
Abstract
Sesquiterpene lactone helenalin is used as an antiphlogistic in European and Chinese folk medicine. The pharmacological activities of helenalin have been extensively investigated, yet insufficient information exists about its metabolic properties. The objectives of the present study were (1) to investigate the in vitro NADPH-dependent metabolism of helenalin (5 and 100 µM) using human and rat liver microsomes and liver cytosol, (2) to elucidate the role of human cytochrome P450 (CYP) enzymes in its oxidative metabolism, and (3) to study the inhibition of human CYPs by helenalin. Five oxidative metabolites were detected in NADPH-dependent human and rat liver microsomal incubations, while two reduced metabolites were detected only in NADPH-dependent human microsomal and cytosolic incubations. In human liver microsomes, the main oxidative metabolite was 14-hydroxyhelenalin, and in rat liver microsomes 9-hydroxyhelenalin. The overall oxidation of helenalin was several times more efficient in rat than in human liver microsomes. In humans, CYP3A4 and CYP3A5 followed by CYP2B6 were the main enzymes responsible for the hepatic metabolism of helenalin. The extrahepatic CYP2A13 oxidized helenalin most efficiently among CYP enzymes, possessing the Km value of 0.6 µM. Helenalin inhibited CYP3A4 (IC50 = 18.7 µM) and CYP3A5 (IC50 = 62.6 µM), and acted as a mechanism-based inhibitor of CYP2A13 (IC50 = 1.1 µM, KI = 6.7 µM, and kinact = 0.58 ln(%)/min). It may be concluded that the metabolism of helenalin differs between rats and humans, in the latter its oxidation is catalyzed by hepatic CYP2B6, CYP3A4, CYP3A5, and CYP3A7, and extrahepatic CYP2A13.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Helenalin is a naturally occurring sesquiterpene lactone of the pseudoguaianolide type found in multiple plants in the Asteracea (Compositae) family, namely Arnica montana L., Arnica chamissonis Less. ssp. foliosa (Nutt.) Maguire, and Centipeda minima (L.) A. Braun and Asch. (Douglas et al. 2004; Leven and Willuhn 1987; Staneva et al. 2011; Todorova et al. 2016; Wu et al. 2012). Phytopharmaceuticals containing helenalin have a long record of use as topical antiphlogistics and analgesics. Accordingly, the European Medicines Agency’s monograph on A. montana approves the use of tinctures and ethanolic extracts prepared from the flowers of A. montana for the treatment of bruises, sprains and localized muscular pain (European Medicines Agency 2014). Preparations made from C. minima, used particularly in traditional Chinese medicine, are applied nasally and orally in the treatment of multiple inflammatory conditions, nasal allergies, nasopharyngeal carcinoma, cough, and headache (Hempen and Fischer 2009; Wu et al. 2012; Zhang et al. 2018).
The molecule of helenalin contains two reactive electrophilic α,β-unsaturated carbonyl moieties, namely α-methylene-γ-butyrolactone and cyclopentenone, which react with the sulfhydryl groups of cell nucleophiles (mainly cysteine thiol groups) via the Michael addition yielding covalent adducts (Buchele et al. 2010; Hall et al. 1977; Schmidt 1997). The presence of the two so-called Michael acceptors conditions to a certain extent the pharmacological activity of helenalin as the removal of either one or both moieties by means of reduction leads to a significant decrease or even complete loss of activity (Hall et al. 1980, 1979; Lee 1973; Lee and Furukawa 1972). Helenalin is a broad-spectrum active compound, featuring in particular anti-inflammatory, antitumor, and antiprotozoal activity (Drogosz and Janecka 2019). In vitro, it acts as a selective inhibitor of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (Lyss et al. 1997) binding directly to Cys38 within the DNA binding domain of NF-κB subunit p65/RelA (Garcia-Pineres et al. 2001; Lyss et al. 1998). In addition, helenalin reduces the level of p65/RelA and induces autophagy cell death (Lim et al. 2012). Helenalin initiates the intrinsic apoptotic pathway by generating reactive oxygen species (ROS) (Berges et al. 2009; Hoffmann et al. 2011; Jang et al. 2013). However, its ability to overcome the protection conferred by the overexpression of anti-apoptotic B-cell lymphoma 2 (Bcl-2) and B-cell lymphoma-extra large (Bcl-xL) mitochondrial proteins suggests a more complex mechanism of action (Dirsch et al. 2001; Hoffmann et al. 2011; Jang et al. 2013). Helenalin has also been found to selectively inhibit the activity of human telomerase (Huang et al. 2005). Moreover, the in vivo hepatoprotective activity of helenalin against liver injury and hepatofibrosis has been recently reported in rats (Lin et al. 2014) and mice (Li et al. 2019).
Although the pharmacological activities of helenalin have been extensively investigated (Drogosz and Janecka 2019; Lin et al. 2014; Li et al. 2019), insufficient information exists regarding its metabolism and pharmacokinetic behavior (Kriplani and Guarve 2020). Helenalin, quantified as helenalin isobutyrate, was found to penetrate in and permeate through the stratum corneum of porcine skin. This penetration was more efficient when different Arnica preparations were used compared to that of the isolated compound dissolved in an ethanol/water (7:3) mixture (Wagner et al. 2004b). Furthermore, the penetration rate seemed to be independent of the content of sesquiterpene lactones, but rather was dependent on the type of formulation (Wagner and Merfort 2007). In a study by Bergamante et al. (2007), helenalin was also found to permeate through porcine skin (1 mm in thickness) placed in a Franz diffusion cell. In addition, the permeation profiles were dependent on the type of formulation with microemulsions being more effective than gel formulations. The binding of helenalin to plasma proteins was investigated by Wagner et al. (2004a). Helenalin isobutyrate was observed to bind to human serum albumin as well as to proteins in human blood plasma and whole blood. This binding did not occur specifically via Cys34 of human serum albumin. Rather, a reaction with other amino acids such as lysine as well as non-covalent interactions with plasma proteins were deemed to contribute to the binding.
Previous in vivo and in vitro studies have reported an inhibitory effect of helenalin on drug-metabolizing enzymes, particularly CYP. Helenalin decreased the hepatic microsomal CYP content and inhibited the activities of aminopyrine demethylase and aniline hydroxylase in mice in vivo and in vitro (Chapman et al. 1989, 1991, 1988) as well as the activity of 7-ethoxyresorufin deethylase in mice in vitro (Chapman et al. 1989). The decrease in the activity of aminopyrine demethylase as well as CYP content was higher in the presence of NADPH (Chapman et al. 1989, 1991). Moreover, exposure to helenalin (intragastric administration of 2.5 mg/kg) decreased CYP content and the activities of NADPH-CYP reductase, aniline hydroxylase, aminopyrine demethylase, and glutathione S-transferase in rat liver (Jodynis-Liebert et al. 2000).
Information concerning the metabolic properties of helenalin in human as well as the role of individual human CYPs involved in its metabolism is not yet available. Therefore, the objective of the present study was to compare the NADPH-dependent metabolism of helenalin in human and rat liver subcellular fractions, to characterize the role of individual human CYPs in the metabolism of helenalin, and to investigate the inhibitory potential of helenalin towards human CYPs.
Materials and methods
Chemicals and reagents
Helenalin ((3aR,5R,5aR,8aR,9S,9aS)-9-hydroxy-5,8a-dimethyl-1-methylidene-3a,4,5,5a,9,9a-hexahydroazuleno[6,7-b]furan-2,8-dione) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA; sc-218579, purity ≥ 97%). Stock solution (10 mM) was prepared in dimethyl sulfoxide (DMSO), aliquoted, and kept at − 80 °C. Coumarin, 7-hydroxycoumarin, resorufin, ethoxyresorufin, pentoxyresorufin, tris(hydroxymethyl)aminomethane (Tris)-HCl, MnCl2, isocitric acid, isocitric acid dehydrogenase, uridine diphosphate glucuronic acid (UDPGA), and 3'-phosphoadenosine-5′-phosphosulfate (PAPS) were all purchased from Sigma Aldrich (Steinheim, Germany). Nicotinamide adenine dinucleotide phosphate (NADP+) was bought from Roche Diagnostics (Mannheim, Germany), KCl and MgCl2 from J.T. Baker (Waltham, MA, USA). NADPH regenerating system contained 178.5 mg NADP+, 645 mg isocitric acid, 340 mg KCl, 240 mg MgCl2, 0.32 mg MnCl2, and 15 U isocitric acid dehydrogenase in a final volume of 200 mL. The mixture was stored at − 80 °C in small aliquots. All chemicals were of the highest purity available from their commercial suppliers. Synthesis and purity of coumarin derivatives 3-(3-benzyloxo)phenyl-7-methoxycoumarin (BPMC), 3-(3-hydroxyphenyl)-6-hydroxycoumarin (HPHC), 3-(3-methoxyphenyl)coumarin (MPC), and 3-(4-trifluoromethylphenyl)-6-methylcoumarin (TFPMC) have been described elsewhere in detail (Juvonen et al. 2019a, 2021; Niinivehmas et al. 2018; Rauhamaki et al. 2018). Methanol (Chromasolv™ LC–MS Ultra, tested for UHPLC-MS) was obtained from Honeywell Riedel-de Haën (Vantaa, Finland); formic acid (LC–MS grade) was purchased from Thermo Fisher Scientific.
Biological material
Human liver cytosol used in this study was prepared from the human liver tissue obtained from the University Hospital of Oulu (Oulu, Finland) as a surplus from cadaveric kidney transplantation donors. The procedure was approved by the Ethics Committee of the Medical Faculty of the University of Oulu (January 21, 1986). Rat liver microsomes and cytosol were prepared from Wistar rats (200–300 g) supplied by the National Laboratory Animal Centre, University of Kuopio. Rat liver tissue was obtained from untreated males used as controls in other experiments, which were approved by the Ethics Committee for Animal Experiments, University of Kuopio (Document 01-38, June 1, 2000). Freshly excised liver specimens were immediately transferred on ice, cut into pieces, snap-frozen in liquid nitrogen, and stored at − 80 °C until subcellular fractions were prepared as described elsewhere (Lang et al. 1981). Protein concentration was determined using the Bradford method (Bradford 1976). The samples were stored at − 80 °C until further use. Pooled mixed gender human liver microsomes were purchased from Sekisui XenoTech (Kansas City, KS, USA). Baculovirus-infected insect cell-expressed human CYP1A1, 1A2, 1B1, 2A6, 2A13, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, 3A5, and 3A7 were purchased from BD Biosciences Discovery Labware (Badford, MA, USA).
Metabolism of helenalin
All incubations proceeded in a final volume of 100 µL. The CYP oxidation incubation mixture was composed of 100 mM potassium phosphate buffer pH 7.4, 5 µM or 100 µM helenalin, 0.4–1.44 g/L microsomal protein, and 20% NAPDH regenerating system. Blank samples lacked either helenalin, microsomes or NADPH. The glucuronidation incubation mixture was composed of 100 mM potassium phosphate buffer pH 7.4, 100 µM helenalin, 0.4–1.44 g/L microsomal protein, 5 mM MgCl2, and 0.5 mM UDPGA. Blank samples lacked either helenalin, microsomes or UDPGA. The sulfonation incubation mixture consisted of 100 mM potassium phosphate buffer pH 7.4, 100 µM helenalin, 1–2.5 g/L cytosolic protein, 5 mM MgCl2, and 10 µM PAPS. Blank samples lacked either helenalin, cytosol or PAPS. The metabolism of helenalin was further investigated under experimental conditions that would allow for simultaneous CYP oxidation and glucuronidation or sulfation. The reactions were initiated by adding the reaction-specific cofactor and allowed to incubate for 60 min at 37 °C in a shaking block incubator (Heidolph Titramax 100, Germany). All reactions were stopped by adding 300 µL of acetonitrile to the incubation mixtures. Samples were centrifuged at 10,000 rpm for 10 min (MiniSpin, Eppendorf, Hamburg, Germany) and supernatants stored at − 20 °C until liquid chromatography-tandem mass spectrometry (LC–MS/MS) analysis.
Oxidation of helenalin by human recombinant CYP enzymes
To determine which CYP forms are involved in the oxidation of helenalin, human recombinant CYP1A1, 1A2, 1B1, 2A6, 2A13, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, 3A5, and 3A7 were used for this purpose. Fifty µL of the incubation mixture in 100 mM potassium phosphate buffer (pH 7.4) contained 5 µM or 100 µM helenalin, 20 nM recombinant CYP, and 20% NADPH regenerating system. Blank samples did not contain either helenalin, recombinant CYP or NADPH. The reaction was started by adding NADPH and incubated for 60 min at 37°. The reaction was stopped by adding 150 µL of acetonitrile. Samples were centrifuged at 10,000 rpm for 10 min and supernatants were stored at − 20 °C until LC–MS/MS analysis.
Oxidation kinetics of helenalin
The standard incubation mixture consisted of 100 mM potassium phosphate buffer pH 7.4, 0–100 µM helenalin, 0.5–1 g/L microsomal protein or 20 nM human recombinant CYP, and 20% NADPH regenerating system. The final volume of microsomal or recombinant CYP incubations was 100 µL or 50 µL, respectively. Blank samples lacked either helenalin, microsomes/recombinant CYP, or NADPH. The incubation mixtures were preincubated at 37 °C for 10 min and the reaction was commenced by the addition of NADPH into the mixtures. The microsomal and recombinant CYP incubations proceeded at 37 °C for 30 and 60 min, respectively. During the incubation, less than 10% of helenalin was consumed in the reaction. The reaction was terminated by adding a threefold volume of acetonitrile to the mixtures. Samples were centrifuged at 10,000 rpm for 10 min and supernatants were stored at − 20 °C until LC–MS/MS analysis. The data were analyzed using GraphPad Software (San Diego, CA) and the Michaelis–Menten equation
in which v is the reaction rate at substrate concentration S, Vmax is the maximal rate of the reaction and Km is the Michaelis constant.
LC–MS/MS analysis
The LC–MS/MS analyses were performed using the Vanquish UHPLC system (Thermo Fisher Scientific, Germering, Germany) coupled with a Q Exactive high-resolution mass spectrometer equipped with heated electrospray ionization (HESI) (Thermo Fisher Scientific, Bremen, Germany). Samples were kept at 10 °C prior to the injection. The chromatographic separation was carried out on a Zorbax Eclipse XDB-C18 column (2.1 × 100 mm, 1.8 µm, Agilent Technologies) maintained at 45 °C. The injection volume was 2 µL. Mobile phases, delivered at 0.4 mL/min, consisted of MilliQ water (A) and methanol (B), both containing 0.1% (v/v) formic acid. The following gradient program was used: 2%→100% B (0–10 min), 100% B (10–14.5 min), 100%→2% B (14.5–14.51 min), and 2% B (14.51–16.5 min). The HESI source was operated in positive ionization mode except for the sulfonation assay samples, which were analyzed in negative ionization mode. The MS parameters were as follows: sheath gas flow rate 20, aux gas flow rate 5, aux gas heat temperature 400 °C, spray voltage 3 kV, and capillary temperature 350 °C. A divert valve was used to direct the LC flow to waste from 0 to 1.3 min. Data acquisition was performed using full scan mode across a mass range of 120.0–1200.0 m/z with the resolution of 70,000 followed by dd-MS2 using normalized collision energy of 30% with isolation window 1.5 m/z and resolution of 17,500. Identification of metabolites was based on their exact masses and fragmentation spectra.
Inhibition of human microsomal and recombinant CYP enzymes by helenalin
The inhibition assays were performed according to Juvonen et al. (2019a). The incubations proceeded in black frame/white well 96-well plates (PerkinElmer) in a final volume of 100 µL. The incubation mixture consisted of 100 mM Tris–HCl buffer (pH 7.4), 0.1–0.2 g/L human liver microsomal protein or 2.5–20 nM human recombinant CYP, 0–100 µM helenalin, 20% NADPH regenerating system, and a corresponding CYP substrate. The CYP substrates were as follows: 1.5 µM or 10 µM coumarin, 10 μM coumarin derivatives (BPMC, HPHC, MPC, and TFPMC), 1 µM ethoxyresorufin, or 0.5 µM pentoxyresorufin. Non-inhibited (100%) samples did not contain helenalin and blank samples lacked either microsomes/recombinant CYP or NADPH. Final DMSO concentration in all incubation mixtures was 1%. The incubation mixtures were preincubated for 5 min at 37 °C and the reaction was initiated by adding NADPH to the wells. Fluorescence was measured every second minute for 40 min using a Victor2 plate reader (PerkinElmer Life Sciences, Turku, Finland) maintained at 37 °C. Excitation and emission wavelengths for coumarin and its derivatives were set to 405 nm and 460 nm, respectively, and for ethoxyresorufin and pentoxyresorufin to 570 nm and 615 nm, respectively. The 7-hydroxycoumarin was used as a standard and surrogate standard for the assays with coumarin and coumarin derivatives, respectively, and resorufin was used as a standard for the ethoxyresorufin and pentoxyresorufin assays. To estimate the half-maximal inhibitory concentration (IC50), the oxidation rates (µM/min) and relative remaining activity at different concentrations of helenalin were calculated and the data were fitted to a sigmoidal dose–response curve using nonlinear regression in GraphPad software. The IC50 was calculated using the equation
where vi stands for the rate at a specific concentration of inhibitor, v0 is the rate of uninhibited reaction, and i is the inhibitor concentration.
The effect of preincubation in the presence of NADPH on IC50 of helenalin in CYP2A13, CYP3A4, and CYP3A5 catalyzed reactions was further studied. The incubation mixture consisting of 100 mM Tris–HCl buffer (pH 7.4), 0–100 µM helenalin, 5–10 nM CYP was preincubated with or without NADPH for 30 min at 37 °C. After preincubation, 10 µM CYP substrate (HPHC for CYP2A13, BPMC for CYP3A4/5) and NADPH were added to the corresponding wells, so that all wells contained 20% NADPH and the final volume was 100 µL. The non-inhibited (100%) samples did not contain helenalin, and blank samples lacked either CYP or NADPH. The fluorescence was measured, and the IC50 calculations in preincubated and non-preincubated samples were performed in the same way as described above.
To further investigate the time- and concentration-dependent inhibition of CYP2A13 by helenalin, the following two-step incubation scheme was employed. Firstly, 0–25 µM helenalin was preincubated with 10 nM CYP2A13 and 20% NADPH in the presence of 100 mM Tris–HCl buffer (pH 7.4) in a final volume of 50 µL for 1–10 min. After preincubation, 10 µL of the mixture was pipetted to 190 µL reaction mixture so that the reaction proceeded in the presence of 100 mM Tris–HCl buffer (pH 7.4), 25 µM substrate (HPHC), and 5% NADPH. Blank samples did not contain helenalin or CYP2A13. Fluorescence was measured in the same way as described above. The inactivation rate constant at an infinite concentration of inhibitor (kinact) and the inhibitor concentration required for a half-maximal rate of inactivation (KI) values were approximated using GraphPad software and the following relationship
where kobs stands for observed inactivation rate constant (ln(% of inactivation)/min) at I, which is the inhibitor concentration.
Results
Metabolism of helenalin in vitro
NADPH-dependent metabolism of helenalin was studied at low 5 µM and high 100 µM concentration in vitro in human and rat liver microsomes and liver cytosol. The former represents the concentration, which could occur after non-toxic or low toxicity causing helenalin exposure (Merrill et al. 1988), while the latter represents the concentration, which could occur after toxic helenalin exposure. Using these two helenalin concentrations, the difference in its metabolism at non-saturating and saturating concentration can be observed. The samples were analyzed by UHPLC-Q-Exactive-MS/MS and metabolites were identified based on their accurate m/z values and fragmentation spectra. A total of seven and five NADPH-dependent metabolites of helenalin were detected in human and rat samples, respectively (Table 1, Figs. 1 and 2).

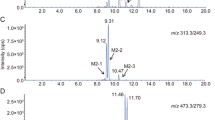
Extracted ion chromatograms of helenalin metabolites (M1-M7) formed via oxidation (m/z 279.1227) or reduction (m/z 265.1434) in human liver microsomes (HLM), rat liver microsomes (RLM), and their corresponding blanks. Liver microsomes were incubated with 100 µM helenalin in the presence of 20% NADPH regenerating system and 100 mM potassium phosphate buffer pH 7.4 for 60 min at 37 °C. Blanks contained only liver microsomes and helenalin, and were devoid of NADPH regenerating system

NADPH-dependent in vitro metabolism of helenalin and proposed molecular structures of its metabolites
Helenalin was eluted at 5.32 min under the experimental conditions and detected as protonated [M + H]+ ion at m/z 263.1277 (C15H19O4). Characteristic product ions, which were observed at m/z 245.1173 [M + H-H2O]+, 227.1065 [M+H–2H2O]+, and 199.1116 [M+H–2H2O–CO]+, resulted from a consecutive loss of H2O and CO. In addition, the product ions with the greatest relative intensities were detected at m/z 123.0807 (C8H11O) and 107.0859 (C8H11). The former was previously assumed to originate from the cleavage of the cycloheptane ring yielding a fragment encompassing the cyclopentenone moiety (Tsai et al. 1969). The formation of the latter is proposed in Fig. S1B. The MS/MS spectrum and the proposed fragmentation pathway of helenalin are shown in Fig. S1.
Oxidative metabolites
Metabolites M1-M5 were formed in the incubations with liver microsomes from both species in the presence of NADPH (Fig. 1). M1-M5 were eluted in a time range from 4.41 to 4.70 min at m/z 279.1223, 279.1227, 279.1226, 279.1232, 279.1229, respectively ([M+H]+, C15H19O5). The molecular weight of M1–M5 was 16 Da mass units higher than that of helenalin (m/z 263.1277 [M+H]+), which indicated that there had been the addition of an oxygen atom. The MS/MS spectra of M2 and M3 could not be acquired, thus their identification was based only on their accurate m/z values. On the other hand, common characteristic product ions at m/z 243.1016, 215.1065, 197.0961, and 187.1118 were observed in the MS/MS spectra of M1, M4, and M5, and resulted from a successive loss of H2O and CO, which was consistent with the fragmentation pathway of helenalin. The MS/MS spectra of M1, M4, and M5 are presented in Fig. S5, Fig. S2A, and Fig. S3A, respectively. Besides the characteristic losses of H2O and CO, the MS/MS spectra of M4 and M5 showed different fragmentation patterns as summarized in Table 1.
The MS/MS spectrum of M4 displayed the fragment ion at m/z 123.0805 with small relative intensity and did not display the product ion at m/z 107.0855, thus we assumed that oxidation might have occurred on the corresponding part of the molecule. The fragmentation of M4 produced a characteristic product ion at m/z 205.0496 (C11H9O4), which could have been generated after the loss of H2O at C14, with the following cycloheptane skeleton cleavage and the lactone ring opening yielding a fragment ion with a carboxylic acid moiety. The following loss of CO2 produced the product ion at m/z 161.0597 (C10H9O2). Skeleton cleavage initiated not only by the cleavage of the lactone moiety, but also by the presence of methylene group on the cycloheptenone ring in a structurally similar sesquiterpene lactone, dehydrocostus lactone, was proposed by Peng et al. (2014). In the case of M4, the methylene group could be formed after the loss of H2O at C14. In addition, the fragment ion at m/z 173.0595 (C11H9O2) could have been formed after the cleavage of the α-methylene-γ-lactone moiety and methyl group at C10, which differed from the fragmentation of helenalin. Therefore, M4 was tentatively determined as 14-hydroxyhelenalin (Fig. 2). The MS/MS spectrum and the proposed fragmentation pathway of M4 are depicted in Fig. S2.
The MS/MS spectrum of M5 displayed product ions at m/z 123.0809 and 107.0861, with the former one also being a base peak in the spectrum. The presence of these two product ions suggested that the oxidation probably did not occur on the corresponding part of the molecule. The cleavage of α-methylene-γ-lactone moiety was followed by the cleavage of a three-carbon fragment from the cycloheptane ring rather than the cleavage of four-carbon fragment as in the case of helenalin, and the product ion at m/z 151.0753 (C9H11O2) was generated. The following loss of CO produced the product ion at m/z 123.0809. Therefore, M5 was tentatively identified as 9-hydroxyhelenalin (Fig. 2). The MS/MS spectrum and the proposed fragmentation pathway of M5 are depicted in Fig. S3.
In human liver microsomes, the major metabolite was M4 (14-hydroxyhelenalin), and in rat liver microsomes, the major metabolite was M5 (9-hydroxyhelenalin) (Table 1, Fig. 3). When human liver microsomes were incubated with 5 µM helenalin, metabolites M1 and M2 were not formed at all, and M3 and M5 were formed in an amount less than 10% compared to the relative amount of the main metabolite M4. When human liver microsomes were incubated with 100 µM helenalin, M5 was formed in an almost equal amount to M4 (70%). The order of relative amounts of the metabolites in rat liver microsomal incubations was M5 >> M4 > M3 > M2 > M1 at 5 µM and M5 >>> M4 = M2 > M1 > M3 at 100 µM helenalin. It can be summarized that the same oxidative metabolites were formed in both human and rat liver microsomal incubations, although the metabolite pattern differed between the species.

The comparison of in vitro oxidation of helenalin by human and rat liver microsomes. Helenalin at the concentrations of 5 μM (a) or 100 μM (b) was incubated with human or rat liver microsomes (0.4 g/L) in the presence of 20% NADPH regenerating system and 100 mM potassium phosphate buffer pH 7.4 for 60 min at 37 °C. The metabolites (M1-M5) were detected by UHPLC-Q-Exactive-MS/MS. The results are presented as average ± SD of two independent experiments. HLM: human liver microsomes; RLM: rat liver microsomes
Reduced metabolites
Metabolites M6 and M7 were formed only in the incubations with human liver cytosol and microsomes in the presence of the NADPH regenerating system. The peak area of M6 was bigger than that of M7. M6 and M7 were detected at 4.97 and 5.88 min at m/z 265.1433 and 265.1427, respectively ([M+H]+, C15H21O4). The molecular weight of M6 and M7 was 2 Da mass units higher compared to that of helenalin (m/z 263.1277 [M+H]+). M6 and M7 produced fragment ions at m/z 247.1327, 229.1222, and 201.1273, thus a similar fragmentation behavior was proved in relation to that of the parent compound in terms of consecutive loss of H2O and CO, which further suggested that M6 and M7 were hydrogenated metabolites of helenalin. The further fragmentation pathway of M6 corresponded with the fragmentation pathway of helenalin, except for the loss of ethene (m/z 201.1273–173.0963) instead of ethyne (m/z 199.1116–173.0959). Therefore, we proposed that the hydrogenation occurred on the carbon–carbon double bond between C11 and C13 (Fig. 2). Thus, M6 was tentatively determined as 11,13-dihydrohelenalin. The MS/MS spectrum and the proposed fragmentation pathway of M6 are depicted in Fig. S4. Given the longer elution time of M7, we suggest that the hydrogenation occurred on the other carbon–carbon double bond (Fig. 2) rather than on a carbonyl group. Therefore, M7 was tentatively identified as 2,3-dihydrohelenalin. The MS/MS spectrum of M7 is presented in Fig. S6.
The incubations of helenalin in the presence of liver microsomes and UDPGA or liver cytosol and PAPS produced no phase II metabolites in either animal species. When the experimental conditions were set to allow for simultaneous oxidation and glucuronidation or sulfation, only metabolites M1-M5 could be observed, not their conjugates.
Oxidation of helenalin by human recombinant CYP enzymes
To elucidate which CYPs are responsible for the oxidation of helenalin in humans, 5 μM and 100 μM helenalin was incubated with human recombinant CYP enzymes and the NADPH regenerating system. Samples were subsequently analyzed by UHPLC-Q-Exactive-MS/MS and metabolites were identified as described above. Several CYP forms, namely CYP2A13, CYP2B6, CYP3A4, CYP3A5, and CYP3A7, were discovered to catalyze the oxidation of helenalin (Fig. 4), although they catalyzed the formation of different metabolites depending on the concentration of helenalin. CYP2A13 was the most active CYP in the incubations with 5 μM helenalin, catalyzing predominantly the formation of M3 and to a lesser extent M4. The second most active enzyme, CYP3A5, was found to oxidize helenalin into three different metabolites, M3, M4, and M5, with M4 being the most abundant. All three members of the CYP3A family were particularly involved in the formation of M4 regardless of the concentration of helenalin. In incubations with 100 μM helenalin, CYP2B6 was the most active form involved mainly in the formation of M5. Interestingly, M1 and M2 were produced exclusively by CYP2B6 and could be detected only when the high concentration of helenalin was employed (Fig. 4).

The oxidation of helenalin by human recombinant CYP enzymes. Helenalin (5 μM and 100 μM) was incubated with individual CYP forms (20 nM) in the presence of 20% NADPH regenerating system and 100 mM potassium phosphate buffer pH 7.4 for 60 min at 37 °C. The metabolites (M1–M5) were detected by UHPLC-Q-Exactive-MS/MS and the results are expressed as the average ± SD of two independent experiments
Kinetic analysis of the oxidation of helenalin by human and rat liver microsomes and human recombinant CYP enzymes
The kinetics of the oxidation of helenalin into three major metabolites, M3, M4, and M5, was studied in incubations containing a wide concentration range of helenalin (0–100 μM), human or rat liver microsomes, and NADPH. Samples were analyzed using UHPLC-Q-Exactive-MS/MS. Nevertheless, given the unavailability of suitable analytical standards, the exact quantification of metabolites concentration was not feasible. For that reason, calculations were performed using respective values of the peak area of the metabolites in the chromatograms as a substitute unit. The data were fitted into the Michaelis–Menten equation (R2 value was > 0.93 for all data sets) and corresponding enzyme kinetic parameters Km, Vmax as well as intrinsic clearance Km/Vmax were calculated (Table 2). The results further indicated a substantial difference between rats and humans in the oxidation of helenalin.
The overall NADPH-dependent oxidation of helenalin was more efficient in rat than in human liver microsomes due to its more efficient oxidation to M5 and M3. However, the oxidation of helenalin to M4 was almost equally efficient in human and rat liver microsomes, as the Clint values were almost equal.
The same experimental approach was adopted for incubations with the four most active human CYPs involved in the oxidation of helenalin, i.e. CYP2A13, 2B6, 3A4, and 3A5 (Table 3). The kinetic parameters of CYP2B6, which produced predominantly M5, could not be determined since the oxidation of helenalin by CYP2B6 did not follow Michaelis–Menten kinetics within the employed 0–100 μM concentration range of helenalin (data not shown). Extrahepatic CYP2A13 showed the highest affinity towards the oxidation of helenalin into M3 and M4, with the Km values at 0.6 μM and 0.8 μM, respectively. Likewise, CYP2A13 proved to be the most efficient in the oxidation of helenalin as characterized by the highest Clint values. Out of the two studied hepatic CYP3A forms, CYP3A5 exhibited the highest Vmax and Clint values for the formation of M4, while the formation of M3 and M5 reached only 14–19% of the efficiency of M4. CYP3A4 was less efficient than CYP3A5 in the formation of M4 (70% of the efficiency expressed as Clint) (Table 3).
Inhibition of human microsomal and recombinant CYP enzymes by helenalin
The inhibition of CYP activities by helenalin was initially screened in incubations with human liver microsomes using several profluorescent CYP substrates to encompass all major xenobiotic-metabolizing CYPs. Enzyme inhibition (IC50 = 76.0 μM) was detected when helenalin was incubated with BPMC, a more inclusive CYP substrate (Table 4). No inhibition was detected with more selective substrates of CYP1 (ethoxyresorufin, MPC, TFPMC), 2A6 (coumarin, MPC), 2B (pentoxyresorufin) or 2C19 (MPC). Therefore, the inhibitory activity was studied in a similar manner with individual human recombinant CYPs. Helenalin was found to inhibit the activity of CYP3A4 (IC50 = 18.7 μM/BPMC) and CYP3A5 (IC50 = 62.6 μM/BPMC), which was dependent on the substrate concentration. The inhibition of these two CYPs provides an explanation for its inhibitory activity in human liver microsomes. Interestingly, helenalin potently inhibited the activity of extrahepatic CYP2A13 (IC50 = 1.6 μM/coumarin, IC50 = 1.1 μM/HPHC). The activity of other CYPs was not affected (Table 5), suggesting selectivity of helenalin for individual CYP forms. Subsequently, the effect of preincubation with NADPH on the inhibition towards the three inhibited CYPs was investigated. The preincubation with NADPH resulted in an 11-fold decrease in the IC50 value for CYP2A13, while the inhibition of CYP3A4 and CYP3A5 was not affected (Table 6). In addition, the inhibition of CYP3A4 proceeded via a competitive mechanism as linear dependency between substrate concentration and IC50 value was observed (Fig. S7).
Time-, concentration- and NADPH-dependent inactivation of CYP2A13 by helenalin
The residual CYP2A13 activity was determined at different concentrations of helenalin (0–25 μM) and different preincubation times with NADPH (1–10 min). The relative remaining activity was expressed as a ln(percentage of the activity of control samples (without helenalin)) for each preincubation time. The inactivation of CYP2A13 showed a clear dependence on both the inhibitor concentration and the preincubation time, suggesting a mechanism-based inhibition of this enzyme by helenalin (Fig. 5a). The observed rate of inactivation (kobs) was determined from the linear regression of the time-course data of each concentration of helenalin and the resulting hyperbolic relationship between kobs values and helenalin concentration was further used to calculate the value of KI = 6.7 μM and the maximal rate of inactivation kinact = 0.58 ln(%)/min indicating that 1.78% of CYP2A13 was inactivated in a minute (Fig. 5b).

Time- and concentration-dependent inactivation of human recombinant CYP2A13 by helenalin. Different helenalin concentrations (0–25 µM) were first incubated with CYP2A13 (10 nM) and NADPH for up to 10 min in 100 mM Tris–HCl (pH 7.4). Then, 10 µL of the first incubation mixture were taken into 190 µL of the second incubation mixture containing 100 mM Tris–HCl (pH 7.4), 25 µM substrate HPHC, and NADPH to determine the CYP2A13 activity. a Shows the time-dependent decrease in the relative remaining activity of CYP2A13 at different helenalin concentrations, slope is the kobs value, b shows the dependency of the inactivation rate of CYP2A13 activity (kobs values) at every helenalin concentration derived from (a) and the estimated KI and kinact value
Discussion
A. montana and C. minima are widely used therapeutical plants that contain a sesquiterpene lactone helenalin. In recent studies, helenalin as well as A. montana and C. minima showed significant health benefits, including anti-inflammatory and anti-cancer properties (Kriplani and Guarve 2020). Moreover, helenalin application for hepatic fibrosis and inflammation treatment has been recently patented (Moujir et al. 2020). The pharmacological effects of helenalin have been studied in vitro as well as in vivo (Drogosz and Janecka 2019; Lin et al. 2014; Li et al. 2019), yet less attention has been devoted to its metabolic properties that can directly affect drug safety, efficacy, and toxicity profiles (Kriplani and Guarve 2020). To our best knowledge, there are no studies that would attempt to comprehensively elucidate the metabolism of helenalin. For this reason, the objective of the present study was to characterize and compare the metabolism of helenalin in vitro using human and rat liver subcellular fractions and human recombinant CYPs as well as to determine the effect of helenalin on the activity of human CYPs. The NADPH-dependent metabolism of helenalin in vitro proceeded via two biotransformation pathways, either oxidation or reduction. As a result, five oxidative metabolites and two reduced metabolites were formed with human liver subcellular fractions, whereas only five oxidative metabolites were formed in the incubations with rat liver subcellular fractions. The metabolism was faster in the rat than in the human samples. Moreover, extrahepatic human CYP2A13 was the oxidizing enzyme with higher affinity than hepatic CYP2B6, CYP3A4, CYP3A5 or CYP3A7. Helenalin was a potent mechanism-based inhibitor of CYP2A13 and a weak competitive inhibitor of CYP3A4. These results prove that helenalin is metabolized by several CYP enzymes, yet it inhibits only the activity of CYP2A13 and CYP3A4.
The NADPH-dependent oxidation of helenalin produced five metabolites (M1-M5) both in human and rat liver microsomes, although important interspecies differences could be observed. Firstly, 14-hydroxyhelenalin (M4) was the predominant metabolite of helenalin in human liver microsomes, while 9-hydroxyhelenalin (M5) was the predominant metabolite in rat liver microsomes. The in vitro oxidation rate of helenalin was faster in rats than in humans because of the higher metabolites formation rate in rat liver microsomes than in human liver microsomes, and because the sum of Clint of the metabolites formed in rats was about three times higher than the one in humans. Therefore, it can be concluded that helenalin is oxidized faster in rats than in humans.
Because the oxidation of helenalin was NADPH-dependent in human liver microsomes, the role of individual CYPs was further elucidated. Extrahepatic CYP2A13 and hepatic CYP2B6, CYP3A4, CYP3A5 and CYP3A7 were found to oxidize helenalin. Interestingly, the most efficient CYP with high affinity was CYP2A13, which is commonly expressed in the respiratory tract with the highest expression in nasal mucosa, followed by lungs and trachea (Su et al. 2000; Zanger and Schwab 2013). Similarly, CYP2A13 was the most efficient oxidizing catalyst among CYPs for scoparone (Fayyaz et al. 2018) or for scopoletin (Juvonen et al. 2019b). CYP3A4 and CYP3A5, on the other hand, were found to be involved in the hepatic metabolism, participating in the formation of M4, the major metabolite found in the incubations with human liver microsomes. In addition, our results suggest different regioselectivity of the two CYP3A forms, and higher efficiency of CYP3A5 in the oxidation of helenalin. The overall higher metabolic capability of CYP3A5 was an interesting finding, as CYP3A5 generally exhibits reduced catalytic activity for xenobiotic or eubiotic substrates as compared to CYP3A4 (Niwa et al. 2019, 2020; Williams et al. 2002). The fetal-predominant CYP3A7 also contributed to the formation of M4 (Fig. 4), which is in agreement with the general understanding of similar substrate selectivity in the CYP3A family (Zanger and Schwab 2013). Helenalin was further found to be oxidized by CYP2B6. The results from the incubations with human recombinant CYPs elucidated that helenalin is oxidized by CYP enzymes in human liver microsomes. In vivo, the effect of CYPs on the metabolism of helenalin is dependent both on the oxidation efficiency of individual CYPs and their amount in the metabolizing tissues, particularly in the liver (Zanger and Schwab 2013).
The reduction pathway of helenalin took place NADPH-dependently both in human liver microsomes and cytosol and yielded two metabolites 11,13- and 2,3-dihydrohelenalin (M6 and M7, respectively). Based on the fragmentation spectra and chromatographic behavior of the reduced metabolites, the reduction took place on the carbon–carbon doubles bond rather than on the carbonyl double bond. Such reductive metabolism has been previously reported for xenobiotics containing α,β-unsaturated ketone moieties (Itoh et al. 2008; Kitamura et al. 2002; Moore et al. 2014; Yu et al. 2013), which are characteristic for the structure of helenalin. In addition, similar carbon–carbon double bond reduction was reported when the metabolism of sesquiterpene lactones alantolactone (Yao et al. 2016) and ixerin Z (Cai et al. 2015) was investigated.
Surprisingly, no phase II metabolites of helenalin, namely glucuronides or sulfates, were detected in either studied species in vitro, although its 6-hydroxyl substituent could potentially be glucuronidated or sulfonated. Earlier, helenalin was found to react with the cysteine thiol group of glutathione, but the conjugation was rather spontaneous and independent of the presence of glutathione S-transferase (Schmidt 2000; Schmidt et al. 1999). Similarly, the glutathione conjugation of structurally similar alantolactone and isoalantolactone has also been deemed to be the result of a chemical reaction rather than a product of enzyme-catalyzed metabolic transformation (Zhou et al. 2018). Our preliminary data (not shown) supported this observation and therefore the glutathione conjugation was not further investigated.
Helenalin has previously been found to decrease the CYP content and to inhibit the activities of both murine and rat CYP enzymes in vitro and in vivo (Chapman et al. 1989, 1991, 1988; Jodynis-Liebert et al. 2000). In the present study, the inhibition of CYP2A13 (IC50 = 1.1 µM), CYP3A4 (IC50 = 18.7 µM), and CYP3A5 (IC50 = 62.6 µM) was observed using human recombinant CYPs. Compared to the other CYP forms, helenalin showed selective inhibition towards CYP2A13. The inhibition of CYP3A4 and CYP3A5 was more efficient at low substrate concentrations and not complete at higher substrate concentrations (Fig. S8). Incomplete inhibition has been also observed in murine liver microsomes, in which the activity of aminopyrine demethylase and 7-ethoxyresorufin deethylase was inhibited to a maximum of 60%, while the activity of aniline hydroxylase was inhibited to a maximum of 30% by 1 mM helenalin (Chapman et al. 1989). Indeed, oral administration of helenalin to rats (2.5 mg/kg in a single dose) resulted in a decrease in aniline hydroxylase (by 51%) and aminopyrine demethylase (by 52%) activities in the liver (Jodynis-Liebert et al. 2000). On the other hand, CYP2E1 protein content was equal in control and helenalin-treated rats receiving 3 mg/kg of helenalin daily for 24 weeks and no signs of hepatotoxicity were observed (Lin et al. 2014). CYP-mediated metabolism of helenalin into a reactive metabolite and the subsequent irreversible CYP inhibition was first suggested by Chapman et al. (1989, 1991) as the decrease in the activity of aminopyrine demethylase and CYP content in mouse liver microsomes was higher in the presence than in the absence of NADPH. In the present study, the preincubation of helenalin with CYP3A4 or CYP3A5 in the presence of NADPH did not alter the IC50 value of helenalin for either form. Rather, helenalin appeared to inhibit the activity of CYP3A4 in a competitive manner. On the other hand, the preincubation of helenalin with CYP2A13 in the presence of NADPH resulted in a significant decrease of the IC50 value of helenalin. The inhibition of CYP2A13 by helenalin was time- and concentration-dependent, suggesting that helenalin acted as a mechanism-based inhibitor of CYP2A13. A mechanism-based inhibition of enzymes in CYP2A family was reported for hirsutinolide-type sesquiterpene lactones isolated from Vernonia cinerea as well as for CYP2A6 by 7-methylcoumarin (Juvonen et al. 2016). The sesquiterpene compounds demonstrated a rather similar effect towards both isoenzymes, CYP2A6 and CYP2A13 (Boonruang et al. 2017). In comparison, helenalin acted as a specific inhibitor of CYP2A13 as the activity of CYP2A6 was not inhibited at all.
Conclusion
Sesquiterpene lactone helenalin is used as an antiphlogistic in European and Chinese folk medicine. Although the pharmacological activities of helenalin have been extensively investigated, insufficient information exists about its metabolic properties. In this study, the in vitro NADPH-dependent metabolism of helenalin using human and rat liver subcellular fractions was studied. Helenalin was oxidized into five metabolites both in human and rat liver microsomes, with 14-hydroxyhelenalin (M4) being the main metabolite in human samples and 9-hydroxyhelenalin (M5) in rat samples. Helenalin was also reduced to 11,13- and 2,3-dihydrohelenalin (M6 and M7, respectively) in human liver microsomes and cytosol, but not in rat liver subcellular fractions. In vitro NADPH-dependent metabolism was more efficient in rat than in human liver microsomes. Human hepatic CYP2B6 oxidized helenalin into M1, M2, and M5, CYP3A4 into M4, CYP3A5 into M3, M4, and M5, CYP3A7 into M4, and lastly, extrahepatic CYP2A13 oxidized helenalin into M3 and M4. CYP2A13 exhibited high affinity and was a more efficient oxidizer of helenalin than the hepatic human CYPs. It is thus concluded that the metabolism of helenalin is faster in rats than in humans, and that in humans it may also be significantly metabolized by extrahepatic CYP2A13. Helenalin acted as a mechanism-based inhibitor of CYP2A13 and a weak competitive inhibitor of CYP3A4. Therefore, various pharmacological/toxicological consequences might be expected, and its internal or nasal application should be thoroughly considered.
References
Bergamante V, Ceschel GC, Marazzita S, Ronchi C, Fini A (2007) Effect of vehicles on topical application of aloe vera and arnica montana components. Drug Deliv 14(7):427–432. https://doi.org/10.1080/10717540701202960
Berges C, Fuchs D, Opelz G, Daniel V, Naujokat C (2009) Helenalin suppresses essential immune functions of activated CD4+ T cells by multiple mechanisms. Mol Immunol 46(15):2892–2901. https://doi.org/10.1016/j.molimm.2009.07.004
Boonruang S, Prakobsri K, Pouyfung P et al (2017) Inhibition of human cytochromes P450 2A6 and 2A13 by flavonoids, acetylenic thiophenes and sesquiterpene lactones from Pluchea indica and Vernonia cinerea. J Enzyme Inhib Med Chem 32(1):1136–1142. https://doi.org/10.1080/14756366.2017.1363741
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1006/abio.1976.9999
Buchele B, Zugmaier W, Lunov O, Syrovets T, Merfort I, Simmet T (2010) Surface plasmon resonance analysis of nuclear factor-kappaB protein interactions with the sesquiterpene lactone helenalin. Anal Biochem 401(1):30–37. https://doi.org/10.1016/j.ab.2010.02.020
Cai W, Zhang JY, Dong LY et al (2015) Identification of the metabolites of Ixerin Z from Ixeris sonchifolia Hance in rats by HPLC-LTQ-Orbitrap mass spectrometry. J Pharm Biomed Anal 107:290–297. https://doi.org/10.1016/j.jpba.2015.01.004
Chapman DE, Roberts GB, Reynolds DJ et al (1988) Acute toxicity of helenalin in BDF1 mice. Fundam Appl Toxicol 10(2):302–312
Chapman DE, Holbrook DJ, Chaney SG, Hall IH, Lee KH (1989) In vitro inhibition of mouse hepatic mixed-function oxidase enzymes by helenalin and alantolactone. Biochem Pharmacol 38(22):3913–3923. https://doi.org/10.1016/0006-2952(89)90668-0
Chapman DE, Holbrook DJ, Chaney SG, Hall IH, Lee KH (1991) In vivo and in vitro effects of helenalin on mouse hepatic microsomal cytochrome P450. Biochem Pharmacol 41(2):229–235. https://doi.org/10.1016/0006-2952(91)90481-j
Dirsch VM, Stuppner H, Vollmar AM (2001) Helenalin triggers a CD95 death receptor-independent apoptosis that is not affected by overexpression of Bcl-x(L) or Bcl-2. Cancer Res 61(15):5817–5823
Douglas JA, Smallfield BM, Burgess EJ et al (2004) Sesquiterpene lactones in Arnica montana: a rapid analytical method and the effects of flower maturity and simulated mechanical harvesting on quality and yield. Planta Med 70(2):166–170. https://doi.org/10.1055/s-2004-815495
Drogosz J, Janecka A (2019) Helenalin—a sesquiterpene lactone with multidirectional activity. Curr Drug Targets 20(4):444–452. https://doi.org/10.2174/1389450119666181012125230
European Medicines Agency (2014) Community herbal monograph on Arnica montana L., flos. EMA/HMPC/198793/2012. In: Committee on Herbal Medicinal Products (HMPC). https://www.ema.europa.eu/en/documents/herbal-monograph/final-community-herbal-monograph-arnica-montana-l-flos_en.pdf. Accessed 14 Sep 2021
Fayyaz A, Makwinja S, Auriola S, Raunio H, Juvonen RO (2018) Comparison of in vitro hepatic scoparone 7-O-demethylation between humans and experimental animals. Planta Med 84(5):320–328. https://doi.org/10.1055/s-0043-119886
Garcia-Pineres AJ, Castro V, Mora G et al (2001) Cysteine 38 in p65/NF-kappaB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J Biol Chem 276(43):39713–39720. https://doi.org/10.1074/jbc.M101985200
Hall IH, Lee KH, Mar EC, Starnes CO, Waddell TG (1977) Antitumor agents. 21. A proposed mechanism for inhibition of cancer growth by tenulin and helenalin and related cyclopentenones. J Med Chem 20(3):333–337. https://doi.org/10.1021/jm00213a003
Hall IH, Lee KH, Starnes CO et al (1979) Anti-inflammatory activity of sesquiterpene lactones and related compounds. J Pharm Sci 68(5):537–542. https://doi.org/10.1002/jps.2600680505
Hall IH, Lee KH, Starnes CO, Muraoka O, Sumida Y, Waddell TG (1980) Antihyperlipidemic activity of sesquiterpene lactones and related compounds. J Pharm Sci 69(6):694–697. https://doi.org/10.1002/jps.2600690622
Hempen C-H, Fischer T (2009) XIII—herbs that transform phlegm and stop coughing. In: Hempen C-H, Fischer T (eds) A materia medica for chinese medicine: plants, minerals, and animal products. Churchill, Livingstone, pp 618–679
Hoffmann R, von Schwarzenberg K, Lopez-Anton N et al (2011) Helenalin bypasses Bcl-2-mediated cell death resistance by inhibiting NF-kappaB and promoting reactive oxygen species generation. Biochem Pharmacol 82(5):453–463. https://doi.org/10.1016/j.bcp.2011.05.029
Huang PR, Yeh YM, Wang TC (2005) Potent inhibition of human telomerase by helenalin. Cancer Lett 227(2):169–174. https://doi.org/10.1016/j.canlet.2004.11.045
Itoh K, Yamamoto K, Adachi M, Kosaka T, Tanaka Y (2008) Leukotriene B4 12-hydroxydehydrogenase/15-ketoprostaglandin Delta 13-reductase (LTB4 12-HD/PGR) responsible for the reduction of a double-bond of the alpha, beta-unsaturated ketone of an aryl propionic acid non-steroidal anti-inflammatory agent CS-670. Xenobiotica 38(3):249–263. https://doi.org/10.1080/00498250701767667
Jang JH, Iqbal T, Min KJ et al (2013) Helenalin-induced apoptosis is dependent on production of reactive oxygen species and independent of induction of endoplasmic reticulum stress in renal cell carcinoma. Toxicol in Vitro 27(2):588–596. https://doi.org/10.1016/j.tiv.2012.10.014
Jodynis-Liebert J, Murias M, Bloszyk E (2000) Effect of sesquiterpene lactones on antioxidant enzymes and some drug-metabolizing enzymes in rat liver and kidney. Planta Med 66(3):199–205. https://doi.org/10.1055/s-2000-8566
Juvonen RO, Kuusisto M, Fohrgrup C et al (2016a) Inhibitory effects and oxidation of 6-methylcoumarin, 7-methylcoumarin and 7-formylcoumarin via human CYP2A6 and its mouse and pig orthologous enzymes. Xenobiotica 46(1):14–24. https://doi.org/10.3109/00498254.2015.1048327
Juvonen RO, Ahinko M, Huuskonen J, Raunio H, Pentikainen OT (2019a) Development of new Coumarin-based profluorescent substrates for human cytochrome P450 enzymes. Xenobiotica 49(9):1015–1024. https://doi.org/10.1080/00498254.2018.1530399
Juvonen RO, Novak F, Emmanouilidou E et al (2019b) Metabolism of scoparone in experimental animals and humans. Planta Med 85(6):453–464. https://doi.org/10.1055/a-0835-2301
Juvonen RO, Jokinen EM, Huuskonen J, Karkkainen O, Raunio H, Pentikainen OT (2021) Molecular docking and oxidation kinetics of 3-phenyl coumarin derivatives by human CYP2A13. Xenobiotica 2021:1–28. https://doi.org/10.1080/00498254.2021.1898700
Kitamura S, Kohno Y, Okamoto Y, Takeshita M, Ohta S (2002) Reductive metabolism of an alpha, beta-ketoalkyne, 4-phenyl-3-butyn-2-one, by rat liver preparations. Drug Metab Dispos 30(4):414–420. https://doi.org/10.1124/dmd.30.4.414
Kriplani P, Guarve K (2020) Recent Patents on Anti-Cancer Potential of Helenalin. Recent Pat Anticancer Drug Discov 15(2):132–142. https://doi.org/10.2174/1574892815666200702142601
Lang MA, Gielen JE, Nebert DW (1981) Genetic evidence for many unique liver microsomal P-450-mediated monooxygenase activities in heterogeneic stock mice. J Biol Chem 256(23):12068–12075
Lee KH (1973) Antitumor agents. V. Effect of epoxidation on cytotoxicity of helenalin-related derivatives. J Pharm Sci 62(6):1028–1029. https://doi.org/10.1002/jps.2600620645
Lee KH, Furukawa H (1972) Antitumor agents. 3. Synthesis and cytotoxic activity of helenalin amine adducts and related derivatives. J Med Chem 15(6):609–611. https://doi.org/10.1021/jm00276a010
Leven W, Willuhn G (1987) Sesquiterpene lactones from Arnica chamissonis less.: VI. Identification and quantitative determination by high-performance liquid and gas chromatography. J Chromatogr A 410:329–342
Li Y, Zeng Y, Huang Q et al (2019) Helenalin from Centipeda minima ameliorates acute hepatic injury by protecting mitochondria function, activating Nrf2 pathway and inhibiting NF-kappaB activation. Biomed Pharmacother 119:109435. https://doi.org/10.1016/j.biopha.2019.109435
Lim CB, Fu PY, Ky N et al (2012) NF-kappaB p65 repression by the sesquiterpene lactone, Helenalin, contributes to the induction of autophagy cell death. BMC Complement Altern Med 12:93. https://doi.org/10.1186/1472-6882-12-93
Lin X, Zhang S, Huang R et al (2014) Helenalin attenuates alcohol-induced hepatic fibrosis by enhancing ethanol metabolism, inhibiting oxidative stress and suppressing HSC activation. Fitoterapia 95:203–213. https://doi.org/10.1016/j.fitote.2014.03.020
Lyss G, Schmidt TJ, Merfort I, Pahl HL (1997) Helenalin, an anti-inflammatory sesquiterpene lactone from Arnica, selectively inhibits transcription factor NF-kappaB. Biol Chem 378(9):951–961. https://doi.org/10.1515/bchm.1997.378.9.951
Lyss G, Knorre A, Schmidt TJ, Pahl HL, Merfort I (1998) The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-kappaB by directly targeting p65. J Biol Chem 273(50):33508–33516. https://doi.org/10.1074/jbc.273.50.33508
Merrill JC, Kim HL, Safe S, Murray CA, Hayes MA (1988) Role of glutathione in the toxicity of the sesquiterpene lactones hymenoxon and helenalin. J Toxicol Environ Health 23(2):159–169. https://doi.org/10.1080/15287398809531103
Moore TW, Zhu S, Randolph R, Shoji M, Snyder JP (2014) Liver S9 fraction-derived metabolites of curcumin analogue UBS109. ACS Med Chem Lett 5(4):288–292. https://doi.org/10.1021/ml4002453
Moujir L, Callies O, Sousa P, Sharopov F, Seca AM (2020) Applications of sesquiterpene lactones: a review of some potential success cases. Appl Sci 10(9):3001
Niinivehmas S, Postila PA, Rauhamaki S et al (2018) Blocking oestradiol synthesis pathways with potent and selective coumarin derivatives. J Enzyme Inhib Med Chem 33(1):743–754. https://doi.org/10.1080/14756366.2018.1452919
Niwa T, Narita K, Okamoto A, Murayama N, Yamazaki H (2019) Comparison of steroid hormone hydroxylations by and docking to human cytochromes P450 3A4 and 3A5. J Pharm Pharm Sci 22(1):332–339. https://doi.org/10.18433/jpps30558
Niwa T, Okamoto A, Narita K et al (2020) Comparison of steroid hormone hydroxylation mediated by cytochrome P450 3A subfamilies. Arch Biochem Biophys 682:108283. https://doi.org/10.1016/j.abb.2020.108283
Peng Z, Wang Y, Gu X, Guo X, Yan C (2014) Study on the pharmacokinetics and metabolism of costunolide and dehydrocostus lactone in rats by HPLC-UV and UPLC-Q-TOF/MS. Biomed Chromatogr 28(10):1325–1334. https://doi.org/10.1002/bmc.3167
Rauhamaki S, Postila PA, Niinivehmas S et al (2018) Structure-activity relationship analysis of 3-phenylcoumarin-based monoamine oxidase B inhibitors. Front Chem 6:41. https://doi.org/10.3389/fchem.2018.00041
Schmidt TJ (1997) Helenanolide-type sesquiterpene lactones–III. Rates and stereochemistry in the reaction of helenalin and related helenanolides with sulfhydryl containing biomolecules. Bioorg Med Chem 5(4):645–653. https://doi.org/10.1016/s0968-0896(97)00003-5
Schmidt TJ (2000) Glutathione adducts of helenalin and 11 alpha,13-dihydrohelenalin acetate inhibit glutathione S-transferase from horse liver. Planta Med 66(2):106–109. https://doi.org/10.1055/s-2000-11123
Schmidt TJ, Lyss G, Pahl HL, Merfort I (1999) Helenanolide type sesquiterpene lactones. Part 5: the role of glutathione addition under physiological conditions. Bioorg Med Chem 7(12):2849–2855. https://doi.org/10.1016/s0968-0896(99)00234-5
Staneva J, Denkova P, Todorova M, Evstatieva L (2011) Quantitative analysis of sesquiterpene lactones in extract of Arnica montana L. by 1H NMR spectroscopy. J Pharm Biomed Anal 54(1):94–99. https://doi.org/10.1016/j.jpba.2010.08.018
Su T, Bao Z, Zhang QY, Smith TJ, Hong JY, Ding X (2000) Human cytochrome P450 CYP2A13: predominant expression in the respiratory tract and its high efficiency metabolic activation of a tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res 60(18):5074–5079
Todorova M, Trendafilova A, Vitkova A, Petrova M, Zayova E, Antonova D (2016) Developmental and environmental effects on sesquiterpene lactones in cultivated Arnica montana L. Chem Biodivers 13(8):976–981. https://doi.org/10.1002/cbdv.201500307
Tsai L, Highet RJ, Herz W (1969) The mass spectra of pseudoguaianolides related to helenalin. J Org Chem 34(4):945–948. https://doi.org/10.1021/jo01256a035
Wagner S, Merfort I (2007) Skin penetration behaviour of sesquiterpene lactones from different Arnica preparations using a validated GC-MSD method. J Pharm Biomed Anal 43(1):32–38. https://doi.org/10.1016/j.jpba.2006.06.008
Wagner S, Kratz F, Merfort I (2004a) In vitro behaviour of sesquiterpene lactones and sesquiterpene lactone-containing plant preparations in human blood, plasma and human serum albumin solutions. Planta Med 70(3):227–233. https://doi.org/10.1055/s-2004-815539
Wagner S, Suter A, Merfort I (2004b) Skin penetration studies of Arnica preparations and of their sesquiterpene lactones. Planta Med 70(10):897–903. https://doi.org/10.1055/s-2004-832613
Williams JA, Ring BJ, Cantrell VE et al (2002) Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab Dispos 30(8):883–891. https://doi.org/10.1124/dmd.30.8.883
Wu P, Su MX, Wang Y et al (2012) Supercritical fluid extraction assisted isolation of sesquiterpene lactones with antiproliferative effects from Centipeda minima. Phytochemistry 76:133–140. https://doi.org/10.1016/j.phytochem.2012.01.003
Yao D, Li Z, Huo C et al (2016) Identification of in vitro and in vivo metabolites of alantolactone by UPLC-TOF-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 1033–1034:250–260. https://doi.org/10.1016/j.jchromb.2016.08.034
Yu L, Jiang Y, Wang L, Sheng R, Hu Y, Zeng S (2013) Metabolism of BYZX in human liver microsomes and cytosol: identification of the metabolites and metabolic pathways of BYZX. PLoS ONE 8(3):e59882. https://doi.org/10.1371/journal.pone.0059882
Zanger UM, Schwab M (2013) Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 138(1):103–141. https://doi.org/10.1016/j.pharmthera.2012.12.007
Zhang X, He J, Huang W et al (2018) Antiviral activity of the sesquiterpene lactones from centipeda minima against influenza a virus in vitro. Natural Prod Commun 13(2):00201
Zhou B, Ye J, Yang N et al (2018) Metabolism and pharmacokinetics of alantolactone and isoalantolactone in rats: Thiol conjugation as a potential metabolic pathway. J Chromatogr B Analyt Technol Biomed Life Sci 1072:370–378. https://doi.org/10.1016/j.jchromb.2017.11.039
Acknowledgements
We thank Hannele Jaatinen for technical assistance during the incubation experiments and Miia Reponen for technical assistance during the LC/MS analyses. We further thank Ivan Vokřál, Ph.D. for valuable comments and Daniel Paul Sampey for the English revision.
Funding
This work was funded by the Charles University Grant Agency (Project GAUK No. 1302120), the Czech Science Foundation (Grant no. 18-09946S), and Charles University (Research Project SVV 260 550). The School of Pharmacy mass spectrometry laboratory is supported by Biocenter Finland and Biocenter Kuopio.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by MŠ, ROJ and SA. The first draft of the manuscript was written by MŠ and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Human liver tissue was obtained from the University Hospital of Oulu (Oulu, Finland) as a surplus from cadaveric kidney transplantation donors. The procedure was approved by the Ethics Committee of the Medical Faculty of the University of Oulu (January 21, 1986). This study was performed in line with the principles of the Declaration of Helsinki. The Wistar rats used in this study were maintained according to European guidelines on the protection of animals used for scientific purposes (Directive 2010/63/EU). The animal experiments were approved by the Ethics Committee for Animal Experiments, University of Kuopio (Document 01-38, June 1, 2000).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Šadibolová, M., Juvonen, R.O., Auriola, S. et al. In vitro metabolism of helenalin and its inhibitory effect on human cytochrome P450 activity. Arch Toxicol 96, 793–808 (2022). https://doi.org/10.1007/s00204-021-03218-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-021-03218-6