
Abstract
Chronic exposure to inorganic arsenic (iAs), a common drinking water and food contaminant, has been associated with an increased risk of type 2 diabetes in population studies worldwide. Several mechanisms underlying the diabetogenic effects of iAs have been proposed through laboratory investigations. We have previously shown that exposure to arsenite (iAs(III)) or its methylated trivalent metabolites, methylarsonite (MAs(III)) and dimethylarsinite (DMAs(III)), inhibits glucose-stimulated insulin secretion (GSIS) in pancreatic islets, without significant effects on insulin expression or insulin content. The goal of the present study was to determine if iAs(III) and/or its metabolites inhibit Ca2+ influx, an essential mechanism that regulates the release of insulin from β cells in response to glucose. We found that in vitro exposures for 48 h to non-cytotoxic concentrations of iAs(III), MAs(III), and DMAs(III) impaired Ca2+ influx in isolated murine pancreatic islets stimulated with glucose. MAs(III) and DMAs(III) were more potent inhibitors of Ca2+ influx than iAs(III). These arsenicals also inhibited Ca2+ influx and GSIS in islets treated with depolarizing levels of potassium chloride in the absence of glucose. Treatment with Bay K8644, a Cav1.2 channel agonist, did not restore insulin secretion in arsenical-exposed islets. Tolbutamide, a KATP channel blocker, prevented inhibition of insulin secretion in MAs(III)- and DMAs(III)-exposed islets, but only marginally in islets exposed to iAs(III). Our findings suggest that iAs(III), MAs(III), and DMAs(III) inhibit glucose-stimulated Ca2+ influx in pancreatic islets, possibly by interfering with KATP and/or Cav1.2 channel function. Notably, the mechanisms underlying inhibition of GSIS by iAs(III) may differ from those of its trivalent methylated metabolites.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Approximately 9% of the global population is diagnosed with diabetes, a condition of chronic hyperglycemia (World Health Organization 2016). The majority of these individuals have type 2 diabetes (T2D), which involves insulin resistance, a result of impaired insulin signaling in insulin-sensitive tissues, followed by insufficient insulin secretion by pancreatic β-cells (Prentki and Nolan 2006). The prevalence of T2D is increasing and is expected to double in the next 20 years if current trends continue (Chen et al. 2012). While obesity is a major factor in the rise of T2D, environmental chemicals, termed “diabetogens”, have also been implicated (Maull et al. 2012; Thayer et al. 2012).
Numerous epidemiological studies from populations around the world have shown inorganic arsenic (iAs), a prevalent drinking water and food contaminant, to be a diabetogen (Maull et al. 2012; Kuo et al. 2017). The pathogenesis of iAs-associated diabetes is not well understood. We have reported that human exposure to iAs is negatively associated with fasting plasma insulin and HOMA-IR (Del Razo et al. 2011), suggesting that insulin secretion by the pancreas may be the primary target for iAs. Several population studies that found iAs exposure to be associated with markers of impaired β-cell dysfunction, but not with insulin resistance also support this mechanism (Del Razo et al. 2011; Gribble et al. 2012; Peng et al. 2015; Rhee et al. 2013). However, results of laboratory studies using animal and in vitro models suggest that exposure to trivalent iAs, arsenite (iAs(III)), can impair both insulin secretion by pancreatic β-cells and insulin signaling in glucose-utilizing tissues (Douillet et al. 2013; Fu et al. 2010; Paul et al. 2007; Zhang et al. 2017). In addition, exposure to iAs has been shown to modify DNA methylation and/or expression of T2D-associated genes in both human population and laboratory models (Martin et al. 2017). Hence, the contributions of these mechanisms to the pathogenesis of iAs-associated diabetes remain unclear.
Our lab has shown that in vitro exposure of isolated murine pancreatic islets to iAs(III) and its trivalent methylated metabolites, methylarsonite (MAs(III)) and dimethylarsinite (DMAs(III)), inhibited glucose-stimulated insulin secretion (GSIS) without altering cell viability, insulin content, or insulin gene expression (Douillet et al. 2013). These findings indicate that arsenicals impair the process of insulin secretion rather than insulin production. Furthermore, methylated trivalent metabolites of iAs(III), MAs(III) and DMAs(III), were more potent inhibitors of GSIS in isolated islets than iAs(III). Thus, the production of methylated metabolites in the pathway of iAs metabolism is likely to play an important role in etiology of iAs-associated diabetes. Yet, the exact mechanisms by which these arsenicals impair GSIS have never been systematically investigated.
Glucose is the primary stimulus for the secretion of insulin by pancreatic β-cells. Upon entry into the β-cell, glucose is metabolized through glycolysis and downstream ATP-producing pathways, including the Krebs cycle and oxidative phosphorylation. The generation of ATP leads to the closure of ATP-dependent potassium (KATP) channels, specifically KIR6.1 or KIR6.2 channels, causing depolarization of the cell membrane. Depolarization initiates the opening of voltage-gated calcium channels, primarily L-type channels (Cav1.2) (Wiser et al. 1999; Barg et al. 2001), leading to an increase in intracellular Ca2+. Ca2+ then activates the exocytotic machinery, enabling fusion of insulin granules with the plasma membrane and the release of insulin into the bloodstream. The fact that insulin secretion does not occur when β-cells are stimulated with glucose in Ca2+-free medium strongly suggests that Ca2+ influx and insulin secretion are tightly linked (Henquin 2000). Furthermore, inhibition of calcium channels with pharmacological agents also inhibits GSIS (Wollheim and Sharp 1981).
In 2008, Díaz-Villaseñor et al. reported that iAs(III) decreased insulin secretion and glucose-stimulated Ca2+ influx in a rat insulinoma Rin-m5F β-cell line (Díaz-Villaseñor et al. 2008). Our study builds on this finding, demonstrating that (1) exposure to iAs(III) or its methylated metabolites inhibits glucose-stimulated Ca2+ influx in isolated murine pancreatic islets, and that (2) KATP and/or Cav1.2 channels are likely targets of these arsenicals in β-cells.
Methods
Islet isolation and treatment
Pancreatic islets were isolated from adult C57BL/6 male mice as previously described (Douillet et al. 2013). Briefly, the pancreas was perfused and digested with 1 mg/ml collagenase P (Roche Diagnostics Crop, Indianapolis, IN) and islets were purified using a Ficoll PM-400 gradient (GE Healthcare, Chicago, IL). After isolation, islets were incubated overnight at 37 °C with 5% CO2 in RPMI 1640 medium with 10% fetal bovine serum, 10 mM HEPES, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Gibco, Waltham, MA). Islets were exposed for 48 h to either iAs(III) (sodium arsenite, > 99% pure, Sigma-Aldrich, St. Louis, MO), MAs(III) (methylarsine oxide, > 98% pure), or DMAs(III) (iododimethylarsine, > 98% pure). The methylated arsenicals were synthetized by Dr. William Cullen’s lab at the University of British Columbia, Canada. Media were changed after 24 h to minimize oxidation of trivalent arsenicals to pentavalency.
Measurement of intracellular calcium
Islets (~ 30–40/run) exposed to arsenicals and control (unexposed) islets were pre-incubated with 2 µM FURA-2AM (Thermo Fisher Scientific) in the presence or absence of the arsenicals for 30 min at 37 °C in KRP buffer (114 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.16 mM MgSO4, 10 mM HEPES, 2.5 mM CaCl2, 0.1% BSA, 25.5 mM NaHCO3, pH 7.2). The islets were then transferred to a Delta T live-cell imaging flow chamber (Bioptechs) and perfused with the same buffer containing 2.5 mM glucose for 5 min, followed by buffer containing 16.7 mM glucose for 8 min at a flow rate of 0.3 ml/min. The imaging chamber was maintained at 37 °C and 5% CO2. For each run, FURA-2 fluorescence at 520 nm wavelength following excitation with 340 nm (F340) or 380 nm (F380) wavelengths (Semrock FURA2-C000 dichroic filter set) was captured every 6 s using an Olympus IX81 inverted microscope and a Hamamatsu ORCA R2 cooled CCD camera. Camera exposure times were kept consistent across runs within the day. Imaging was controlled by Velocity (PerkinElmer, Coventry, England). The ratio (F340/F380), a proxy for calcium concentrations, was calculated for each run using ImageJ. Ratio values were normalized to (i.e., divided by) the average ratio value after 5 min in 2.5 mM glucose. After plotting ratio vs. time, the area under the curve for 8 min, starting when 16.7 mM glucose/KCl entered the perfusion system, was calculated in Excel and assessed for statistical significance.
Insulin secretion assay
Islets exposed to arsenicals or control islets (15 islets/well) were transferred into 12-well culture plates containing a glucose-free buffer (114 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.16 mM MgSO4, 20 mM HEPES, 2.5 mM CaCl2, 0.2% bovine serum albumin, and 25.5 mM NaHCO3, all from Sigma-Aldrich, St. Louis, MO, pH 7.4) for 1 h at 37 °C and 5% CO2, followed by a 1-h incubation with 2.5 mM glucose (Sigma-Aldrich) and a 1-h incubation with 16.7 mM glucose and/or insulin secretagogues (potassium chloride, tolbutamide, and Bay K8644) (all from Sigma-Aldrich). Medium from each incubation step was frozen and stored for insulin analysis. Insulin concentrations were determined using Rat/Mouse Insulin ELISA kit (MilliporeSigma, Burlington, MA). Based on the manufacturer’s information, the limit of sensitivity for this assay is 0.2 ng/mL (35 pM) using a 10 µL sample size.
Cell viability assay
After the 48-h exposure, control and exposed islets were incubated in phenol-free RPMI islet medium with 0.5 mg/ml MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) for 1 h at 37 °C. The formed formazan was dissolved in DMSO and absorbance read at 570 nm with a background correction of 630 nm (Stýblo et al. 2002).
Quantitative PCR
Total RNA was collected from islets (100/treatment) after stimulation with 16.7 mM glucose using RNeasy kits (Qiagen, Valencia, CA) and analyzed by a Nanodrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA). cDNA was prepared with the iScript cDNA synthesis kit (Biorad, Hercules, CA). Gene expression was quantified using PowerUp SYBR green master mix (Thermo Fisher Scientific, Waltham, MA) and primers for the alpha subunit in Cav1.2 channels, Cacna1c, and 18S, purchased through Thermo Fisher Scientific (Waltham, MA). Primer sequences for Cacna1c were CAGCCCAGAAAAGAAACAGG (forward) and GGATTCTCCATCGGCTGTAA (reverse); primer sequences for 18S were CGCGGTTCTATTTTGTTGGT (forward) and AGTCGGCATCGTTTATGGTC (reverse) (Santulli et al. 2015). Target gene expression was normalized to the 18S gene. PCRs were performed in a LightCycler 480 II (Roche, Indianapolis, IN).
Statistical analysis
Data are presented as mean ± SEM for multiple biological replicates where one replicate represents islets isolated from one mouse. Significance was determined using one-way ANOVAs followed by post hoc Dunnett’s test comparing samples treated with arsenicals to untreated controls. Analyses were conducted in JMP (SAS Institute, RTP, NC).
Results
Effects of trivalent arsenicals on GSIS
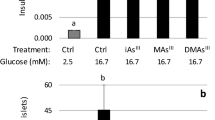
Following the design of our published study (Douillet et al. 2013), we first re-examined the effects of trivalent arsenicals on GSIS in isolated pancreatic islets. We confirmed that 48-h exposures to 2 µM iAs(III), 0.5 µM MAs(III), and 0.5 µM DMAs(III) significantly inhibit GSIS (Fig. 1a), but are not cytotoxic to the islets (Fig. 1b).

Glucose-stimulated insulin secretion in control islets and in islets exposed to trivalent arsenicals. Isolated pancreatic islets were exposed to arsenicals for 48 h and stimulated with 2.5 mM glucose for 1 h followed by 16.7 mM glucose for 1 h. Islets incubated in As-free medium for 48 h were used as controls. Insulin secretion (a) and cell viability measured via MTT assay (b) are shown as mean + SEM for N = 5 biological replicates (assay done in triplicate with 15 islets per replicate). *p < 0.05, the amount of insulin secreted by As-treated islets is significantly different from that secreted by control islets
Effects of trivalent arsenicals on calcium influx
Calcium influx was examined in islets exposed to a range of iAs(III), MAs(III), and DMAs(III) concentrations and in control, unexposed islets. Here, the FURA-2 signal was measured in islets at 2.5 mM glucose followed by stimulation with 16.7 mM (Fig. 2). In control islets, introduction of 16.7 mM glucose into the flow system was followed by a sharp increase in Ca2+ influx that plateaued after around 3 min. Ca2+ influx was inhibited in islets exposed to arsenicals as indicated by a smaller or delayed increase in the fluorescent signal (Fig. 2a–c) and a smaller area under the curve (Fig. 2d). The inhibition of Ca2+ influx by the arsenicals did not follow a typical dose–response pattern, but MAs(III) was clearly the most potent inhibitor, significantly suppressing Ca2+ influx at concentrations as low as 0.05 µM. There were no differences in the intracellular Ca2+ levels between control and exposed islets prior to stimulation with 16.7 mM glucose. For subsequent experiments, we used concentrations of arsenicals that caused significant decreases in intracellular calcium levels after stimulation with 16.7 mM glucose.

Glucose-stimulated calcium influx in control islets and in islets exposed to trivalent arsenicals. Isolated pancreatic islets were exposed to arsenicals for 48 h and stimulated with 16.7 mM glucose (indicated by arrow). Levels of intracellular calcium are shown in time course profiles as an average of 5–10 biological replicates for islets exposed to a iAs(III) at 0 (black solid line), 0.5 µM (dashed line), 1 µM (dotted line), and 2 µM (gray solid line); b MAs(III) at 0 (black solid line), 0.05 µM (dashed line), 0.1 µM (dotted line), and 0.5 µM (gray solid line); c DMAs(III) at 0 (black solid line), 0.05 µM (dashed line), 0.1 µM (dotted line), and 0.5 µM (gray solid line). Areas under the curve (d) calculated from time of glucose injection to end of run are shown as mean ± SEM for N = 5–10 biological replicates, 30–40 islets per replicate. *p < 0.05 compared to control; §p < 0.05 from 0.1 μM
Effects of trivalent arsenicals on Cav1.2 channels
Inhibition of KATP channels by increased levels of ATP causes depolarization of the β-cell membrane, stimulating the opening of L-type voltage-gated calcium channels (Cav1.2) (Wiser et al. 1999; Barg et al. 2001). To determine if the inhibition of calcium influx by arsenicals was due to inhibition of events upstream of membrane depolarization, we stimulated the islets with depolarizing levels of potassium chloride (35 mM KCl) in the absence of glucose. Treatment with KCl stimulated both Ca2+ influx and insulin secretion in control islets (Fig. 3). KCl-stimulated Ca2+ influx was significantly decreased in islets exposed to 0.5 µM DMAs(III); the decrease was marginally significant in islets exposed to 2 µM iAs(III) (p = 0.1) and 0.5 µM MAs(III) (p = 0.08) (Fig. 3a, b). All three arsenicals significantly decreased KCl-stimulated insulin secretion (Fig. 3c).

KCl-stimulated calcium influx in control pancreatic islets and islets exposed to trivalent arsenicals. Isolated pancreatic islets were exposed to arsenicals for 48 h and stimulated with 35 mM potassium chloride (KCl), a potent insulin secretagogue. Levels of intracellular calcium (a) were measured over time and are shown in time course profiles of an average of 4 biological experiments; the arrow indicates injection of KCl. Area under the curve (b) was calculated from time of KCl injection to end of run and shown as mean ± SEM of N = 4 biological replicates, 30–40 islets per replicate. Insulin secretion (c) was measured in the absence of glucose (basal) and after 1-h stimulation with KCl. N = 4 biological replicates (each assay done in triplicate, 15 islets per replicate) *p < 0.05 compared to control
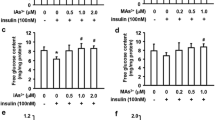
To test if arsenicals inhibit insulin secretion by affecting calcium influx through Cav1.2 channels, we measured GSIS in control and arsenical-exposed islets treated with Bay K8644, a Cav1.2 channel-specific agonist. Bay K8644 prolongs the time Cav1.2 channels are in the open state, thus potentiating insulin secretion (Panten et al. 1985). Control islets stimulated with 16.7 mM glucose and 2 µM Bay K8644 for an hour secreted more insulin than those stimulated with 16.7 mM glucose alone (Fig. 4). However, stimulation with glucose and Bay K8644 did not rescue the inhibition of insulin secretion in islets exposed to arsenicals although arsenical-exposed islets stimulated with glucose and Bay K8644 secreted more insulin than arsenical-exposed islets stimulated only with glucose.

Bay K8644-stimulated insulin secretion and Cav1.2 channel expression in control islets and in islets exposed to trivalent arsenicals. Isolated pancreatic islets were exposed to arsenicals for 48 h and stimulated for 1 h each with 2.5 mM glucose, 16.7 mM glucose, and 16.7 mM glucose with 2 µM Bay K8644. Mean ± SEM of N = 3–4 biological replicates (each assay done in triplicate, 15 islets per replicate) are shown. *p < 0.05 for comparison of arsenical-treated islets to unexposed controls. ǂp < 0.05 for the comparison of 16.7 mM glucose and 16.7 mM glucose + 2 µM Bay K8644
Given that treatment with a Cav1.2 channel-specific agonist did not restore GSIS, we evaluated mRNA levels of Cav1.2α1, the pore-forming subunit of Cav1.2 channel, in arsenical-exposed islets to determine if arsenicals were affecting the transcription Cav1.2 channels. There were no statistically significant differences in Cav1.2α1 mRNA levels between control islets and islets exposed to arsenicals (Fig. 5).

Expression of the alpha subunit of L-type calcium channels in control islets and in islets exposed to trivalent arsenicals. Isolated pancreatic islets were exposed to arsenicals for 48 h and stimulated for 1 h each with 2.5 mM glucose and 16.7 mM glucose. Relative quantitative value (RQV) of the expression of the alpha subunit of Cav1.2 channels (Cacna1c) after GSIS is shown as mean ± SEM for N = 4 biological replicates
Effects of trivalent arsenicals on the KATP channel
Closure of KATP channels by ATP prevents influx of potassium ions and triggers membrane depolarization. To test if arsenicals affect KATP channel-associated membrane depolarization, we measured insulin secretion from control and arsenical-exposed islets after glucose stimulation and treatment with tolbutamide, a KATP channel blocker. Treatment with tolbutamide further increased insulin secretion in glucose-stimulated control islets and islets exposed to 2 µM iAs(III), 0.5 µM MAs(III) and 0.5 µM DMAs(III) (Fig. 6). These exposures inhibited insulin secretion in islets stimulated with glucose but had no significant effects on insulin secretion in glucose-stimulated islets treated with tolbutamide. The inhibitory effect of 2 µM iAs(III) was only marginally significant (p = 0.11).

Tolbutamide-stimulated insulin secretion in control islets and in islets exposed to trivalent arsenicals. Isolated pancreatic islets were exposed to arsenicals for 48 h and stimulated for 1 h each with 2.5 mM glucose, 16.7 mM glucose, and 16.7 mM glucose + 200 µM tolbutamide. Mean ± SEM of N = 3–4 biological replicates (each assay done in triplicate, 15 islets per replicate) are shown. *p < 0.05 for comparison of arsenical-treated islets to unexposed controls. ǂp < 0.05 for the comparison of 16.7 mM glucose and 16.7 mM glucose + 200 µM tolbutamide
Discussion
Exposure to iAs has been associated with increased prevalence or incidence of T2D in many populations around the world (Maull et al. 2012; Kuo et al. 2017). Experimental studies show that iAs(III) and its methylated trivalent metabolites inhibit insulin secretion in isolated pancreatic islets and cultured β-cells (Douillet et al. 2013; Fu et al. 2010; Díaz-Villaseñor et al. 2008; Dover et al. 2017). Some mechanisms for this inhibition have been proposed. For example, chronic exposure to iAs(III) has been shown to upregulate Nrf2, an oxidative stress-sensitive transcription factor, and Nrf2-dependent transcription of antioxidant enzymes in rat insulinoma INS-1 β-cells and other cell types (Lau et al. 2013). Fu et al. proposed that stimulation of antioxidant defenses after iAs(III) exposure suppressed reactive oxygen species that are thought to be involved in the regulation of GSIS (Fu et al. 2010). Dover et al. found that exposure to iAs(III) and MAs(III), but not DMAs(III), impaired mitochondrial respiration in glucose-stimulated INS-1 832/13 cells (Dover et al. 2017), suggesting an impairment in ATP production, one of the critical steps in GSIS regulation. In 2008, Díaz-Villaseñor et al. showed that exposure of rat insulinoma (RINm5F) cells to 0.5–2 µM iAs(III) reduced glucose-stimulated Ca2+ oscillations and increased calpain-10 activity and SNAP-25 proteolysis, proposed mechanisms involved in granule exocytosis (Díaz-Villaseñor et al. 2008).
In this study, we found that iAs(III) and its methylated trivalent metabolites inhibited glucose-stimulated Ca2+ influx in isolated pancreatic islets at concentrations that also inhibit GSIS, but do not impair islet viability. We also found that DMAs(III), and to a lesser extent iAs(III) and MAs(III), inhibited Ca2+ influx and insulin secretion in islets stimulated with a membrane depolarizing concentration of KCl, suggesting that arsenicals affect insulin-regulating mechanisms downstream from membrane depolarization. Furthermore, stimulation with the Cav1.2 channel agonist, Bay K8644, did not restore GSIS in islets exposed to the arsenicals, and the 48-h exposure did not suppress the Cav1.2 mRNA expression. These data suggest that iAs(III), MAs(III) and DMAs(III) may target Cav1.2 channels, thus disrupting the Ca2+-dependent steps in the assembly and/or exocytosis of insulin vesicles.
Since treatment with a Cav1.2 channel agonist did not restore GSIS in arsenical-exposed islets, arsenicals may be interfering with the Cav1.2 channel by modifying its trafficking and/or by inhibiting activity/conductance of the Cav1.2 channels within the plasma membrane of β-cells. Cav1.2 channel expression at the plasma membrane and its activity are regulated by various proteins, including its auxiliary subunits, kinases, and exocytotic proteins. Auxiliary subunits of calcium channels regulate channel translocation to the membrane or channel activity (Dolphin 2012). In β-cells, the α2 subunit is bound to the δ subunit through a disulfide bond (Dolphin 2012). This α2–δ complex then binds to core α1 subunits and can increase plasma membrane expression of Cav1.2, leading to increased glucose-stimulated calcium influx (Dolphin 2012; Yang and Berggren 2006; Gao et al. 2000). Since trivalent arsenicals bind readily to thiols, they could be interfering with the formation of the α2–δ complex and inhibiting channel translocation.
Cav1.2 channel trafficking and activity are also regulated by various kinases (reviewed in Yang and Berggren 2006). In islets and β-cells, inhibition of Ca2+/calmodulin-dependent kinase II decreased insulin secretion, paralleled by a decrease in calcium influx (Dadi et al. 2014). Protein kinase B (Akt) upregulates Cav1.2 channels in excitable cells; phosphorylation by Akt promotes Cav1.2 stabilization and increases trafficking to the plasma membrane (Catalucci et al. 2009; Viard et al. 2004). Akt kinases are expressed in β-cells and have been reported to be a positive regulator of insulin secretion (Cui et al. 2012). We have previously shown that iAs(III) and its trivalent methylated metabolites, MAs(III) and DMAs(III), inhibit the phosphorylation/activation of Akt and the Akt-mediated signaling in adipocytes (Paul et al. 2007) and hepatocytes (Zhang et al. 2017). Thus, the inhibition of Akt phosphorylation could contribute to the inhibition of Ca2+ influx in the islets exposed to trivalent arsenicals.
Cav1.2 channel activity is also regulated by exocytotic proteins. For example, syntaxin 1A was found to decrease the amplitude of the Ca2+ current through the channel and modify the intrinsic kinetic properties of the channel (e.g., rate of activation and inactivation) (Wiser et al. 1996; Atlas 2001). Conversely, another exocytotic protein, SNAP-25, modifies the kinetic properties of L-type channels by increasing the rate of inactivation without affecting current amplitude (Atlas 2001). There are numerous regulators of Cav1.2 channel activity and expression. Measurement of calcium flux across the Cav1.2 channel via the gold-standard patch clamp technique would be needed to determine if arsenicals impair flow of Ca2+ through channels or modify expression of Cav1.2 channel at the membrane.
Tolbutamide is similar to KCl in that it stimulates membrane depolarization. However, tolbutamide does so by blocking KATP channels at the SUR1 subunit, a mechanism that is more physiologically relevant than modifying ion levels with high concentrations of KCl (Ashfield et al. 1999). In this study, exposure to iAs(III), MAs(III), and DMAs(III) had little or no effects on insulin secretion from glucose-stimulated islets treated with tolbutamide, suggesting that these arsenicals may prevent membrane depolarization by inhibiting the closure of KATP channels. The KATP channel has two subunits that regulate channel closure: ATP interacts with the Kir6.2 subunit and tolbutamide interacts with the sulfonylurea receptor protein 1 (SUR1) (Babenko et al. 1999). Since GSIS was restored with tolbutamide exposure, these arsenicals may be interfering with ATP–KATP channel interactions at the Kir6.2 subunit, effects which could be overcome by tolbutamide stimulation. Suppression of ATP activation of KATP channels by trivalent arsenicals could also explain why Bay K8644-stimulation did not restore insulin secretion. During glucose stimulation, the membrane potential of the β-cell membrane oscillates between depolarizing and non-depolarizing levels, thus Cav1.2 channels oscillate between open and closed states, leading to oscillations in intracellular Ca2+ levels during glucose stimulation (Henquin 2009). If arsenicals are inhibiting KATP closure, the membrane is spending less time at a depolarizing voltage, leading to less time Cav1.2 channels are open and lower calcium influx. Thus, although Bay K8644 maintains open Cav1.2 channels, there may be fewer open Cav1.2 channels in arsenical-exposed islets than unexposed islets due to the impairments in membrane depolarization, leading to less insulin secretion despite Bay K8644 treatment.
The restoration of GSIS by tolbutamide in arsenical-exposed islets appears inconsistent with the inhibition of Ca2+ influx and/or downstream mechanisms. However, tolbutamide has been shown to stimulate insulin secretion apart from its effects on the KATP channel (Barg et al. 1999). SURs have been also found in insulin granules and are thought to contribute to the acidification of insulin granules, a priming step needed for exocytosis (Eliasson et al. 2003). Thus, tolbutamide may be able to restore insulin secretion via this Ca2+-independent pathway.
Tolbutamide did not restore GSIS in iAs(III)-exposed islets to the same extent as in islets exposed to methylated arsenicals, suggesting that other actions of iAs(III) in the β-cell may be more important for inhibiting insulin secretion. Studies have found that iAs(III) modifies F-actin remodeling in vitro (Izdebska et al. 2013, 2014; Qian et al. 2005). In the β-cell, iAs(III) could be interfering with the remodeling of β-actin microfilaments that are needed for insulin granule transport, priming, and release (Wang and Thurmond 2009). If iAs(III) reduces the availability of releasable insulin granules, it is conceivable that tolbutamide stimulation would not be able to overcome the effects of iAs(III). Future studies should investigate this potential mechanism.
Conclusion
We found that iAs(III) and its methylated trivalent metabolites (MAs(III) and DMAs(III)) impair glucose-stimulated Ca2+ influx in isolated pancreatic islets, at concentrations that also inhibit GSIS but do not affect islet viability. The trivalent methylated metabolites of iAs have been previously shown to be more potent inhibitors of GSIS than iAs(III). Here, we show that one of these metabolites, MAs(III), is more potent than iAs(III) as an inhibitor of glucose-stimulated Ca2+ influx in islets. DMAs(III), which is as potent as MAs(III) in inhibiting GSIS, was less effective than MAs(III) as inhibitor of Ca2+ influx. Thus, DMAs(III) may be targeting other mechanisms involved in the regulation GSIS in β-cells. Future studies should focus on the identification of these targets to further characterize the role of iAs metabolism in the development of arsenic-associated diabetes.
References
Ashfield R, Gribble FM, Ashcroft SJ, Ashcroft FM (1999) Identification of the high-affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes 48:1341–1347
Atlas D (2001) Functional and physical coupling of voltage-sensitive calcium channels with exocytotic proteins: ramifications for the secretion mechanism. J Neurochem 77:972–985
Babenko AP, Gonzalez G, Bryan J (1999) The tolbutamide site of SUR1 and a mechanism for its functional coupling to KATP channel closure. FEBS Lett 459:367–376
Barg S, Renström E, Berggren PO, Bertorello A, Bokvist K, Braun M, Eliasson L, Holmes WE, Köhler M, Rorsman P et al (1999) The stimulatory action of tolbutamide on Ca2+-dependent exocytosis in pancreatic beta cells is mediated by a 65-kDa mdr-like P-glycoprotein. Proc Natl Acad Sci USA 96:5539–5544
Barg S, Ma X, Eliasson L, Galvanovskis J, Göpel SO, Obermüller S, Platzer J, Renström E, Trus M, Atlas D et al (2001) Fast exocytosis with few Ca(2+) channels in insulin-secreting mouse pancreatic B cells. Biophys J 81:3308–3323
Catalucci D, Zhang DH, DeSantiago J et al (2009) Akt regulates L-type Ca2+ channel activity by modulating CaValpha1 protein stability. J Cell Biol 184(6):923–933
Chen L, Magliano DJ, Zimmet PZ (2012) The worldwide epidemiology of type 2 diabetes mellitus–present and future perspectives. Nat Rev Endocrinol 8:228–236
Cui X, Yang G, Pan M, Zhang X-N, Yang S-N (2012) Akt signals upstream of L-type calcium channels to optimize insulin secretion. Pancreas 41:15–21
Dadi PK, Vierra NC, Ustione A, Piston DW, Colbran RJ, Jacobson DA (2014) Inhibition of pancreatic β-Cell Ca2+/calmodulin-dependent protein kinase II reduces glucose-stimulated calcium influx and insulin secretion, impairing glucose tolerance. J Biol Chem 289:12435–12445
Del Razo LM, García-Vargas GG, Valenzuela OL, Castellanos EH, Sánchez-Peña LC, Currier JM, Drobná Z, Loomis D, Stýblo M (2011) Exposure to arsenic in drinking water is associated with increased prevalence of diabetes: a cross-sectional study in the Zimapán and Lagunera regions in Mexico. Environ Health 10:73
Díaz-Villaseñor A, Burns AL, Salazar AM, Sordo M, Hiriart M, Cebrián ME, Ostrosky-Wegman P (2008) Arsenite reduces insulin secretion in rat pancreatic beta-cells by decreasing the calcium-dependent calpain-10 proteolysis of SNAP-25. Toxicol Appl Pharmacol 231:291–299
Dolphin AC (2012) Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat Rev Neurosci 13:542–555
Douillet C, Currier J, Saunders J, Bodnar WM, Matousek T, Stýblo M (2013) Methylated trivalent arsenicals are potent inhibitors of glucose stimulated insulin secretion by murine pancreatic islets. Toxicol Appl Pharmacol 267:11–15
Dover EN, Beck R, Huang MC, Douillet C, Wang Z, Klett EL, Stýblo M (2017) Arsenite and methylarsonite inhibit mitochondrial metabolism and glucose-stimulated insulin secretion in INS-1 832/13 β cells. Arch, Toxicol
Eliasson L, Ma X, Renström E, Barg S, Berggren P-O, Galvanovskis J, Gromada J, Jing X, Lundquist I, Salehi A et al (2003) SUR1 regulates PKA-independent cAMP-induced granule priming in mouse pancreatic B-cells. J Gen Physiol 121:181–197
Fu J, Woods CG, Yehuda-Shnaidman E, Zhang Q, Wong V, Collins S, Sun G, Andersen ME, Pi J (2010) Low-level arsenic impairs glucose-stimulated insulin secretion in pancreatic beta cells: involvement of cellular adaptive response to oxidative stress. Environ Health Perspect 118:864–870
Gao B, Sekido Y, Maximov A, Saad M, Forgacs E, Latif F, Wei MH, Lerman M, Lee JH, Perez-Reyes E et al (2000) Functional properties of a new voltage-dependent calcium channel alpha(2)delta auxiliary subunit gene (CACNA2D2). J Biol Chem 275:12237–12242
Gribble MO, Howard BV, Umans JG, Shara NM, Francesconi KA, Goessler W, Crainiceanu CM, Silbergeld EK, Guallar E, Navas-Acien A (2012) Arsenic exposure, diabetes prevalence, and diabetes control in the Strong Heart Study. Am J Epidemiol 176:865–874
Henquin JC (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49:1751–1760
Henquin JC (2009) Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 52:739
Izdebska M, Gagat M, Grzanka D, Grzanka A (2013) Ultrastructural localization of F-actin using phalloidin and quantum dots in HL-60 promyelocytic leukemia cell line after cell death induction by arsenic trioxide. Acta Histochem 115:487–495
Izdebska M, Klimaszewska-Wiśniewska A, Lewandowski D, Nowak JM, Gagat M, Grzanka A (2014) Arsenic trioxide preferentially induces nonapoptotic cell deaths as well as actin cytoskeleton rearrangement in the CHO AA8 cell line. Postępy Higieny i Medycyny Doświadczalnej 68:1492–1500
Kuo C-C, Moon KA, Wang S-L, Silbergeld E, Navas-Acien A (2017) The association of arsenic metabolism with cancer, cardiovascular disease, and diabetes: a systematic review of the epidemiological evidence. Environ Health Perspect 125:087001
Lau A, Whitman SA, Jaramillo MC, Zhang DD (2013) Arsenic-mediated activation of the Nrf2-Keap1 antioxidant pathway. J Biochem Mol Toxicol 27:99–105
Martin EM, Stýblo M, Fry RC (2017) Genetic and epigenetic mechanisms underlying arsenic-associated diabetes mellitus: a perspective of the current evidence. Epigenomics 9:701–710
Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, Pi J, Silbergeld EK, Stýblo M, Tseng C-H, Thayer KA et al (2012) Evaluation of the association between arsenic and diabetes: a National Toxicology Program Workshop Review. Environ Health Perspect 120:1658–1670
Panten U, Zielmann S, Schrader M-T, Lenzen S (1985) The dihydropyridine derivative, Bay K 8644, enhances insulin secretion by isolated pancreatic islets. Naunyn-Schmiedeberg’s Arch Pharmacol 328:351–353
Paul DS, Harmon AW, Devesa V, Thomas DJ, Stýblo M (2007) Molecular mechanisms of the diabetogenic effects of arsenic: inhibition of insulin signaling by arsenite and methylarsonous acid. Environ Health Perspect 115:734–742
Peng Q, Harlow SD, Park SK (2015) Urinary arsenic and insulin resistance in US Adolescents. Int J Hyg Environ Health 218:407–413
Prentki M, Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J Clin. Invest 116:1802–1812
Qian Y, Liu KJ, Chen Y, Flynn DC, Castranova V, Shi X (2005) Cdc42 regulates arsenic-induced NADPH oxidase activation and cell migration through actin filament reorganization. J Biol Chem 280:3875–3884
Rhee SY, Hwang Y-C, Woo J, Chin SO, Chon S, Kim YS (2013) Arsenic exposure and prevalence of diabetes mellitus in Korean Adults. J Korean Med Sci 28:861–868
Santulli G, Pagano G, Sardu C, Xie W, Reiken S, D’Ascia SL, Cannone M, Marziliano N, Trimarco B, Guise TA et al (2015) Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J Clin Invest 125:1968–1978
Stýblo M, Drobná Z, Jaspers I, Lin S, Thomas DJ (2002) The role of biomethylation in toxicity and carcinogenicity of arsenic: a research update. Environ Health Perspect 110:767–771
Thayer KA, Heindel JJ, Bucher JR, Gallo MA (2012) Role of environmental chemicals in diabetes and obesity: a National Toxicology Program Workshop Review. Environ Health Perspect 120:779–789
Viard P, Butcher AJ, Halet G et al (2004) PI3 K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat Neurosci 7:939–994
Wang Z, Thurmond DC (2009) Mechanisms of biphasic insulin-granule exocytosis—roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci 122:893–903
Wiser O, Bennett MK, Atlas D (1996) Functional interaction of syntaxin and SNAP-25 with voltage sensitive L- and N-type Ca2 + channels. EMBO J 15:4100–4110
Wiser O, Trus M, Hernández A, Renström E, Barg S, Rorsman P, Atlas D (1999) The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc Natl Acad Sci USA 96:248–253
Wollheim CB, Sharp GW (1981) Regulation of insulin release by calcium. Physiol Rev 61:914–973
World Health Organization (2016) Global report on diabetes. WHO Library Cataloguing-in-Publication Data. http://apps.who.int/iris/bitstream/10665/204871/1/9789241565257_eng.pdf
Yang S-N, Berggren P-O (2006) The role of voltage-gated calcium channels in pancreatic β-cell physiology and pathophysiology. Endocr Rev 27:621–676
Zhang C, Fennel EMJ, Douillet C, Stýblo M (2017) Exposures to arsenite and methylarsonite produce insulin resistance and impair insulin-dependent glycogen metabolism in hepatocytes. Arch Toxicol 91:3811–3821
Funding
This work was supported by a grant from the National Institutes of Environmental Health sciences [R01ES022697] to M.S., a National Research Service Award from the National Institute of Environmental Health Sciences [T32 ES007126] to M.H., and the UNC Nutrition Obesity Research Center funded by the National Institute of Diabetes and Digestive and Kidney Diseases [DK056350].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Huang, M., Douillet, C. & Stýblo, M. Arsenite and its trivalent methylated metabolites inhibit glucose-stimulated calcium influx and insulin secretion in murine pancreatic islets. Arch Toxicol 93, 2525–2533 (2019). https://doi.org/10.1007/s00204-019-02526-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-019-02526-2