AS, Li CH, Zamudio AV et al (2017) YY1 is a structural regulator of enhancer–promoter loops. Cell 171(7):1573-1588.e28. https://doi.org/10.1016/j.cell.2017.11.008
Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S (2017) Heat shock proteins and cancer. Trends Pharmacol Sci 38(3):226–256. https://doi.org/10.1016/j.tips.2016.11.009
Zeng C, Matsuda K, Jia WH et al (2016) Identification of susceptibility loci and genes for colorectal cancer risk. Gastroenterology 150(7):1633–1645. https://doi.org/10.1053/j.gastro.2016.02.076
Zhang Y, Wong CH, Birnbaum RY et al (2013) Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature 504(7479):306–310. https://doi.org/10.1038/nature12716
Zhang B, Jia WH, Matsuda K et al (2014) Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet 46(6):533–542. https://doi.org/10.1038/ng.2985
Zhao Y, Wei L, Shao M et al (2017) BRCA1-associated protein increases invasiveness of esophageal squamous cell carcinoma. Gastroenterology 153(5):1304-1319.e5. https://doi.org/10.1053/j.gastro.2017.07.042
Acknowledgements
We are grateful to every patient, their families, and referring physicians that have participated in these studies, and the numerous members of our laboratories for their valuable contributions over many years.
Funding
This work was supported by the Program of National Science Fund for Distinguished Young Scholars of China (NSFC-81925032), Key Program of National Natural Science Foundation of China (NSFC-82130098), and Natural Science Foundation of Hubei Province (2019CFA009) for Xiaoping Miao; National Natural Science Foundation of China (NSFC-82103929, NSFC-82273713), Fundamental Research Funds for the Central Universities (WHU:2042022kf1205), Knowledge Innovation Program of Wuhan (whkxjsj011), and Translational Medicine and Interdisciplinary Research Joint Fund of Zhongnan Hospital of Wuhan University (ZNJC202207) for Jianbo Tian; Youth Program of National Natural Science Foundation of China (NSFC-82003547) and Fundamental Research Funds for the Central Universities (WHU: 2042022kf1031) for Ying Zhu.
Author information
Authors and Affiliations
Contributions
JT, BL, and XM were the overall principal investigators in this study who conceived the study and obtained financial support, YZ, HL, JT, BL, and XM were responsible for the study design and supervised the entire study. BL, ZL, and FZ performed statistical analyses, interpreted the results, and drafted the initial manuscript. BL, LF, YC, CC, GL, MZ, JH, CN, YL, WW, HG, YL, SC, HL, SY, HZ, and WT performed laboratory experiments, TY, JL, CH, XY, BX, YZ, RZ, HL, JT, BL, and XM were responsible for patient recruitment and sample preparation. All authors approved the final report for publication.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lu, Z., Fan, L., Zhang, F. et al. HSPA12A was identified as a key driver in colorectal cancer GWAS loci 10q26.12 and modulated by an enhancer–promoter interaction. Arch Toxicol 97, 2015–2028 (2023). https://doi.org/10.1007/s00204-023-03494-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-023-03494-4