
Abstract
The liver is a vulnerable target for amphetamine toxicity, but the mechanisms involved in the drug’s hepatotoxicity remain poorly understood. The purpose of the current research was to characterize the mode of death elicited by four amphetamines and to evaluate whether their combination triggered similar mechanisms in immortalized human HepG2 cells. The obtained data revealed a time- and temperature-dependent mortality of HepG2 cells exposed to 3,4-methylenedioxymethamphetamine (MDMA, ecstasy; 1.3 mM), methamphetamine (3 mM), 4-methylthioamphetamine (0.5 mM) and d-amphetamine (1.7 mM), alone or combined (1.6 mM mixture). At physiological temperature (37 °C), 24-h exposures caused HepG2 death preferentially by apoptosis, while a rise to 40.5 °C favoured necrosis. ATP levels remained unaltered when the drugs where tested at normothermia, but incubation at 40.5 °C provoked marked ATP depletion for all treatments. Further investigations on the apoptotic mechanisms triggered by the drugs (alone or combined) showed a decline in BCL-2 and BCL- XL mRNA levels, with concurrent upregulation of BAX, BIM, PUMA and BID genes. Elevation of Bax, cleaved Bid, Puma, Bak and Bim protein levels was also seen. To the best of our knowledge, Puma, Bim and Bak have never been linked with the toxicity induced by amphetamines. Time-dependent caspase-3/-7 activation, but not mitochondrial membrane potential (∆ψm) disruption, also mediated amphetamine-induced apoptosis. The cell dismantling was confirmed by poly(ADP-ribose)polymerase proteolysis. Overall, for all evaluated parameters, no relevant differences were detected between individual amphetamines and the mixture (all tested at equieffective cytotoxic concentrations), suggesting that the mode of action of the amphetamines in combination does not deviate from the mode of action of the drugs individually, when eliciting HepG2 cell death.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
It has been widely recognized that 3,4-methylenedioxymethamphetamine (MDMA, Ecstasy) is hepatotoxic to humans, in spite of this toxicity being random and associated with variable clinical outcomes, ranging from asymptomatic elevation of hepatic enzymes (ALT, AST, alkaline phosphatase) to lethal liver failure (Andreu et al. 1998; Dykhuizen et al. 1995; Ellis et al. 1996; Henry et al. 1992; Khakoo et al. 1995). The underlying mechanisms are not yet fully understood, but a plethora of factors has been proposed to justify these effects, including hyperthermia, drug–drug interactions from polydrug abuse, altered efflux of neurotransmitters, oxidation of biogenic amines, impairment of mitochondrial function, direct effect of reactive metabolites, immune responses and activation of apoptotic pathways (Antolino-Lobo et al. 2011; Carvalho et al. 2010, 2012).
Our group’s previous studies have supported the fact that hepatotoxicity can be exacerbated by the combination of MDMA with other amphetamines, which are either intentionally ingested or present in ecstasy pills as contaminants. This toxicity was shown to occur even for combinations of the drugs at concentrations that individually produce no detectable effects (Dias da Silva et al. 2013b, d). In addition, we have shown that temperatures higher than those physiologically normal, such as those reached in vivo after amphetamine intake, further enhance the hepatotoxicity of amphetamines, as well as of their mixtures (Dias da Silva et al. 2013d). The impairment of thermoregulation elicited by amphetamines, combined with excessive dancing and inadequate fluid intake, as well as the hot, overcrowded venues where the drugs intake often occurs has been shown to induce dramatic increases in body temperature, with reported values ranging between 40.0 and 43.3 °C (Armenian et al. 2012; Coore 1996; Green et al. 2003; Henry et al. 1992). Amphetamine-induced hyperthermia has been associated with the most serious detrimental health effects of these drugs and has been linked to a high number of intoxications (Fineschi et al. 1999; Henry et al. 1992; Milroy 2011; Strobbe et al. 2007; Vanden Eede et al. 2012; Wirth et al. 2007). In fact, high body temperature has been described as one of the most aggressive physiological consequences of amphetamines in acute poisonings and has been recurrently pointed out as a cause for life-threatening complications, such as acute liver failure, rhabdomyolysis, disseminated intravascular coagulation, acidosis and multiple organ failure (Armenian et al. 2012; Henry et al. 1992; Kalant 2001; Kendrick et al. 1977; Patel et al. 2005; Screaton et al. 1992).
The hepatotoxicity of amphetamines has been associated with an increase in apoptosis, and the mechanisms involved in this effect include a decrease in the levels of anti-apoptotic genes and related proteins, such as Bcl-2 and Bcl-XL (De Miglio et al. 2000; Jayanthi et al. 2001; Montiel-Duarte et al. 2002; Upreti et al. 2011), an overexpression of pro-apoptotic factors, like Bax and Bad, Bid and p53 (De Miglio et al. 2000; Genc et al. 2003; Jayanthi et al. 2001; Cerretani et al. 2011), the release of cytochrome c (cyt c), SMAC/DIABLO and apoptosis inducing factor (AIF) from the mitochondria (Beauvais et al. 2011; Montiel-Duarte et al. 2002; Cerretani et al. 2011), the activation of caspase-3 (Montiel-Duarte et al. 2002), the proteolysis of poly(ADP-ribose)polymerase (PARP) (Montiel-Duarte et al. 2002), the condensation of nuclei chromatin (Montiel-Duarte et al. 2002) and the accumulation of oligonucleosomal fragments in the cytoplasm (Montiel-Duarte et al. 2002). Upreti et al. (2011) also demonstrated that MDMA activates c-Jun N-terminal protein kinase and p38 kinase. These protein kinases phosphorylate the anti-apoptotic Bcl-2 protein, so, indirectly, their activation may promote apoptosis in MDMA-exposed tissues.
Conversely, numerous clinical hepatotoxic descriptions of amphetamines correlate with morphological evidence of acute necrosis, rather than apoptosis, particularly in cases of intoxicated patients who died or required liver transplantations (Brauer et al. 1997; Garbino et al. 2001; Ibranyi and Schonleber 2003; Kamijo et al. 2002).
It is clear from the aforementioned studies that the relative contribution of apoptosis and/or necrosis to amphetamine-induced liver injury remains controversial, as evidence for both types of cell death has been reported in hepatocytes. In addition to the uncertainties surrounding the precise mechanisms of cytotoxic action of individual amphetamines is the question of whether the mode of action of these drugs remains unaltered when they are combined. Does the simple combination of these drugs impact on the mode of cell death? Furthermore, taking into account the substantial increase in body temperature that often occurs with amphetamine abuse, would the mechanism of cell death, for both the individual and the mixed amphetamines, also be affected in a hyperthermic situation?
In order to address these questions and provide new insights into the mechanisms underlying the cytotoxicity linked to amphetamine intoxications, the present study attempted to delineate the mechanisms of cell death under normothermic (37 °C) and hyperthermic (40.5 °C) conditions caused by MDMA, methamphetamine (METH), 4-methylthioamphetamine (4-MTA), d-amphetamine (d-AMP) and their mixture in HEPG2 cells. For that, we evaluated the mode of HepG2 cell death (apoptosis or necrosis) and the cellular energetic status following drug treatments. Additionally, the relative mRNA expressions of BCL-2, BCL- XL , BAX, BIM, BAD, PUMA and BID, as well as levels of PARP/cleaved PARP, Bak, Puma, Bim (EL, S and L isoforms), Bid/cleaved Bid and Bax proteins were assessed at 37 °C (the temperature condition showing higher apoptosis rates). Ultimately, and since the tested drugs induced differential expression of anti- and pro-apoptotic BCL-2 family members, we also tested impairment of mitochondrial permeability.
In previous work, we evaluated the cytotoxic effects of a mixture of the four amphetamines here tested using the MTT assay and demonstrated that all four amphetaminic mixture components acted in an additive way to induce the overall mixture effect. Also, as the toxicity of the mixture could be accurately predicted by the mathematical model of concentration addition, it was suggested that all tested drugs acted via a similar mechanism of action (Dias da Silva et al. 2013b). In the work presented here, it was important to select test concentrations (of individual amphetamines and mixture) that produced an equal level of cell death, i.e. were equipotents. In doing so, we ensured an easy and direct comparison between individual drugs and their mixture in the outcomes evaluated. Based on the additivity interactions shown previously, it was expected that all chemicals would have the same mechanism of action, and therefore, the equipotency of all tested drugs, as well as of the mixture, should be maintained in all studied endpoints. A deviation from those would suggest a difference in mechanism of action, rather than a difference in potency.
Overall, a better understanding of the cellular and molecular mechanisms involved in amphetamine toxicity should help to generate more adequate therapeutic approaches to lessen the long-term consequences of amphetamine use.
Materials and methods
Test drugs
All chemicals used were of analytical grade. d-AMP (sulphate salt) was purchased from Tocris Bioscience (Bristol, UK), and (+)-METH hydrochloride (98 % purity) was purchased from Sigma-Aldrich, Co. (St. Louis, MO, USA). MDMA (HCl salt) was extracted and purified from high purity MDMA tablets provided by the Portuguese Criminal Police Department, at REQUIMTE/Toxicology Laboratory, Biological Sciences Department of Faculty of Pharmacy, University of Porto. The obtained salts were purified and fully characterized by nuclear magnetic resonance (NMR) and mass spectrometry (MS) methodologies. 4-MTA hydrochloride (racemic mixture) was generously provided by Dr. David Nichols (Purdue University, West Lafayette, IN).
All chemicals were used as supplied. Stock solutions were prepared in ultra purified water and kept at −20 °C. Dilutions were freshly prepared before each experiment and were at least 20 times more concentrated than the highest concentration tested, so that media dilution was avoided.
HepG2 cells culture
HepG2 cells were kindly provided by Dr. Maryam Modarai from UCL School of Pharmacy, London, UK. HepG2 cells were routinely maintained in 75 cm2 canted-neck tissue culture flasks (Helena Biosciences, Gateshead, UK) in minimum essential medium alpha (MEM Alpha) with GlutMAX supplemented with 10 % foetal bovine serum (FBS), 1 % antibiotic (5,000 U/ml penicillin, 5,000 μg/ml streptomycin), 0.5 % fungizone (250 μg/ml amphotericin B) and 0.0015 % human transferrin (4 mg/ml) in a humidified incubator, at 37 °C, with 5 % CO2. Cells were subcultured at approximately 70 % confluence over a maximum of 10 passages and regularly tested negative for Mycoplasma. All the reagents used in routine cell culture, including 0.05 % trypsin/1 mM EDTA and Hanks balanced salt solution (HBSS) without Ca and Mg were obtained from GIBCO, Invitrogen Corporations (Paisley, UK), unless stated otherwise.
Mixture design
The tested mixture was planned as described before (Dias da Silva et al. 2013b). The composition of the mixture was based on parameters derived from previous experimental studies, using the tetrazolium-based (MTT) viability assay (Dias da Silva et al. 2013b) and is shown in Table 1. The ratio between the four amphetamines was based on the concentrations eliciting 50 % of the maximal mortality in the MTT assay (EC50), after 48 h incubations at 37 °C. This mixture design ensures that all mixture components are present at equipotent concentrations, thus preventing non-proportional individual contributions.
Drug treatments
The individual drugs and the mixture were tested at concentrations that induced 25 % of the maximum effect observed in the MTT reduction assay (EC25), i.e. 1.3 mM MDMA, 3 mM METH, 0.5 mM 4-MTA, 1.7 mM d-AMP and 1.6 mM mixture (Dias da Silva et al. 2013b) (Table 1). This effect level was selected as it corresponds to some degree of toxicity by moderately decreasing cell metabolic competence/viability, allowing the assessment of mechanisms of action and avoiding high levels of mortality.
Neutral red uptake for cell viability assessment
The neutral red (NR) viability assay is based on the ability of viable cells to incorporate and subsequently retain the supravital dye neutral red (3-amino-m-dimethylamino-2-methylphenazine hydrochloride; Sigma-Aldrich).
8 × 104 cells were seeded per well on the 96-well plates and allowed to attach. After 24 h, the culture medium was replaced by treatment solutions (six replicates), freshly prepared in cell culture medium. Each plate also included a negative control. After the incubation periods (6, 12, 24 and 48 h), at 37 °C (physiological temperature) or 40.5 °C (hyperthermic conditions), the culture medium was removed and the plate was rinsed with HBSS. 100 μl medium containing neutral red (50 μg/ml) was added to each well. After 2 h of incubation at 37 °C, the medium was removed and the plate was rinsed twice with HBSS. Then, 200 μl lysis solution, containing 1 % glacial acetic acid, 50 % ethanol and 49 % H2O, was added to each well and shaken for 15 min in the dark. The absorbance (Abs) of the coloured solution was measured with a multi-well plate reader (Labsystems Multiskan, Multisoft Primary EIA V.1.3-0, Basingstoke, UK) at 540 nm and is directly correlated to the number of cells with an intact membrane and unimpaired lysosomal activity. To reduce inter-experimental variability, data were normalized on a plate-by-plate basis and scaled between 0 % (negative controls) and 100 % effect (positive controls consisting of culture media containing 1 %Triton X-100). Results were graphically presented as percentage of cell death versus time (h).
ImageStream multi-spectral imaging flow cytometry
In order to distinguish between live, necrotic, and early and late apoptotic cells, the combination propidium iodide (PI)/fluorescein-labelled Annexin V (AnV-FITC) was used.
The type of cell death elicited by amphetamines, isolated or in mixture, on HepG2 cells, and the influence of temperature and time of exposure were evaluated by seeding 1 × 106 cells onto 10 cm2 petri dishes and left to attach for 24 h. Then, the cells were exposed to the test solutions and incubated at 37 °C or at 40.5 °C, for 24 or 48 h. At the end of the treatment periods, the cells were rinsed twice with HBSS and trypsinized for 5 min with 300 μl 0.05 % Trypsin–EDTA, at 37 °C. The cell culture media were kept and processed since adherent cells undergoing apoptosis may detach from the culture surface. Cells were washed in cold HBSS at 500×g for 5 min (pre-cooled centrifuge, at 4 °C), and the supernatant was discarded. The cell pellet was resuspended in approximately 100 μl of 1x annexin-binding buffer (BD Pharmingen, Oxford, UK). Cell counting was carried out using a Neubauer chamber and the density of the cell suspension determined. Final cell density was then adjusted to ~1 × 107 cells per ml. The samples were stained with 5 μl AnV-FITC and 5 μl PI (BD Pharmigena. Oxford, UK). After gently vortexing, cells were incubated for 15 min, at room temperature (RT, 25 °C), in the dark. All the cell preparation procedures were carried out with care to maintain cellular integrity. The samples were placed on ice, and the analysis was performed within 1 h on the ImageStream multi-spectral imaging flow cytometer (Amnis Corporation, Seattle, WA). The system provides both photometric and morphometric classifications, as it enables visualization of individual cells passing through the flow chamber, offering additional information regarding morphology and spatial distribution of each cell. At least 5–10 × 104 images were collected for each sample, and images were divided into four subgroups: live cells (unstained), cells at early apoptotic state (stained with AnV-FITC), cells at late apoptotic state (stained by both AnV-FITC and PI) and necrotic cells (stained with PI). Debris, including cell doublets and clumps, was eliminated from the data as the machine gates around only single cells for analysis. In flow cytometry, spectral overlap between stains is a potential source of error. To account for this, we performed compensation in line with the standard procedure used for the Imagestream (Ortyn et al. 2006). Briefly, a non-treated sample was divided into 3 tubes corresponding to negative control (AnV−/PI−, without staining), AnV-positive control (AnV +/PI − , stained only with AnV) and PI-positive control (AnV−/PI+, stained only with PI). The samples for each individual stain were run separately, and results acquired for 1,000 events were processed using the compensation matrix feature of the ImageStream data analysis and exploration software (IDEAS 5.0). The compensation matrix was validated according to the matrix coefficient intensity plots.
ATP content
Cellular ATP levels can be rapidly detected through a bioluminescent luciferase-based assay, since luciferase catalyses the formation of light from the reaction of ATP and luciferin. Briefly, 1 × 106 cells were seeded onto 10 cm2 petri dishes to obtain confluent monolayers 24 h later. On the day of the experiment, the media were gently aspirated and the cells were incubated for 24 h with MDMA, METH, 4-MTA, d-AMP and mixture dilutions prepared in fresh medium, at 37 °C or at 40.5 °C. Then, the cells were rinsed twice with HBSS and scrapped/precipitated with 5 % perchloric acid (HClO4, w/v). After centrifugation for 5 min at 16,000×g (4 °C), the supernatants were kept frozen at −80 °C until further quantification. The pellet was used for protein quantification. The thawed acidic supernatant was neutralized with an optimized equal volume of 0.76 M potassium bicarbonate (KHCO3) and centrifuged for 5 min at 16,000×g (4 °C). The ATP contents were measured in triplicate in 96-well white plates (Greiner CELLSTAR® from Sigma), using an ATP determination kit (Invitrogen Corporations) following the manufacturer instructions. ATP levels are linearly related to the emitted light intensity that was determined by using a luminescence microplate reader (FLUOstar Optima, BMG Labtech GmbH) and compared to a standard curve performed with each experiment. The results were normalized to the total protein amount, and the final results were expressed as nmol ATP.mg−1 protein.
Protein determination
Protein was quantified through the Bradford assay (Bradford 1976). The absorbance measurements were taken at 595 nm using a multi-well plate reader (Labsystems Multiskan, Basingstoke, UK) and were compared with a bovine serum albumin (BSA, Sigma) standard curve to assess protein amount.
Cell treatment for RNA isolation
HepG2 cells were seeded at a density of 1 × 106 cells per 10 cm2 petri dish and grown for 24 h. The cell culture media were then replaced by selected dilutions of MDMA, METH, 4-MTA, d-AMP and mixture. Non-treated cells were used as controls. Following the dosing period (1, 4, 8, 16, 24 and 48 h at 37 °C), the cells were harvested by the addition of 300 μl 0.05 % trypsin–EDTA and incubated at 37 °C for 5 min. The cell suspension was centrifuged at 500×g for 5 min, and the supernatant was removed. The pellet was immediately frozen in liquid nitrogen and stored at −80 °C until further use for RNA isolation.
Isolation and quantification of total RNA
Total RNA was isolated from cell suspensions using the Nucleospin RNA II kit (Macherey–Nagel, Abgene, Epson, UK) following the manufacturer instructions, and the obtained RNA pellets were solubilized in RNAse/DNAse free water to prevent traces of genomic contamination. The concentration and purity of the final preparations were determined after measuring the absorbances at 230, 260 and 280 nm, by calculating the 260/230 and 260/280 absorbance ratios, respectively (NanoDrop 1000-v3.7 spectrophotometer, Thermo Fisher Scientific).
Reverse transcription
Approximately 2.5 μg of total RNA was reverse transcribed into cDNA using the following protocol: 7 μl 5x RT buffer (Promega, Southampton, UK), 4 μl 10 mM dNTP mix (Agilent Technologies), 1 μl RNAse inhibitor (Promega), 1 μl hexamer random primers (Invitrogen), 2.5 μg RNA and 15 μl of RNAse/DNAse free water (Sigma-Aldrich) were mixed and heated at 62 °C for 10 min. The samples were then cooled on ice for 2 min before adding 2 μl M-MLV reverse transcriptase (Promega). The reaction was then allowed to proceed for another 90 min at 42 °C. The reverse transcribed products were stored at −80 °C until further use.
Primer design and quantitative real-time PCR (qPCR)
Primers for qPCR were designed using Beacon designer 5.1 software (Premier Biosoft International, Palo Alto USA) and purchased as high quality, purified OliGold Primers from Eurogentec Ltd. (Hampshire, UK). The sequences of the primers used are presented in Table 2.
cDNA was used in qPCR performed on the iCycler iQ qPCR detection system with the iCycler v3.1. Software (Bio-Rad Laboratories, Hertfordshire, UK). For a total volume of 20 μl per reaction, 0.8 μl cDNA and 10 μl 1x iQ SYBR Green Supermix (Bio-Rad) were mixed with 9.2 μl of an adequate dilution of forward and reverse primers mixture (1 μM stock) in a 96-well low profile PCR plate (Bio-rad). Optimized primer concentrations are listed in Table 2. Melting curves were generated for every primer pair following every qPCR to ensure that a single amplification product was obtained. Amplification was optimized for each gene with regard to temperature, and primer and template concentration (data not shown). PCR conditions were considered optimized when PCR efficiency ranged between 98 and 100 %. Relative quantification was performed using the comparative threshold cycle method. GAPDH, HPRT1 and β-ACTIN were tested as internal controls (reference genes). As neither gene’s expression changed with the treatments, HPRT1 was used to normalize gene expression throughout.
Data analysis for qPCR
Expression data were analysed in terms of relative quantification. HPRT1 was used as reference gene. Data were normalized and relatively compared to the control group according to the ΔΔCt model with the following formulas (Livak and Schmittgen 2001):
The expression ratio of the treatments compared to the controls is expected as 2−ΔΔCt. Results were displayed as fold increase over controls standardized such that values obtained in controls were set to 1.
Immunoblotting
HepG2 cells were seeded at a density of 1x106 cells per 10 cm2 petri dish and cultured for 24 h. Then, cells were incubated with the test drugs for 24 h (PARP/cleaved PARP detection) or 12 h (all other proteins), at 37 °C. After the incubation period, HepG2 cells were lysed by incubating for 20 min with a freshly prepared ice-cold Laemmli sample buffer (Laemmli 1970). Western blotting analysis was performed as described previously (Silva et al. 2010). The used antibodies (Cell Signalling Technology, Inc.) are described in Table 3. The luminescent signal was captured by ChemiDoc XRS System (Bio-Rad). To ensure that the exact same amount of protein was evenly loaded into the SDS-acrylamide gels, the membranes were stripped of the antibody and re-probed with anti-β-Actin antibody.
Stripping and re-probing of membranes
Membranes were incubated at 50 °C with stripping buffer (0.5 M Tris–HCl (pH 6.8), 20 % SDS, 0.8 % β-mercaptoethanol) for 45 min with gentle agitation. The blots were then rinsed with TBS-T, re-blocked and re-probed with anti-β-Actin antibody as described above.
Mitochondrial integrity by TMRE inclusion
Assessment of mitochondrial integrity was evaluated by measuring the tetramethylrhodamine ethyl ester (TMRE) inclusion. TMRE is a cell permeable fluorescent dye that specifically stains viable mitochondria, being accumulated in proportion to mitochondrial membrane potential (∆ψm) (Scaduto and Grotyohann 1999). Briefly, 8 × 104 HepG2 cells were seeded onto 96-well black plates (Greiner CELLSTAR® from Sigma). After 24 h, the media were gently aspirated and the cells were exposed to MDMA, METH, 4-MTA, d-AMP, as well as mixture solutions prepared in fresh medium at 37 °C or at 40.5 °C. At the end of the 24 h treatment period, cells were rinsed twice with HBSS and incubated at 37 °C with 100 μl of 2 μM TMRE (Sigma), for 30 min. As TMRE is a non-water-soluble powder, a 2 mM stock solution was initially prepared in DMSO (Merck) and stored in the dark. The final 2 μM concentration was prepared in fresh culture media, shortly before usage (the final concentration of DMSO did not exceed 0.5 %). Afterwards, the media were gently aspirated and replaced by 0.2 % BSA in HBSS. Fluorescence was measured at 37 °C on a fluorescence microplate reader (FLUOstar Optima, BMG Labtech GmbH) set to 544 nm excitation and 590 nm emission wavelengths. The data obtained for the individual drug and mixture treatments were normalized on a plate-by-plate basis to the values of the respective controls and calculated as the percentage of control conditions at 37 °C or 40.5 °C. This normalization is required to minimize the variability among the individual plate readings and has been extensively described elsewhere (Rajapakse et al. 2004).
Measurement of caspase-3/7 activity
Caspase-3/7 activation assay was performed using a Caspase-Glo 3/7 assay kit according to the manufacturer’s instructions (Promega, Southampton, UK). Briefly, HepG2 cells were seeded onto Greiner Bio-One CELLSTAR® 96-well white plates (Fisher, UK) at a density of 104 cells/well. After 24 h, the cells were exposed to the 4 tested amphetamines, individually or in mixture at 37 °C. Each plate also included a blank (medium alone), a negative control (untreated cells) and a positive control (15 μM Mitomycin C, MMC, derived from Streptomyces caespitosus, Sigma-Aldrich). All the treatments were run at least in triplicate. Following the dosing period (1, 4, 8, 16, 24, 36 and 48 h), Caspase-Glo 3/7 reagent was added to each well. The plate was then incubated in the dark at RT, for 1 h, and the luminescence was measured with a multi-well plate-reading luminometer (FLUOstar Optima, BMG Labtech GmbH). Luminescence is proportional to the amount of caspase activity present.
Sulforhodamine B assay
As the different treatments and time points may influence the cellular density per well in the caspase 3/7 assay, the sulforhodamine B (SRB, Sigma-Aldrich) assay was run in parallel for each treatment and used for normalization of the cell number. Briefly, after the testing periods, the assay was finished by placing a clear 96-well plate on ice, for 1 min. After gently removing the experimental media, the cells were fixed with 200 μl of 10 % trichloroacetic acid, for 20 min and rinsed 5 times with tap water. After air-drying, 50 μl 4 % SRB was added to each well and incubated for 10 min. The cells were then washed with 1 % acetic acid until the solution ran clear, to remove unbound dye. After air-drying in the dark, the bound dye was solubilized with 100 μl of 10 mM Tris solution (pH 10.5). Absorbances were measured at 510 nm, directly in the plate, using a multi-well plate reader (Labsystems Multiskan, Basingstoke, UK). The data obtained was correlated with the cell number of each well by means of a calibration curve previously plotted.
Statistical analysis
All assays were performed in at least four independent experiments (with exception of the cytometry assay performed in two), and SD values were always <10 %. Data achieved were expressed as mean ± SEM (standard error of the mean). All statistical calculations were performed using the GraphPad Prism version 5.0a for Mac OS X (GraphPad Software, San Diego California, USA). Normality of the data distribution was assessed by three tests (Kolmogorov–Smirnov normality test, D’Agostino and Pearson omnibus normality test and Shapiro–Wilk normality test). The control values (raw data) obtained for hyperthermic and normothermic incubations in the determination of the mitochondrial integrity were compared by the Student’s unpaired t test in order to investigate potential effects associated with the increase in temperature alone. All the other statistical comparisons were estimated by the nonparametric method of Kruskal–Wallis [one-way analysis of variance (ANOVA) on ranks] followed by the Dunn’s multiple comparison test. p values lower than 0.05 were considered statistically significant.
Results
Viability assessment by NR lysosomal uptake
In order to corroborate the previously published cytotoxicity of MDMA, METH, 4-MTA, d-Amp and mixture in HEPG2 cells (Dias da Silva et al. 2013b) obtained in the MTT assay and confirm that the concentrations selected here were, in fact, equieffective and corresponded to 25 % toxicity effects, we evaluated the viability of HEPG2 cells in response to the amphetamines and their mixture in the NR lysosomal uptake assay.
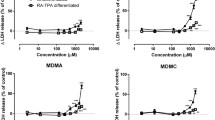
The data presented in Fig. 1 (white bars) indicate a time-dependent increase in cytotoxicity, when the HepG2 cells were exposed at 37 °C to the 4 tested amphetamines, alone and combined. At 37 °C, MDMA cytotoxicity was observed after 12 h of treatment, and 4-MTA cytotoxicity was observed after 24 h of treatment. All other treatments elicited cytotoxicity only after 48 h of treatment. Conversely, when the incubation temperature was raised to 40.5 °C, all the drugs (alone or combined) induced cytotoxicity after 6 h of treatment. Overall, we observed that, at 48 h, under normothermia (the temperature used to determine the equieffective cytotoxic concentrations in the MTT assay), the cytotoxicity determined by the NR assay showed effect levels that were similar to those obtained in the MTT (Dias da Silva et al. 2013b), confirming that, at the selected concentrations, the cytotoxic effects of the individual amphetamines and the mixture are similar. Accordingly, no substantial disparities were observed when comparing responses elicited by individual amphetamines with those obtained from the mixture. The noted exceptions were for MDMA (at 40.5 °C after 48 h, p < 0.05) and METH (at 37 °C after 24 h, p < 0.05).

Time-course of HepG2 cell death, as indicated by NR lysosomal inclusion, after exposure to MDMA (a), METH (b), 4-MTA (c) and d-AMP (d), individually or in mixture (e), at 37 °C (white bars) or 40.5 °C (black bars). The compounds were tested at concentrations that induce 25 % of the maximum effect observed in the MTT reduction assay (EC25), i.e. 1.3 mM MDMA, 3 mM METH, 0.5 mM 4-MTA, 1.7 mM d-AMP and 1.6 mM mixture (Dias da Silva et al. 2013b). Data (mean ± SEM) presented as % cell death relative to controls are from five independent experiments, run in six replicates. Statistical comparisons were made by Kruskal–Wallis test followed by the Dunn’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001 show statistically significant differences versus controls at 37 °C. × p<0.05, ×× p < 0.01 and ××× p < 0.001, indicate significant differences versus controls at 40.5 °C. # p < 0.05, ## p < 0.01 and ### p < 0.001 show statistically significant differences versus 37 °C. + p<0.05 indicates differences between individual treatments and the mixture at same incubation time and temperature (color figure online)
Differential contributions of necrosis and apoptosis to HepG2 cell death elicited by amphetamines
To investigate which mechanisms mediate the cytotoxic effects of the four amphetamines and their combination, we analysed, by flow cytometry, the contribution of apoptosis and necrosis to the observed hepatocyte death. HepG2 cells had been exposed to individual and combined drugs at different time points (24 h and 48 h) and different temperatures (37 and 40.5 °C). After incubation, the cells were stained with AnV-FITC and PI and evaluated using the ImageStream multi-spectral flow cytometer (Amnis Corporation, Seattle, WA). On the ImageStream, each cell was concomitantly imaged in dark field (488 nm laser side-scatter), green fluorescence (500–550 nm, annexin V channel), orange fluorescence (550–600 nm, PI channel) and bright field (660–720 nm). Based on the total intensities of AnV-FITC and PI staining, the cells were grouped into live (AnV−/PI−), early apoptotic (AnV+/PI−), late apoptotic (AnV+/PI+) and necrotic (AnV−/PI+) populations (Fig. 2A). Of note is the fact that positive ANV/PI double staining is not always specific to cells in late stages of apoptosis and may also be present in cells undergoing necrosis, so they cannot be distinguished based solely on fluorescence descriptions. A unique aspect of the ImageStream flow cytometry system is that it allows visual examination of the images taken, allowing the differentiation between advanced apoptotic and necrotic populations, based on their specific morphology. Cells in late apoptosis exhibit shrunken, irregular shape and condensed (pyknosis), fragmented nuclei (karyorrhexis). Necrotic cells are larger, with large, unfragmented nuclei that uniformly stain with PI. Additionally, apoptotic cells exhibit lower bright field intensity and greater granularity compared to necrotic cells (Fig. 2A). So, based on these morphological features, the population gates were adjusted, enabling a better resolution between necrotic and late apoptotic cells.

Imaging flow cytometry-based cellular classification according to fluorescent staining and morphological examination of HepG2 cells using ImageStream multi-spectral flow cytometer (Amnis Corporation, Seattle, WA). a After categorizing HepG2 cells into live (AnV−/PI−), early apoptotic (AnV+/PI−), late apoptotic (AnV+/PI+) and necrotic populations (AnV−/PI+) using fluorescence intensities, the identities of cell populations were confirmed using the “virtual cell sorting” function of the IDEAS data analysis software that analyses the data from the ImageStream, by inspecting the bright field, fluorescence, and dark field (laser side-scatter) imagery obtained from each cell. These population gates were adjusted based on the morphology of cells, enabling a better resolution of necrotic from late apoptotic cells. b Representative analysis of gated cells to show procedure for classification. PI = Propidium iodide; AnV = Annexin V; Composite image = an overlay of AnV, PI, and bright field
Figure 3 depicts the relative numbers (% of cell population) of live, early apoptotic, late apoptotic and necrotic HepG2 cells at the time of staining. The vast majority of untreated cells (negative controls) cultivated at 37 °C were viable after 24-h exposures (75.5 ± 0.6 %). Usually, the mortality in the controls is not as high (up to 15 %). The reason for the relatively elevated amount of cells undergoing cell death processes might be related with the basal levels within the cell line population, the after-staining waiting time before measurement or the damage to the plasma membrane caused during the preparation procedures, which could have occurred, despite all the care taken during technique execution.

Effect of both temperature (37 or 40.5 °C) and incubation time (24 or 48 h) on AnV/PI staining of HepG2 cells after exposure to MDMA, METH, 4-MTA and d-AMP, individually or in mixture. The treatments were tested at concentrations that induced 25 % of the maximum cytotoxic effect observed in the MTT reduction assay (EC25), i.e. 1.3 mM MDMA, 3 mM METH, 0.5 mM 4-MTA, 1.7 mM d-AMP and 1.6 mM mixture (Dias da Silva et al. 2013b). HepG2 population was separated into live (AnV−/PI−), early apoptotic (AnV+/PI−), late apoptotic (AnV+/PI+), and necrotic cells (AnV−/PI+), according to cellular staining with AnV-FITC and PI and cell morphology. Results are mean ± SEM from two independent experiments, run in triplicate. Statistical comparisons were made by using Kruskal–Wallis test followed by the Dunn’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001 show statistically significant differences versus controls at the same incubation time and temperature.× p<0.05,×× p < 0.01 represent statistical comparisons between individual drugs and the mixture at the same incubation time and temperature. # p < 0.05, ### p < 0.01 and ### p < 0.001 show statistical comparison versus the 37 °C incubation temperature. ++ p < 0.01 and +++ p < 0.001, represent statistically significant differences versus the 24 h incubation time
The viable fraction in controls slightly decreased with the increase in temperature to 40.5 °C (68.4 ± 2.8 % at 24 h) and time of incubation to 48 h (70.75 ± 5.15 % at 37 °C), reaching the highest drop when cells were incubated at 40.5 °C for 48 h (57.15 ± 0.35 %). The number of living cells suffered a substantial decline when HepG2 cells were treated with all the amphetamines, alone or combined (p < 0.05 vs. control). Analysis of cell populations revealed that the distribution of the cells between the population sets (live, early apoptotic, late apoptotic and necrotic cells) varied with temperature and incubation time. An overall evaluation indicated that, at 37 °C, with the increase in incubation time from 24 to 48 h, there was a decrease in cell viability that was mostly counterbalanced by an increase in cells in early and/or late stages of apoptosis; while at 40.5 °C, the decrease in cell viability predominantly gave place to a significant rise in the necrotic cell population. A more obvious tendency occurred regarding the temperature increment from 37 to 40.5 °C as, along with the decrease in cell viability, there was a noteworthy expansion in the number of cells undergoing necrosis, clearly observed for all treatments (p < 0.05), for both 24 and 48 h incubations.
In general, no significant differences were detected when HepG2 cells were exposed to amphetamines individually or in mixture. Notwithstanding, some exceptions occurred at 37 °C, with early apoptosis more evident in the mixture relative to the MDMA and METH individual incubations after 24 h, and late apoptosis less evident in the mixture relative to the individual treatments with MDMA, METH, and 4-MTA at 48 h incubations. This indicates that, although these drugs lead to the same cytotoxic outcome, the underlying mechanisms may differ slightly.
Intracellular energetic levels
Research evidence indicates that the cell death fate, either by apoptosis or necrosis, is a condition determined by intracellular ATP levels, as apoptosis is an extremely controlled phenomenon involving a number of energy-dependent processes, such as enzymatic hydrolysis (e.g. caspase activation), chromatin condensation, contraction and membrane blebbing (Eguchi et al. 1997; Zamaraeva et al. 2005).
Results displayed in Fig. 4 suggest there were no significant differences in cellular ATP between controls and drug treatments, when HepG2 cells were exposed at normothermia (37 °C). Nonetheless, the increase in the temperature of incubation to 40.5 °C prompted significant alterations in energy storages for all amphetaminic exposures (p < 0.05). No statistically significant differences were observed between the mixture and the drugs alone under the same temperature conditions.

Effect of MDMA, METH, 4-MTA, d-AMP and the mixture on ATP intracellular storages in HepG2 cells, at 37 °C (white bars) or 40.5 °C (black bars), after 24 h exposure. HepG2 cells were exposed to amphetamines, for 24 h, at the following equieffective concentrations (EC25 MTT Assay, after 48 h at 37 °C): 1.3 mM MDMA, 3 mM METH, 0.5 mM 4-MTA, 1.7 mM d-AMP and 1.6 mM mixture (Dias da Silva et al. 2013b). Results from at least four independent experiments are presented as mean ± SEM. Statistical comparisons were made by using Kruskal–Wallis test followed by the Dunn’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001 represent statistical differences versus controls within the same incubation temperature (37 or 40.5 °C). # p < 0.05, ### p < 0.01 and ### p < 0.001 show statistical comparison between different incubation temperatures
Expression profiles of BCL-2 family genes
Our results revealed that apoptosis plays a highly significant role in the mechanism of cell death provoked by the amphetamines in HepG2 cells, particularly at a physiological temperature (37 °C). With the aim of exploring the specific pathways responsible for initiating apoptosis in the presence of the test substances, early intracellular events linked to apoptotic phenomena, such as the regulation of pro- and anti-apoptotic factors, were studied under the same experimental settings. BCL-2 family genes were found to be differentially expressed in HEPG2 cells after incubation with equieffective cytotoxic concentrations of individual and combined amphetamines at 37 °C. Several time points (1, 4, 8, 16, 24 and 48 h) were tested (data not shown) and the earliest incubation period exhibiting statistically significant differences from untreated controls was at 8 h. As shown in Fig. 5, a remarkable down-regulation of the anti-apoptotic BCL-2 and BCL- XL genes was observed for all treatments (p < 0.001). Accordingly, a significant increase in the expression rates of the genes coding for pro-apoptotic factors, BAX, BIM, PUMA and BID was also found, with slight variations among the different drugs (Fig. 5). No alteration in the expression pattern of BAD was detected, with the exception of HepG2 cells exposed to MDMA (p < 0.05).

Gene expression of pro-apoptotic and anti-apoptotic regulators involved in HepG2 cell death elicited by individual and combined amphetamines. qPCR analysis of BCL-2, BCL- XL , BAX, BIM, BAD, PUMA and BID mRNA levels was performed after incubation of HepG2 cells at 37 °C, for 8 h with 1.3 mM MDMA, 3 mM METH, 0.5 mM 4-MTA, 1.7 mM d-AMP and 1.6 mM of mixture (concentrations inducing 25 % of the maximum effect observed in the MTT reduction assay, EC25) (Dias da Silva et al. 2013b). Expression of each gene was normalized to HPRT1 (reference gene) and compared to the control group, according to the 2−ΔΔCt model. Control values were set to 1 (dashed red line), so values higher than 1 are indicative of up-regulation and values lower than 1 reveal down-regulation. Results are at least from four independent experiments and are given as mean ± SEM. Statistical comparisons were made using Kruskal–Wallis test followed by the Dunn’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001 show statistically significant differences versus controls. × p<0.05 indicates significant differences between treatments with individual drugs and the mixture
Individual and mixture results revealed great concordance, as no significant differences were observed, with exception of BAX in MDMA-treated cells (p < 0.05).
Protein expression analysis
In order to confirm at the protein level, the increased expressions observed at the gene level, the levels of pro-apoptotic BCL-2 family markers were investigated at 37 °C, after 12 h treatments by immunoblotting. Results showed an increase in the levels of pro-apoptotic proteins in HepG2 cells, when these were treated with the test amphetamines. Increases in Puma and Bax proteins were more prominent for treatments with the mixture, while the levels of Bim and Bid were mostly elevated with d-AMP incubations. Similarly, an increase in Bak levels in relation to those present in untreated controls was also observed for all treatments (Fig. 6).

Immunoblotting analysis of the effect of MDMA, METH, 4-MTA, d-AMP and their mixture on the expression of pro-apoptotic markers. a Western blotting of poly (ADP-ribose) polymerase (PARP)/cleaved PARP, Bak, Puma, Bim (EL, S and L isoforms), Bid/cleaved Bid and Bax protein expressions performed after incubation of HepG2 cells at 37 °C, for 24 h (PARP/cleaved PARP) or 12 h (all other proteins), with 1.3 mM MDMA, 3 mM METH, 0.5 mM 4-MTA, 1.7 mM d-AMP and 1.6 mM mixture (concentrations inducing 25 % of the maximum effect observed in the MTT reduction assay, EC25) (Dias da Silva et al. 2013b). β-Actin was used as an internal loading control. b The bar graph represents fold increase in protein expression over controls (mean ± SEM, n ≤ 3) normalized against an actin loading control
Nuclear PARP, a protein involved in DNA repair, is one of the main cleavage targets of caspase-3 and caspase-7 in vivo. PARP inactivation has been proposed to prevent depletion of ATP, which is thought to be required for the latest events of apoptosis. Our data suggest that, at 24 h, there are signs of PARP cleavage, as given by the reduction in the intensity of the PARP (116 kDa) band and the presence of an 89 kDa fragment (corresponding to the cleaved protein). This was particularly evident for d-AMP (Fig. 6).
In spite of the exceptions noted above, an overall analysis of these data did not show significant differences between the individual and mixture treatments (Fig. 6).
Mitochondrial integrity assessed by TMRE inclusion
Our data on the accumulation of TMRE inside HepG2 cells exposed to test amphetamines for 24 h revealed only slight, insignificant (p > 0.05) declines in ∆ψm, when incubations were carried out at 37 °C (Fig. 7). However, considerable deviations from controls were obtained in hyperthermic conditions (p < 0.01), as observed by the decline in TMRE mitochondrial inclusion, indicating significant mitochondrial impairment. No significant differences were identified (p > 0.05) between control incubations at 37 °C (40702 ± 12264 fluorescence units) and at 40.5 °C (44255 ± 10562 fluorescence units), showing that the increase in temperature alone had no impact on ∆ψm. Moreover, at 40.5 °C, mixture results presented relevant dissimilarities from those obtained with MDMA, METH and 4-MTA (p < 0.05). For d-AMP, no significant differences were observed between the individual drug and the mixture.

Effect of MDMA, METH, 4-MTA, d-AMP and mixture on mitochondrial membrane potential in HepG2 cells, at 37 °C (white bars) or 40.5 °C (black bars). HepG2 cells were exposed to amphetamines, for 24 h, at the following equieffective concentrations (EC25 MTT Assay): 1.3 mM MDMA, 3 mM METH, 0.5 mM 4-MTA, 1.7 mM d-AMP and 1.6 mM mixture (Dias da Silva et al. 2013b). Shown are the mean ± SEM from four independent experiments, with six replicates each, expressed as percentage of controls (fluorescence of non-treated cells) for 37 or 40.5 °C incubations. The red dashed line indicates the control values, set to 100 %. Statistical comparisons were made using the Kruskal–Wallis test followed by Dunn’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001 show statistically significant differences versus controls, at the same incubation temperature. × p<0.05, ×× p < 0.01 indicate differences between the individual treatments and the mixture, at the same incubation temperature. # p < 0.05, ### p < 0.01 and ### p < 0.001 show statistical differences versus 37 °C incubations
Measurement of caspase-3/-7 activity
The purpose of the current determination was to define the contribution of caspase-3/7 activity to the extent of observed cell death and identify the onset and peak of this molecular event for each treatment, in order to identify possible differences between the mechanisms underlying the cytotoxicity of individual and combined amphetamines.
As observed in Fig. 8, MDMA and METH induced a short, but significant increase in enzyme activity from 8–16 h of incubation (maximal effects of 1.34 ± 0.14-fold increase and 1.36 ± 0.21-fold increase, respectively), which remained more or less constant after 16 h of exposure (p < 0.001). Earlier and more pronounced effects were observed when HepG2 cells were exposed to 4-MTA and to d-AMP, as significant effects were seen as early as 1 h (4-MTA) and 4 h (d-AMP) of incubation. A peak in the effects of these drugs was achieved at 16 h (5.08 ± 0.36-fold increase) and 24 h (3.91 ± 0.69-fold increase) for 4-MTA and d-AMP, respectively.

Time-course of caspase-3/-7 activation in HepG2 cells exposed to MDMA (a), METH (b), 4-MTA (c), d-AMP (d), individually and in combination (e). The compounds were tested at concentrations that induce 25 % of the maximum effect observed in the MTT reduction assay (EC25), i.e. 1.3 mM MDMA, 3 mM METH, 0.5 mM 4-MTA, 1.7 mM d-AMP and 1.6 mM mixture (Dias da Silva et al. 2013b). Caspase-3/-7 activation was measured using a caspase-Glo 3/7 assay kit (Promega) after 1, 4, 8, 16, 24, 36 or 48 h of incubation, at 37 °C. Results are from five independent experiments performed in six replicates and are presented as mean ± SEM. Control values are set to 0 (dashed red line). Statistical comparisons were made using the Kruskal–Wallis test followed by Dunn’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001 show statistically significant differences versus controls (color figure online)
There were no statistically significant differences between the mixture and any other treatment, for any of the incubation periods. In fact, mixture effects were clearly seen at 8 h of treatment (p < 0.05) and seem to lie between all individual responses, being more pronounced at 16 h (2.16 ± 0.40-fold increase).
Discussion
Years of clinical and experimental evidence have demonstrated hepatotoxicity after amphetamine exposure in human abusers and laboratory animals (Andreu et al. 1998; Beitia et al. 1999; Brncic et al. 2006; Khakoo et al. 1995; Montiel-Duarte et al. 2002). Accordingly, our in vitro findings in the human hepatoma HepG2 cell line indicate that all tested amphetamines and their combination promote time-(6–48 h) and temperature-(37 and 40.5 °C) dependent mortality, when evaluated by the NR reuptake assay. The neutral red uptake assay provides a quantitative estimation of the number of viable cells in a culture based on lysosomal membrane damage, while the MTT assay, which was used in some of our previous studies to determine cytotoxicity (Dias da Silva et al. 2013b), reflects the number of viable cells by measuring cellular metabolic activity via mitochondrial oxidoreductase enzymes. Under normothermic conditions, the NR assay demonstrated similar sensitivity to the MTT reduction assay at 48 h, for all of our tested drugs, further confirming their cytotoxic profile (Dias da Silva et al. 2013b).
Several lines of investigation suggest that either necrosis or apoptosis mediates the reported amphetamine-related toxicity. Previous reports have shown that amphetamine designer drugs can induce apoptosis in human serotonergic JAR cells (Simantov and Tauber 1997), rat cortical neurons (Cunha-Oliveira et al. 2006; Stumm et al. 1999; Warren et al. 2007), immortalized mesencephalic neurons (Cadet et al. 1997; Kanthasamy et al. 2006), rat cerebellar granule cells (Jimenez et al. 2004), striatal glutamic acid decarboxylase (GAD)-containing neurons (Jayanthi et al. 2004) and in mice striatal dopaminergic neurons (Krasnova et al. 2005; Zhu et al. 2005), among others. Capela et al. (2012) have additionally demonstrated MDMA-induced programmed neuronal death, involving both the mitochondria machinery and the extrinsic cell death pathway in primary cultures of hippocampus obtained from rat embryos. Albeit the neuronal system being the most widely studied, apoptosis has also been demonstrated in cultured hepatic stellate cells (Montiel-Duarte et al. 2002, 2004) and in freshly isolated rat hepatocytes exposed to amphetamines (Montiel-Duarte et al. 2002). Conversely, post-mortem reports of intoxicated abusers describe cases of human livers exhibiting clear necrotic effects, from foci of individual necrotic cells to massive hepatic necrosis (Milroy et al. 1996), which seems to indicate that apoptosis is not the only mechanism of cell death attributed to amphetamines (Pontes et al. 2008). In addition to liver necrosis, which is accompanied by fatty degeneration and inflammation, Sano et al. (2009) have also observed small foci of myocyte necrosis, providing further evidence of the extension of this cell death process to other organ systems.
A classical definition of necrosis, based on morphological criteria, presumes an unregulated phenomenon involving damage of membrane integrity, cellular disruption and swelling of cytoplasmic organelles; whereas apoptosis is characterized by organized plasma membrane blebbing, cell shrinkage and typical modifications of nuclear morphology, such as chromatin condensation and fragmentation.
With the current investigation, we expected to provide new insights into the putative mechanisms underlying the reported cellular death observed here and in previous studies (Dias da Silva et al. 2013b, c, d), under physiological and hyperthermic settings. Additionally, we expected to provide new understandings of the way amphetamines work when in combination. Effectively, a few studies aimed at establishing the type of interaction between amphetamines have already been carried out (Dias da Silva et al. 2013a, b, c, d), and support the hypothesis that these drugs act in a similar way to prompt the overall mixture cytotoxicity. However, the mechanisms underlying such additive interactions are yet to be fully understood.
We, therefore, evaluated the rate of necrosis versus apoptosis in HepG2 cells following incubation with these drugs, under different time and temperature conditions. Our findings indicate that in the absence of heat stress (at 37 °C), HepG2 cells preferentially exhibited death with apoptotic characteristics, with an increase in early apoptotic cells, especially evident in the incubations with 4-MTA, d-AMP and the mixture. This agrees well to the observed increases in caspase 3/7 activities at the same incubation conditions. The extent of hepatocellular apoptosis is dependent on the duration of exposure to the tested drugs, being more pronounced after 48 h incubations.
Apoptosis is a highly effective cell-suicide programme, which entails the orchestrated interplay of a range of promoter and suppressor agents, many of which belong to the BCL-2 family, which comprises a number interrelated proteins that regulate mitochondrial or intrinsic pathway of apoptosis via their pro-apoptotic and pro-survival (antiapoptotic) activity. Pro-apoptotic BCL-2 factors are also involved in the enhancement of the extrinsic death receptor pathway. So far, approximately 20 BCL-2 family members have been identified and are classically divided into three subfamilies: Bcl-2 pro-survival subfamily (including Bcl-2 and Bcl-xL), Bax-like pro-apoptotic subfamily (comprises Bax and Bak) and BH3-only pro-apoptotic subfamily. These proteins are crucial controllers of the permeabilization of the outer membrane of the mitochondria, which promotes the release of cyt c and other apoptotic factors to the cytosol, activating the downstream effector caspase-3/-7 cascade. As this is considered an irreversible point in the programmed cell death, these proteins play a pivotal role in deciding whether a cell lives or dies. Pro- and anti-apoptotic BCL-2 members may be regulated not only by post-translational mechanisms but also at the genomic level, by a change in gene expression profiles. To ascertain to what extent pro-apoptotic and anti-apoptotic BCL-2 family regulators may account for triggering apoptosis in HepG2 cells after exposure to amphetamines, the expression of some apoptosis-regulating genes was investigated by qPCR. The results presented herein clearly show the significant downregulation of BCL-2 and BCL- XL gene expressions in response to amphetamine treatment. Other authors have already reported a decreased expression of these anti-apoptotic markers in the rat liver after exposure to MDMA (Cerretani et al. 2011; Montiel-Duarte et al. 2002; Upreti et al. 2011) and d-AMP (De Miglio et al. 2000), but this correlation was never reported for hepatotoxicity provoked by METH and 4-MTA, nor for amphetamine combinations.
The proteins encoded by these anti-apoptotic genes have the role of preventing the activation of a proteolytic caspase cascade that condemns cells to death, through direct interactions with the pro-apoptotic members (Cory et al. 2003; Garcia-Saez 2012; Gross et al. 1999). Indeed, it had become evident that it is the imbalance between the opposing BCL-2 family members, in favour of pro-apoptotic regulators, rather than the proteins themselves, that controls the release of downstream effector caspases. Accordingly, we observed increases in the BAX/BCL-2 ratios with all drug treatments (MDMA 4.78; METH 7.34; 4-MTA 9.54; d-AMP 11.06; mixture 9.18; where control value was set to 1).
In accordance with results obtained in other cellular systems for METH (Genc et al. 2003; Pereira et al. 2006; Shao et al. 2012) and d-AMP (De Miglio et al. 2000; Krasnova et al. 2005), anti-survival Bax was found to be differentially expressed in HepG2 cells after amphetamine treatment. Notwithstanding, as far as we know, this is the first study reporting Bax overexpression induced by MDMA and 4-MTA. Also, this is the first time that an association between Bak overexpression and apoptosis has been reported for our test drugs. It is conceivable that in our cellular model, upon a death stimulus, Bax, which is present in the cell in the form of cytosolic monomers, translocates to the mitochondria. Once there, Bax and Bak, the latter being constitutively bound to the outer membrane of the organelle, suffer conformational changes, integrate into the membrane and oligomerize (Nechushtan et al. 2001). The formed oligomers provoke the permeabilization of the outer mitochondrial membrane, permitting the release of several crucial pro-death factors from the mitochondrial intermembrane space into the cytoplasm, which in turn activate the effector caspase-3 and -7 that execute apoptosis (Susin et al. 1999; Verhagen and Vaux 2002).
Under normal cellular conditions, the BH3-only regulators (e.g. Bid, Bim and Puma) have no activity or exist only at residual levels. Upon stimuli, such as that induced by amphetamines, they are transcriptionally upregulated (similarly to what was observed with Bim, Bad, Puma and Bid) or activated by post-translational modifications, such as dephosphorylation, subcellular translocation or proteolysis (as with Bim and Bid). To the best of our knowledge, Puma and Bim have never been linked with the toxicity induced by amphetamines. Increased Bad has been associated with apoptosis induced by METH in mouse neocortex (Jayanthi et al. 2001) and rat striatum (Beauvais et al. 2011), and upregulation of Bid has been reported in rat liver exposed to MDMA (Cerretani et al. 2011) and in mouse brain exposed to METH (Deng et al. 2007; Jayanthi et al. 2001), but not in hepatocytes. When activated, these proteins govern apoptosis either by directly activating Bax/Bak or neutralizing the role of pro-survival members, such as Bcl-2, Bcl-xL. Upon BAD dephosphorylation, a heterodimer with Bcl-2 and Bcl-xL is formed, inactivating these anti-apoptotic markers, and thus promoting Bax/Bak-triggered apoptosis. On the other hand, BAD phosphorylation allows the formation of BAD homodimers, leaving Bcl-2 and Bcl-xL free to inhibit apoptosis.
In the particular case of Bid, it was observed that its expression was increased at both transcript and protein levels under our experimental conditions. This p22 protein is generally confined to the cytosol of the cell in an inactive form. When activated, caspase-8 mediates its cleavage into one major p15 and two minor p13 and p11 fragments, and leads to the translocation of the p15-truncated form (tBID) to the mitochondrial outer membrane (Li et al. 1998; Luo et al. 1998; Yin et al. 1999). Since the apical caspase-8 is activated by cell surface death receptors, such as Fas and tumour necrosis factor (TNF), it is plausible that extrinsic signalling pathways mediate, at least in part, the apoptotic responses triggered by amphetamines in HepG2 hepatocytes. Supporting this possibility are the results presented by Capela et al. (2012) that demonstrated programmed cell death of primary cultures of hippocampal neurons exposed to MDMA and 2,5-dimethoxy-4-iodoamphetamine with significant increases in both caspase-8 and caspase-3 activities induced by both drugs. Also, the intrinsic triggering of the programmed cell death cannot be excluded, as other studies have already linked the activation of caspase 9 to the cytotoxicity elicited by amphetamines. For example, the activation of this apoptosis mitochondrial pathway executioner has been reported after SH-SY5Y human neuroblastoma cell line exposure to MDMA and 4-MTA (Montgomery et al. 2010). In addition, d-Amp induced the activation of caspase-9 in rat cortical neurons and METH promoted the cleavage of this caspase in rat brain (Jayanthi et al. 2004) and in an immortalized rat striatal cell line (Deng et al. 2002). It can also be argued that following translocation of tBID (involved in the extrinsic pathway), the Bax/Bak oligomerization promotes cyt c efflux from the mitochondria that proteolytically activates caspase-9.
Regardless of the programmed cell death being triggered by the activation of cell surface death receptors (extrinsic apoptosis pathways) or in response to signals instigated from inside the cell (intrinsic apoptosis pathways), the signals of such diverse sources seem to ultimately converge to activate common downstream mechanisms, specifically the apoptosis executioner caspase cascade. Among effector caspases that are proteolytically activated are the caspase-3 and -7, two cysteine-aspartic acid proteases that, following activation, cleave a specific set of protein substrates, including procaspases themselves and PARP, resulting in the spreading of apoptotic death stimulus and eventually in the execution of cell suicide. Our results seem to indicate that apoptosis induced by amphetamines in HepG2 cells occurred primarily through extrinsic and/or mitochondrial pathways. This was further confirmed by the observed activation of caspases-3 and-7 and cleavage of cellular components, like PARP, in the presence of the test substances.
The mitochondria have been shown to play a major role in apoptosis by mediating and propagating death signals originated from the inside (intrinsic apoptotic pathway) or outside (extrinsic apoptotic pathway) of the cell. Most apoptotic events involve the dissipation of the mitochondrial inner transmembrane potential (∆ψm), which is crucial for maintaining the physiological function of the mitochondrial respiratory chain. A significant loss in ∆ψm impairs oxidative phosphorylation, which depletes the cells of energy and promotes the release of apoptotic factors, with subsequent death. We did not detect any disruption of mitochondrial membrane potential when HepG2 cells were exposed to amphetamines, at 37 °C. Notwithstanding, the importance of mitochondrial dysfunction in hepatocyte programmed death is unclear. Despite the loss of ∆ψm being allegedly an early apoptotic event, there is emerging evidence advocating that this may not be the case. Depending on the cell system under investigation and the apoptotic stimuli, the loss of ∆ψm may not be required for cyt c release (Ly et al. 2003). These conflicting data may also be attributed to the distinct abilities of different fluorochromes used in the different studies to accurately discriminate changes in ∆ψm.
From our results, it was also clear that the increase in temperature (from 37 to 40.5 °C) rendered cells more vulnerable to necrosis, for both time conditions (24 and 48 h). This suggests that there is a change in the mode of cell death with the increase in temperature. At physiological temperature, amphetamines led preferentially to apoptosis, but under hyperthermia, cellular death was mostly due to necrosis and not apoptosis. It is known that hyperthermia aggravates the cytotoxicity of amphetamines in several study models including primary hepatocytes (Carvalho et al. 1997, 2001; Pontes et al. 2008), neuronal cells (Capela et al. 2006) and skeletal muscle (Duarte et al. 2005). We observed that a 3.5 °C increment was sufficiently detrimental to provoke premature death of HepG2 cells by necrosis, even in controls, advocating that hyperthermia per se is deleterious enough to trigger necrotic pathways. Necrotic effects were additionally enhanced with the widening of drug exposure period (from 24 to 48 h). Previous studies have shown that high temperatures induce a pro-oxidant state by triggering the formation of reactive oxygen and nitrogen species with subsequent production of oxidative/nitrosative stress, which depletes HepG2 cells from glutathione and, particularly, from ATP (Dias da Silva et al. 2013c ). In the present study, the vulnerability of the cells exposed at hyperthermia was further exacerbated with the exposure to amphetamines, as necrosis was greatly increased in the treatment groups in comparison to controls. Supporting our observations, a previous study performed by Pontes et al. (2008), which evaluated the apoptotic/necrotic features induced by MDMA in primary cultured rat hepatocytes incubated at 37 or 40.5 °C for 24 h, showed that the cell death occurred preferentially through a necrotic pathway at the higher incubation temperature.
Intracellular ATP levels corroborate these results. Although energy contents slightly decrease in HepG2 cells treated with amphetamines at 37 °C, the ATP decline only presented statistically substantial differences (p < 0.05) when cells where exposed at 40.5 °C. Actually, as observed in this study, when temperature was set to physiological conditions (37 °C), cells exposed to amphetamines preferentially died by apoptosis. This programmed process is highly regulated and requires energy for a number of ATP-dependent stages (Richter et al. 1996; Zamaraeva et al. 2005), such as caspase activation (Li et al. 1997), chromatin condensation (Kass et al. 1996) and blebbing (Barros et al. 2003). On the other hand, it has been proven that the exhaustion of ATP cellular storages redirects the type of death from apoptosis to necrosis (Leist et al. 1997; Nicotera et al. 1998). This is in agreement with our results, considering that hyperthermia (40.5 °C) per se was capable of reducing cellular ATP and that this effect was further exacerbated in the presence of amphetamines. While in the present study we did not observe a significant decrease in ATP levels at 37 °C; in previous studies, these amphetamines were able to reduce the intracellular HepG2 energetic levels on their own, but only at higher concentrations (Dias da Silva et al. 2013c). However, also in this previous study, the most significant ATP reductions were attained at hyperthermic incubation conditions (Dias da Silva et al. 2013c). Supporting our observations, studies with primary hepatocytes incubated with MDMA at 37 and 40 °C showed that a significant ATP depletion occurred only at the highest incubation temperature (Pontes et al. 2008).
Apoptosis is a physiologically occurring programmed and targeted way of cell death that normally provides benefits to the organism. In contrast, despite investigations suggesting that necrosis occurrence and progression might be tightly controlled (Golstein and Kroemer 2007; Proskuryakov et al. 2003), it almost always implies negative consequences. Contrary to apoptosis, in which apoptotic bodies containing cytosol, condensed nuclear material and organelles are ultimately engulfed by phagocytes without causing any damage; cells undergoing necrosis suffer early plasma membrane rupture, loss of selective permeability, dilatation of cytoplasmic organelles, particularly mitochondria and uncontrolled leakage of cellular contents into the extracellular space. This results in injury of neighbouring cells and severe inflammatory reaction in the corresponding tissue, which could become chronic and cause irreparable damage. Given the fact that the higher temperature used in our experiment (40.5 °C) coincides with the body temperatures that recreational abusers may attain after ecstasy consumption, our results support the hypothesis that amphetamine-induced thermodysregulation, which is also exacerbated by the hot and overcrowded venues were consumption often occurs, is one of the key causes of necrosis-mediated acute liver failure.
Overall, no major differences were observed between the independently tested amphetamines and their mixture for any of the investigated endpoints. This supports the assumption that these compounds, tested at equal effect concentrations, behave similarly and do not interact with each other in any unexpected way. In other words, this supports previous observations which showed that, when combined, these amphetamines act in an additive manner (Dias da Silva et al. 2013b).
The tested concentrations used in the present study are in the range of concentrations used in several mechanistic in vitro studies (Simantov and Tauber 1997; Stumm et al. 1999; Carvalho et al. 2004a, b; Capela et al. 2006). They are higher than concentrations commonly found in human abusers. However, it should be noted that high interindividual variations of blood levels in cases of severe and even fatal intoxications have been observed. For MDMA, for example, blood concentrations can be as high as 70 μM (De Letter et al. 2004, 2006). In such cases, the autopsy findings have shown that the tissue levels of the drug can be up to 30 times higher than blood concentrations (De Letter et al. 2006; Garcia-Repetto et al. 2003).
In conclusion, the cell death mechanism (apoptosis or necrosis) induced by amphetamines, alone and in mixture, depends on both temperature and duration of exposure. It is possible that under less hostile temperature settings, psychostimulant amphetamines promote programmed hepatocyte death involving both the mitochondrial machinery, (through the synchronized interaction between BCL-2 members), and the extrinsic cell death, possibly by activating caspase-8. Although upstream mechanisms were not totally clarified, our results evidenced that apoptosis common pathways were eventually activated, after HepG2 cells exposure to amphetamines. Further approaches aimed at elucidating the involvement of the mitochondrial and, particularly, extrinsic receptor pathway should be considered in the future. We stress the importance of these results for an improved knowledge of the molecular mechanisms involved in amphetamines toxicity, which should aid in implementing novel therapeutic approaches with a view to avoid or attenuate the acute and/or chronic consequences of ecstasy misuse.
References
Andreu V, Mas A, Bruguera M, Salmeron JM, Moreno V, Nogue S, Rodes J (1998) Ecstasy: a common cause of severe acute hepatotoxicity. J Hepatol 29(3):394–397
Antolino-Lobo I, Meulenbelt J, van den Berg M, van Duursen MB (2011) A mechanistic insight into 3,4-methylenedioxymethamphetamine (“ecstasy”)-mediated hepatotoxicity. Vet Q 31(4):193–205. doi:10.1080/01652176.2011.642534
Armenian P, Mamantov TM, Tsutaoka BT, Gerona RR, Silman EF, Wu AH, Olson KR (2012) Multiple MDMA (Ecstasy) overdoses at a rave event: a case series. J Intensive Care Med. doi:10.1177/0885066612445982
Barros LF, Kanaseki T, Sabirov R, Morishima S, Castro J, Bittner CX, Maeno E, Ando-Akatsuka Y, Okada Y (2003) Apoptotic and necrotic blebs in epithelial cells display similar neck diameters but different kinase dependency. Cell Death Differ 10(6):687–697. doi:10.1038/sj.cdd.4401236
Beauvais G, Atwell K, Jayanthi S, Ladenheim B, Cadet JL (2011) Involvement of dopamine receptors in binge methamphetamine-induced activation of endoplasmic reticulum and mitochondrial stress pathways. PLoS ONE 6(12):e28946. doi:10.1371/journal.pone.0028946
Beitia G, Cobreros A, Sainz L, Cenarruzabeitia E (1999) 3,4-Methylenedioxymethamphetamine (ecstasy)-induced hepatotoxicity: effect on cytosolic calcium signals in isolated hepatocytes. Liver 19(3):234–241
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brauer RB, Heidecke CD, Nathrath W, Beckurts KT, Vorwald P, Zilker TR, Schweigart U, Holscher AH, Siewert JR (1997) Liver transplantation for the treatment of fulminant hepatic failure induced by the ingestion of ecstasy. Transpl Int 10(3):229–233
Brncic N, Kraus I, Viskovic I, Mijandrusic-Sincic B, Vlahovic-Palcevski V (2006) 3,4-methylenedioxymethamphetamine (MDMA): an important cause of acute hepatitis. Med Sci Monit 12(11):CS107–CS109
Cadet JL, Ordonez SV, Ordonez JV (1997) Methamphetamine induces apoptosis in immortalized neural cells: protection by the proto-oncogene, bcl-2. Synapse 25(2):176–184. doi:10.1002/(SICI)1098-2396(199702)25:2<176::AID-SYN8>3.0.CO;2-9
Capela JP, da Araujo SC, Costa VM, Ruscher K, Fernandes E, Bastos MD, Dirnagl U, Meisel A, Carvalho F (2012) The neurotoxicity of hallucinogenic amphetamines in primary cultures of hippocampal neurons. Neurotoxicology. doi:10.1016/j.neuro.2012.09.005
Capela JP, Ruscher K, Lautenschlager M, Freyer D, Dirnagl U, Gaio AR, Bastos ML, Meisel A, Carvalho F (2006) Ecstasy-induced cell death in cortical neuronal cultures is serotonin 2A-receptor-dependent and potentiated under hyperthermia. Neuroscience 139(3):1069–1081. doi:10.1016/j.neuroscience.2006.01.007
Carvalho F, Remiao F, Soares ME, Catarino R, Queiroz G, Bastos ML (1997) d-Amphetamine-induced hepatotoxicity: possible contribution of catecholamines and hyperthermia to the effect studied in isolated rat hepatocytes. Arch Toxicol 71(7):429–436
Carvalho M, Remiao F, Milhazes N, Borges F, Fernandes E, Carvalho F, Bastos ML (2004a) The toxicity of N-methyl-alpha-methyldopamine to freshly isolated rat hepatocytes is prevented by ascorbic acid and N-acetylcysteine. Toxicology 200(2–3):193–203. doi:10.1016/j.tox.2004.03.016
Carvalho M, Remiao F, Milhazes N, Borges F, Fernandes E, Monteiro Mdo C, Goncalves MJ, Seabra V, Amado F, Carvalho F, Bastos ML (2004b) Metabolism is required for the expression of ecstasy-induced cardiotoxicity in vitro. Chem Res Toxicol 17(5):623–632. doi:10.1021/tx049960f
Carvalho M, Carmo H, Costa VM, Capela JP, Pontes H, Remiao F, Carvalho F, Bastos ML (2012) Toxicity of amphetamines: an update. Arch Toxicol 86(8):1167–1231. doi:10.1007/s00204-012-0815-5
Carvalho M, Carvalho F, Bastos ML (2001) Is hyperthermia the triggering factor for hepatotoxicity induced by 3,4-methylenedioxymethamphetamine (ecstasy)? An in vitro study using freshly isolated mouse hepatocytes. Arch Toxicol 74(12):789–793
Carvalho M, Pontes H, Remiao F, Bastos ML, Carvalho F (2010) Mechanisms underlying the hepatotoxic effects of ecstasy. Curr Pharm Biotechnol 11(5):476–495
Cerretani D, Bello S, Cantatore S, Fiaschi AI, Montefrancesco G, Neri M, Pomara C, Riezzo I, Fiore C, Bonsignore A, Turillazzi E, Fineschi V (2011) Acute administration of 3,4-methylenedioxymethamphetamine (MDMA) induces oxidative stress, lipoperoxidation and TNFalpha-mediated apoptosis in rat liver. Pharmacol Res 64(5):517–527. doi:10.1016/j.phrs.2011.08.002
Coore JR (1996) A fatal trip with ecstasy: a case of 3,4-methylenedioxymethamphetamine/3,4- methylenedioxyamphetamine toxicity. J R Soc Med 89(1):51P–52P
Cory S, Huang DC, Adams JM (2003) The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 22(53):8590–8607. doi:10.1038/sj.onc.1207102
Cunha-Oliveira T, Rego AC, Cardoso SM, Borges F, Swerdlow RH, Macedo T, de Oliveira CR (2006) Mitochondrial dysfunction and caspase activation in rat cortical neurons treated with cocaine or amphetamine. Brain Res 1089(1):44–54. doi:10.1016/j.brainres.2006.03.061
De Letter EA, Bouche MP, Van Bocxlaer JF, Lambert WE, Piette MH (2004) Interpretation of a 3,4-methylenedioxymethamphetamine (MDMA) blood level: discussion by means of a distribution study in two fatalities. Forensic Sci Int 141(2–3):85–90. doi:10.1016/j.forsciint.2003.12.015
De Letter EA, Piette MHA, Lambert WE, Cordonnier JACM (2006) Amphetamines as potential inducers of fatalities: a review in the district of Ghent from 1976–2004. Med Sci Law 46(1):37–65
De Miglio MR, Muroni MR, Simile MM, Calvisi DF, Tolu P, Deiana L, Carru A, Bonelli G, Feo F, Pascale RM (2000) Implication of Bcl-2 family genes in basal and D-amphetamine-induced apoptosis in preneoplastic and neoplastic rat liver lesions. Hepatology 31(4):956–965. doi:10.1053/he.2000.5411
Deng X, Cai NS, McCoy MT, Chen W, Trush MA, Cadet JL (2002) Methamphetamine induces apoptosis in an immortalized rat striatal cell line by activating the mitochondrial cell death pathway. Neuropharmacology 42(6):837–845
Deng X, Ladenheim B, Jayanthi S, Cadet JL (2007) Methamphetamine administration causes death of dopaminergic neurons in the mouse olfactory bulb. Biol Psychiatry 61(11):1235–1243. doi:10.1016/j.biopsych.2006.09.010
Dias da Silva D, Carmo H, Silva E (2013a) Mixtures of 3,4-methylenedioxymethamphetamine (ecstasy) and its major human metabolites act additively to induce significant toxicity to liver cells even when combined at low, non-cytotoxic concentrations. J Appl Toxicol. doi:10.1002/jat.2885
Dias da Silva D, Carmo H, Silva E (2013b) The risky cocktail: what combination effects can we expect between ecstasy and other amphetamines? Arch Toxicol 87(1):111–122. doi:10.1007/s00204-012-0929-9
Dias da Silva D, Silva E, Carmo H (2013c) Combination effects of amphetamines under hyperthermia—the role played by oxidative stress. J Appl Toxicol. doi:10.1002/jat.2889
Dias da Silva D, Silva E, Carmo H (2013d) Cytotoxic effects of amphetamine mixtures in primary hepatocytes are severely aggravated under hyperthermic conditions. Toxicol In Vitro 27(6):1670–1678. doi:10.1016/j.tiv.2013.04.010
Duarte JA, Leao A, Magalhaes J, Ascensao A, Bastos ML, Amado FL, Vilarinho L, Quelhas D, Appell HJ, Carvalho F (2005) Strenuous exercise aggravates MDMA-induced skeletal muscle damage in mice. Toxicology 206(3):349–358. doi:10.1016/j.tox.2004.07.012
Dykhuizen RS, Brunt PW, Atkinson P, Simpson JG, Smith CC (1995) Ecstasy induced hepatitis mimicking viral hepatitis. Gut 36(6):939–941
Eguchi Y, Shimizu S, Tsujimoto Y (1997) Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res 57(10):1835–1840
Ellis AJ, Wendon JA, Portmann B, Williams R (1996) Acute liver damage and ecstasy ingestion. Gut 38(3):454–458
Fineschi V, Centini F, Mazzeo E, Turillazzi E (1999) Adam (MDMA) and Eve (MDEA) misuse: an immunohistochemical study on three fatal cases. Forensic Sci Int 104(1):65–74
Garbino J, Henry JA, Mentha G, Romand JA (2001) Ecstasy ingestion and fulminant hepatic failure: liver transplantation to be considered as a last therapeutic option. Vet Hum Toxicol 43(2):99–102
Garcia-Repetto R, Moreno E, Soriano T, Jurado C, Gimenez MP, Menendez M (2003) Tissue concentrations of MDMA and its metabolite MDA in three fatal cases of overdose. Forensic Sci Int 135(2):110–114. doi:10.1016/S0379-0738(03)00179-8
Garcia-Saez AJ (2012) The secrets of the Bcl-2 family. Cell Death Differ 19(11):1733–1740. doi:10.1038/cdd.2012.105
Genc K, Genc S, Kizildag S, Sonmez U, Yilmaz O, Tugyan K, Ergur B, Sonmez A, Buldan Z (2003) Methamphetamine induces oligodendroglial cell death in vitro. Brain Res 982(1):125–130
Golstein P, Kroemer G (2007) Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 32(1):37–43. doi:10.1016/j.tibs.2006.11.001
Green AR, Mechan AO, Elliott JM, O’Shea E, Colado MI (2003) The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”). Pharmacol Rev 55(3):463–508
Gross A, McDonnell JM, Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13(15):1899–1911
Henry JA, Jeffreys KJ, Dawling S (1992) Toxicity and deaths from 3,4-methylenedioxymethamphetamine (“ecstasy”). Lancet 340(8816):384–387
Ibranyi E, Schonleber J (2003) Acute liver failure caused by Ecstasy. Orv Hetil 144(29):1455–1456
Jayanthi S, Deng X, Bordelon M, McCoy MT, Cadet JL (2001) Methamphetamine causes differential regulation of pro-death and anti-death Bcl-2 genes in the mouse neocortex. FASEB J 15(10):1745–1752
Jayanthi S, Deng X, Noailles PA, Ladenheim B, Cadet JL (2004) Methamphetamine induces neuronal apoptosis via cross-talks between endoplasmic reticulum and mitochondria-dependent death cascades. FASEB J 18(2):238–251. doi:10.1096/fj.03-0295com
Jimenez A, Jorda EG, Verdaguer E, Pubill D, Sureda FX, Canudas AM, Escubedo E, Camarasa J, Camins A, Pallas M (2004) Neurotoxicity of amphetamine derivatives is mediated by caspase pathway activation in rat cerebellar granule cells. Toxicol Appl Pharmacol 196(2):223–234. doi:10.1016/j.taap.2003.12.017
Kalant H (2001) The pharmacology and toxicology of “ecstasy” (MDMA) and related drugs. CMAJ 165(7):917–928
Kamijo Y, Soma K, Nishida M, Namera A, Ohwada T (2002) Acute liver failure following intravenous methamphetamine. Vet Hum Toxicol 44(4):216–217
Kanthasamy A, Anantharam V, Ali SF, Kanthasamy AG (2006) Methamphetamine induces autophagy and apoptosis in a mesencephalic dopaminergic neuronal culture model: role of cathepsin-D in methamphetamine-induced apoptotic cell death. Ann N Y Acad Sci 1074:234–244. doi:10.1196/annals.1369.022
Kass GE, Eriksson JE, Weis M, Orrenius S, Chow SC (1996) Chromatin condensation during apoptosis requires ATP. Biochem J 318(Pt 3):749–752
Kendrick WC, Hull AR, Knochel JP (1977) Rhabdomyolysis and shock after intravenous amphetamine administration. Ann Intern Med 86(4):381–387
Khakoo SI, Coles CJ, Armstrong JS, Barry RE (1995) Hepatotoxicity and accelerated fibrosis following 3,4-methylenedioxymetamphetamine (“ecstasy”) usage. J Clin Gastroenterol 20(3):244–247
Krasnova IN, Ladenheim B, Cadet JL (2005) Amphetamine induces apoptosis of medium spiny striatal projection neurons via the mitochondria-dependent pathway. FASEB J 19(7):851–853. doi:10.1096/fj.04-2881fje
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P (1997) Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med 185(8):1481–1486
Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94(4):491–501
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91(4):479–489
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25(4):402–408. doi:10.1006/meth.2001.1262
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94(4):481–490
Ly JD, Grubb DR, Lawen A (2003) The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis 8(2):115–128
Milroy CM (2011) “Ecstasy” associated deaths: what is a fatal concentration ? Analysis of a case series. Forensic Sci Med Pathol 7(3):248–252. doi:10.1007/s12024-010-9220-7
Milroy CM, Clark JC, Forrest AR (1996) Pathology of deaths associated with “ecstasy” and “eve” misuse. J Clin Pathol 49(2):149–153
Montgomery T, Sitte H, McBean G (2010) 4-Methylthioamphetamine (4-MTA) induces mitochondrial-dependent apoptosis in SH-SY5Y cells independently of dopamine and noradrenaline transporters. BMC Pharmacol 10(Suppl 1):A22
Montiel-Duarte C, Ansorena E, Lopez-Zabalza MJ, Cenarruzabeitia E, Iraburu MJ (2004) Role of reactive oxygen species, glutathione and NF-kappaB in apoptosis induced by 3,4-methylenedioxymethamphetamine (“Ecstasy”) on hepatic stellate cells. Biochem Pharmacol 67(6):1025–1033. doi:10.1016/j.bcp.2003.10.020
Montiel-Duarte C, Varela-Rey M, Oses-Prieto JA, Lopez-Zabalza MJ, Beitia G, Cenarruzabeitia E, Iraburu MJ (2002) 3,4-Methylenedioxymethamphetamine (“Ecstasy”) induces apoptosis of cultured rat liver cells. Biochim Biophys Acta 1588(1):26–32
Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ (2001) Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol 153(6):1265–1276
Nicotera P, Leist M, Ferrando-May E (1998) Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett 102–103:139–142
Ortyn WE, Hall BE, George TC, Frost K, Basiji DA, Perry DJ, Zimmerman CA, Coder D, Morrissey PJ (2006) Sensitivity measurement and compensation in spectral imaging. Cytometry A 69(8):852–862. doi:10.1002/cyto.a.20306
Patel MM, Belson MG, Longwater AB, Olson KR, Miller MA (2005) Methylenedioxymethamphetamine (ecstasy)-related hyperthermia. J Emerg Med 29(4):451–454. doi:10.1016/j.jemermed.2005.05.007
Pereira FC, Lourenco ES, Borges F, Morgadinho T, Ribeiro CF, Macedo TR, Ali SF (2006) Single or multiple injections of methamphetamine increased dopamine turnover but did not decrease tyrosine hydroxylase levels or cleave caspase-3 in caudate-putamen. Synapse 60(3):185–193. doi:10.1002/syn.20285
Pontes H, Sousa C, Silva R, Fernandes E, Carmo H, Remiao F, Carvalho F, Bastos ML (2008) Synergistic toxicity of ethanol and MDMA towards primary cultured rat hepatocytes. Toxicology 254(1–2):42–50. doi:10.1016/j.tox.2008.09.009
Proskuryakov SY, Konoplyannikov AG, Gabai VL (2003) Necrosis: a specific form of programmed cell death? Exp Cell Res 283(1):1–16
Rajapakse N, Silva E, Scholze M, Kortenkamp A (2004) Deviation from additivity with estrogenic mixtures containing 4-nonylphenol and 4-tert-octylphenol detected in the E-SCREEN assay. Environ Sci Technol 38(23):6343–6352
Richter C, Schweizer M, Cossarizza A, Franceschi C (1996) Control of apoptosis by the cellular ATP level. FEBS Lett 378(2):107–110
Sano R, Hasuike T, Nakano M, Kominato Y, Itoh H (2009) A fatal case of myocardial damage due to misuse of the “designer drug” MDMA. Leg Med (Tokyo) 11(6):294–297. doi:10.1016/j.legalmed.2009.09.003
Scaduto RC Jr, Grotyohann LW (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 76(1 Pt 1):469–477. doi:10.1016/S0006-3495(99)77214-0
Screaton GR, Singer M, Cairns HS, Thrasher A, Sarner M, Cohen SL (1992) Hyperpyrexia and rhabdomyolysis after MDMA (“ecstasy”) abuse. Lancet 339(8794):677–678
Shao X, Hu Z, Hu C, Bu Q, Yan G, Deng P, Lv L, Wu D, Deng Y, Zhao J, Zhu R, Li Y, Li H, Xu Y, Yang H, Zhao Y, Cen X (2012) Taurine protects methamphetamine-induced developmental angiogenesis defect through antioxidant mechanism. Toxicol Appl Pharmacol 260(3):260–270. doi:10.1016/j.taap.2012.03.003
Silva E, Kabil A, Kortenkamp A (2010) Cross-talk between non-genomic and genomic signalling pathways–distinct effect profiles of environmental estrogens. Toxicol Appl Pharmacol 245(2):160–170. doi:10.1016/j.taap.2010.02.015
Simantov R, Tauber M (1997) The abused drug MDMA (Ecstasy) induces programmed death of human serotonergic cells. FASEB J 11(2):141–146
Strobbe L, de Jaer CP, Louwerse ES, Rozendaal FW (2007) Fatal ecstasy intoxication in a 22-year-old man. Ned Tijdschr Geneeskd 151(30):1690–1694
Stumm G, Schlegel J, Schafer T, Wurz C, Mennel HD, Krieg JC, Vedder H (1999) Amphetamines induce apoptosis and regulation of bcl-x splice variants in neocortical neurons. FASEB J 13(9):1065–1072
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397(6718):441–446. doi:10.1038/17135
Upreti VV, Moon KH, Yu LR, Lee IJ, Eddington ND, Ye X, Veenstra TD, Song BJ (2011) Increased oxidative-modifications of cytosolic proteins in 3,4-methylenedioxymethamphetamine (MDMA, ecstasy)-exposed rat liver. Proteomics 11(2):202–211. doi:10.1002/pmic.201000203
Vanden Eede H, Montenij LJ, Touw DJ, Norris EM (2012) Rhabdomyolysis in MDMA intoxication: a rapid and underestimated killer “Clean” ecstasy, a safe party drug? J Emerg Med 42(6):655–658. doi:10.1016/j.jemermed.2009.04.057
Verhagen AM, Vaux DL (2002) Cell death regulation by the mammalian IAP antagonist Diablo/Smac. Apoptosis 7(2):163–166
Warren MW, Larner SF, Kobeissy FH, Brezing CA, Jeung JA, Hayes RL, Gold MS, Wang KK (2007) Calpain and caspase proteolytic markers co-localize with rat cortical neurons after exposure to methamphetamine and MDMA. Acta Neuropathol 114(3):277–286. doi:10.1007/s00401-007-0259-9
Wirth I, Correns A, Pragst F, Schmeling A (2007) Fatal MDMA-intoxication with postmortem diagnosis of hyperthermia. Arch Kriminol 220(5–6):141–151
Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, Korsmeyer SJ (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400(6747):886–891. doi:10.1038/23730
Zamaraeva MV, Sabirov RZ, Maeno E, Ando-Akatsuka Y, Bessonova SV, Okada Y (2005) Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell Death Differ 12(11):1390–1397. doi:10.1038/sj.cdd.4401661
Zhu JP, Xu W, Angulo JA (2005) Disparity in the temporal appearance of methamphetamine-induced apoptosis and depletion of dopamine terminal markers in the striatum of mice. Brain Res 1049(2):171–181. doi:10.1016/j.brainres.2005.04.089
Acknowledgments
4-MTA hydrochloride (racemic mixture) was a kind gift from Dr. David Nichols (Purdue University, West Lafayette, IN). The authors would like to thank Prof. John Sumpter and Dr. Chris Parris (Brunel University, London) for assistance with the flow cytometry. Dr. Sibylle Ermler (University of Brunel, London) is gratefully acknowledged for insightful discussions and useful suggestions. Thanks are also due to Diogo Fernandes (University of Brunel, London) and Maria Riverso (UCL School of Pharmacy, University of London) for technical assistance with Western blotting methodology. This work was supported by the Portuguese Research Council Fundação para a Ciência e para a Tecnologia (FCT) [SFRH/BD/45617/2008 to D.D.S. and Pest-C/EQB/LA0006/2011] and cofounded by the European Community financial support Programa Operacional Factores de Competitividade do Quadro de Referência Estratégico Nacional (QREN POFC).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Dias da Silva, D., Carmo, H., Lynch, A. et al. An insight into the hepatocellular death induced by amphetamines, individually and in combination: the involvement of necrosis and apoptosis. Arch Toxicol 87, 2165–2185 (2013). https://doi.org/10.1007/s00204-013-1082-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-013-1082-9