
Abstract
Salmonella, a Gram-negative bacterium that infects humans and animals, causes diseases ranging from gastroenteritis to severe systemic infections. Here, we discuss various strategies used by Salmonella against host cell defenses. Epithelial cell invasion largely depends on a Salmonella pathogenicity island (SPI)-1-encoded type 3 secretion system, a molecular syringe for injecting effector proteins directly into host cells. The internalization of Salmonella into macrophages is primarily driven by phagocytosis. After entering the host cell cytoplasm, Salmonella releases many effectors to achieve intracellular survival and replication using several secretion systems, primarily an SPI-2-encoded type 3 secretion system. Salmonella-containing vacuoles protect Salmonella from contacting bactericidal substances in epithelial cells and macrophages. Salmonella modulates the immunity, metabolism, cell cycle, and viability of host cells to expand its survival in the host, and the intracellular environment of Salmonella-infected cells promotes its virulence. This review provides insights into how Salmonella subverts host cell defenses for survival.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Salmonella species are among the most prevalent food-borne and facultative intracellular pathogen affecting humans and animals, including livestock and poultry. Hitherto, two species have been identified, i.e., Salmonella bongori, commonly found in cold-blooded animals, and Salmonella enterica, predominantly isolated from warm-blooded animals (Fookes et al. 2011). Salmonella enterica serovars are associated with four distinct clinical syndromes, i.e., typhoid fever, caused by S. enterica subsp. enterica serovar Typhi (S. Typhi); paratyphoid fever, caused by S. enterica subsp. enterica serovar Paratyphi (S. Paratyphi); gastroenteritis, caused by nontyphoidal Salmonella (NTS) serovars in immunocompetent individuals; and bacteremia, caused by NTS serovars in immunocompromised individuals (Gal-Mor 2019). Many virulence-related gene clusters exist in the genome of Salmonella, known as Salmonella pathogenicity islands (SPIs), such as SPI-1, 2, 4, 6, and 19. Most SPIs of Salmonella encode the structural proteins and secreted effectors of secretion systems (Bao et al. 2020). It should be noted that T6SS gene clusters have been identified within pathogenicity islands SPI-6, 19, 20, 21, and 22, which are differentially distributed among Salmonella serotypes and encode different T6SSs in terms of functions (Hernandez et al. 2020). SPI-1-encoded type 3 secretion system (T3SS1) is mainly used by Salmonella to invade host cell when it is outside of host cell. Following invasion into epithelial cells, Salmonella survives in vesicles called Salmonella-containing vacuoles (SCVs), and SPI-2-encoded T3SS (T3SS2) is largely used for the intracellular replication when the pathogen is inside SCVs.
The SPI-4-encoded type 1 secretion system (T1SS) and T3SS1 trigger the attachment of Salmonella to the intestinal epithelium. Salmonella can thus effectively invade intestinal epithelial cells, preferentially in the terminal ileum by injecting an array of effector proteins via T3SS1 and type 6 secretion system (T6SS) (Sana et al. 2016; Bao et al. 2020). One of the primary targets invaded by Salmonella are M cells (a specialized intestinal epithelial cell type), which are interspersed among enterocytes and cover Peyer’s patches containing lymphoid follicles (Griffin and McSorley 2011). Salmonella utilizes various effectors secreted from the T3SS2, a T6SS, and a T4SS to evade immune responses and survive within the SCVs in host cells (Bao et al. 2020). Macrophages play a crucial role in facilitating systemic infection by Salmonella. Once the pathogen traverses the epithelial layer, it is phagocytosed and persists within the SCVs of macrophages. Although NTS infections typically remain localized in the intestinal mucosa and mesenteric lymph nodes, other S. enterica serovars can disseminate throughout the body via the bloodstream or lymphatic system, leading to the colonization of the liver, spleen, kidney, and bone marrow, eventually giving rise to typhoid fever and bacteremia when not controlled during the initial stages of infection (Ruby et al. 2012; Orf and Cunnington 2015). The processes whereby S. Typhi causes systemic infection in the host is shown in Fig. 1.

Processes whereby Salmonella enterica subsp. enterica serovar Typhi causes systemic infection in its host. (1) Ingested Salmonella enters the host esophagus through contaminated food and water. (2) Salmonella survives the highly acidic environment in the stomach. (3) Salmonella colonizes the intestines of the host. (4) Salmonella invades the M cells and enterocytes of the intestines. (5) Salmonella released from M cells and enterocytes are engulfed by phagocytes and disseminated through the lymphatic and circulatory systems. (6) Salmonella colonizes internal organs such as the liver, spleen, and kidneys. (7) Increased numbers of Salmonella enter blood vessels from different infected organs, leading to lethal bacteremia
Extensive research has been conducted on interactions between Salmonella and host cells. Nonetheless, the precise pathogenic mechanisms underlying Salmonella infection still require further investigation. In this review, we aim to explore the survival and immune evasion strategies employed by Salmonella to gain a better understanding of the pathogenic processes of this intracellular pathogen.
Survival of Salmonella in host intestine
To infect host, Salmonella must survive in the host intestines and establish its growth advantage. Salmonella employs a strategy that involves outcompeting intestinal commensal microbes to colonize the intestines of humans and animals, which is achieved by injecting effector proteins into prokaryotic cells through T6SS to eliminate intestinal microbes (Sana et al. 2016; Sibinelli-Sousa et al. 2020; Amaya et al. 2022). Inside the gut, Salmonella takes advantage of the host-derived secondary metabolic products induced by inflammation, such as thiosulfate, tetrathionate, ethanolamine, and nitrate. Inflammation triggers phagocytes (such as neutrophils and macrophages) to generate superoxide anion radicals that oxidize hydrogen sulfide to thiosulfate and tetrathionate (Winter and Baumler 2011). Simultaneously, the membranes of the dead intestinal cells produce ethanolamine. Salmonella can grow on ethanolamine as a carbon source while using tetrathionate for anaerobic respiration. Similar to tetrathionate, host-derived superoxide anion radicals react with inflammation-induced nitric oxide to form nitrate, which promotes the intestinal growth of Salmonella as an electron acceptor (Behnsen et al. 2015). Salmonella can also grow using metabolites produced by other intestinal microorganisms, such as lactic acid and propionic acid (Gillis et al. 2018; Shelton et al. 2022).
Entry of Salmonella into host cells
Adhesion of Salmonella on host cells
Salmonella uses a variety of fimbrial and nonfimbrial structures to adhere to host cells (Hassuna et al. 2017). Fimbrial adhesins include type 1 fimbriae (Fim), plasmid-encoded fimbriae (Pef), long polar fimbriae (Lpf), thin aggregative fimbriae (Agf). Nonfimbrial structures include autotransporter adhesins, including ShdA, MisL, and SadA, and T1SS-secreted adhesins, such as BapA, SiiE. These Salmonella adhesins bind to corresponding receptors on host cells to adhere to host cells (Bao et al. 2020). The four main fimbrial operons—fim, pef, lpf, and agf—are all associated with virulence; however, mutants lacking only one of these operons show little reduction in virulence. Additionally, S. Typhi has a 27 kDa outer membrane protein T2544/ PagN that binds to laminin with high affinity and plays a vital role in bacterial adhesion to the host (Ghosh et al. 2011).
T3SS1-independent invasion into nonphagocytic cells
Rck, PagN, and HlyE are invasins that play pivotal roles in the invasion process of Salmonella. Salmonella expresses an outer membrane protein, Rck, which interacts with the epidermal growth factor receptor on the plasma membrane of host cells, activating host cell signaling pathways that lead to actin polymerization and bacterial entry (Wiedemann et al. 2016). Additionally, Rck-mediated infection increases the number of S-phase (DNA replication phase) cells, creating an environment conducive to Rck-mediated bacterial entry (Mambu et al. 2020). Moreover, Rck provides resistance to complement killing by preventing the formation of membrane attack complexes (Heffernan et al. 1992). The outer membrane protein PagN, a hemagglutinin of S. enterica serovar Typhimurium (S. Typhimurium), contributes to invasion of epithelial cells by utilizing heparinated proteoglycan as a receptor (Lambert and Smith 2008, 2009). HlyE is a pore-forming hemolysin present in the serovars Typhi and Paratyphi A, and hlyE mutants are impaired in their ability to invade. Moreover, the heterologous expression of HlyE in S. Typhimurium improves its colonization in deep organs of mice. Therefore, HlyE can play a vital role in invasion, but the precise mechanism whereby a hemolysin enhances the invasion of intracellular pathogens remains unknown (Fuentes et al. 2008). Other invasins should be existed as there exists many unidentified genes encoding functionally unknown proteins in the genome of Salmonella.
T3SS1-dependent invasion
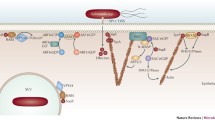
The T3SS1, encoded by SPI-1 and its secreted effectors, promotes the invasion of Salmonella into host cells, particularly epithelial cells (Fig. 2). For example, the effectors SopE, SopE2, SopB, SopD, SipA, and SipC secreted by T3SS1 all contribute to the invasion of host cells by Salmonella, mainly due to their ability to facilitate the rearrangement of the actin cytoskeleton of host cells (Clark et al. 2011). Although the primary mode of the entry of Salmonella into phagocytes is phagocytosis, the effector proteins required for Salmonella to enter monocytes and macrophages are highly similar to those required for entry into epithelial cells (Di Martino et al. 2019).

Salmonella effectors secreted by its T3SS1 promote the invasion of host cells
SipA and SipC work cooperatively to promote the invasion of Salmonella into host cells by directly binding to actin adjacent to the host cell membrane, with SipC being a component of T3SS1 (Fig. 2). The C-terminus of SipC possesses an F-actin-binding site, leading to F-actin polymerization and nucleation (LaRock et al. 2015), facilitating the formation of F-actin filament bundles. SipA enhances the action of SipC by stimulating actin polymerization, inhibiting the depolymerization of F-actin caused by actin-depolymerization factor (ADF) and/ or cofilin, and preventing the severing of F-actin caused by gelsolin (Dai et al. 2004; McGhie et al. 2004).
Unlike SipA and SipC, SopE, SopE2, and SopB promote the internalization of Salmonella into host cells by indirectly acting on actin (Fig. 2). The guanine nucleotide exchange factors SopE and SopE2 can transfer GTP to Rho family guanosine triphosphatases (GTPases), such as Cdc42, Rac1, and RhoG. GTP-bound GTPases can then induce actin cytoskeleton rearrangement and stimulate cell membrane ruffling, which promotes the invasion of Salmonella into cells. However, after Salmonella enters the cell, the effector SptP—a GTPase-activating protein—can normalize cell morphology as it hydrolyzes GTP on GTPases into GDP.
SopB and SopD act together to promote the scission of the host plasma membrane, driving the invasion of Salmonella into host cells, generating SCVs (Fig. 2). SopB, an inositol phosphate phosphatase, dephosphorylates phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), thus forming phosphatidylinositol 5-phosphate (PI(5)P), leading to plasma membrane fission (Terebiznik et al. 2002). Moreover, SopB increases the amounts of phosphatidylinositol 3-phosphate in the membranes of bacteria-containing vacuoles, thereby promoting SCV maturation and intracellular replication of Salmonella (Hernandez et al. 2004; Mallo et al. 2008). SopD also drives the scission of the cell plasma membrane by promoting the inactivation of Rab10 and consequent recruitment of Dynamin-2 (Boddy et al. 2021). Furthermore, the phosphatase activity of SopB activates SH3-containing guanine nucleotide exchange factor (SGEF), a guanosine nucleotide exchange factor for RhoG, resulting in actin rearrangements that facilitate bacterial internalization (Bakowski et al. 2010).
Phagocytosis of Salmonella by host phagocytic cells
Salmonella can be phagocytosed by typical phagocytes such as macrophages and neutrophils. However, macrophages offer a relatively more permissive environment for Salmonella survival and replication than provided by neutrophils (Tyrkalska et al. 2016). Phagocytosis of Salmonella can occur through opsonin-independent mechanisms, wherein phagocytes recognize the surface structural determinants of Salmonella via pattern recognition receptors (PRRs). Alternatively, opsonin-dependent phagocytosis occurs when Salmonella is labeled with opsonins such as antibodies or complements (Drecktrah et al. 2006), facilitating recognition by Fc receptors or complement receptors on phagocytes, leading to phagocytosis.
Several PRRs, including scavenging receptors (CD36), mannose receptors (CD206), and toll-like receptors (TLRs), have been reported to promote the phagocytosis of Salmonella. Overexpression of CD36 and TLR4 increases the number of bacteria phagocytosed into cells (Wan et al. 2018). The activation of PRRs not only enhances the phagocytosis of Salmonella but also initiates immune signaling pathways involved in antigen processing, cytokine/ chemokine production, and bacterial killing. After being phagocytosed by macrophages, Salmonella reduces the negative electric charge on the macrophage surface. This effect reverses the galvanotaxis direction of macrophages, keeping the macrophages containing Salmonella migrating to lymphatic drainage, bloodstream, or both (Sun et al. 2019).
Immune evasion by Salmonella
Inhibition of autophagy
Numerous pathogenic bacteria, including S. Typhimurium, Shigella flexneri, Burkholderia pseudomallei, Listeria monocytogenes, Streptococcus pyogenes, Mycobacterium tuberculosis, and Legionella peumophila, reportedly induce autophagy. In mice, the autophagy of intestinal epithelial cells has been shown to prevent the spread of invasive S. Typhimurium (Benjamin et al. 2013). In the case of Salmonella-infected macrophages, canonical autophagy, xenophagy (selective autophagy against bacteria), and LC3-associated phagocytosis (LAP) can be triggered. Autophagosomes and vesicles transport Salmonella to lysosomes, limiting its growth and replication (Wang et al. 2022). Salmonella growth increases in cells with lower than normal lysosomal activity and decreases in cells with higher than normal lysosomal activity (McGourty et al. 2012). During Salmonella-induced LAP, LC3 is recruited to the single membrane of the phagosome, unlike canonical autophagy, wherein LC3 marks double membranes of autophagosomes (Masud et al. 2019a). Notably, LAP induced by S. Typhimurium is mediated by the host protein Rubicon, which typically acts as a negative regulator of canonical autophagy and as an inducer of LAP (Masud et al. 2019b). It is believed that LAP is the primary autophagy pathway directed against Salmonella under macrophage-mediated immune defense conditions (Masud et al. 2019b).
Intracellular pathogens like Salmonella can employ a common strategy for entering a vacuolar environment during host cell invasion, with Salmonella surviving in an acidic and nutrient-deficient compartment known as SCV (Lahiri et al. 2010). One study suggested that SCVs divide along with Salmonella replication, resulting in an SCV containing a single Salmonella. Additionally, Salmonella infection reduces the number of lysosomes in host macrophages. Therefore, it has been inferred that the number of acidic lysosomes in cells infected with Salmonella is insufficient to cope with increasing SCV numbers (Eswarappa et al. 2010). Studies have shown live salmonellae to exist in SCVs, which avoid fusion with lysosomes. SopB reduces the membrane surface charge of nascent SCVs by reducing the levels of negatively charged PI(4,5)P2 and phosphatidylserine, resulting in the dissociation of numerous host-cell proteins involved in endocytic trafficking from SCVs, and this effect inhibits SCV–lysosome fusion (Bakowski et al. 2010). Although SCVs exhibit lysosomal characteristics, they contain fewer hydrolytic enzymes. The effector SifA forms a stable complex with sphingosine kinase interacting protein and Rab9 in infected cells that disrupts the retrograde transport of hydrolytic enzymes to lysosomes by the mannose-6-phosphate receptor, thereby reducing the bactericidal effect of lysosomes (McGourty et al. 2012). Therefore, Salmonella exploits T3SS-secreted effectors to avoid the fusion of SCVs with lysosomes and reduce the levels of hydrolytic enzymes in lysosomes (Fig. 3).

Salmonella inhibits the autophagic clearance of host cells
Cellular stress and infection can lead to the formation of ubiquitinated aggregates in both nonimmune and immune cells. These ubiquitinated aggregates are recognized and bound by the autophagy receptor p62 (sequestosome 1), which directly interacts with LC3, facilitating the degradation of ubiquitinated protein aggregates via autophagy. Ubiquitination structures form during intracellular replication of Salmonella, and xenophagy pathways selectively bind ubiquitinated Salmonella or SCVs to autophagosomes via autophagy receptor proteins (Fig. 3). The T3SS2-delivered effector SseL has a unique role in deubiquitinating ubiquitin aggregates on the surface of SCVs, reducing the recruitment of autophagy markers (p62 and LC3), and, in turn, limiting autophagy against Salmonella (Mesquita et al. 2012). Bacterial invasion often damages the host cell membrane, leading to acute amino acid starvation in the cytoplasm and triggering xenophagy. However, in Salmonella-infected cells, membrane integrity and cytoplasmic amino acid levels return to normal, favoring the reactivation of mTOR on the SCV surface and Salmonella’s escape from autophagy (Tattoli et al. 2012). In conclusion, Salmonella has specific ways to combat the xenophagy and LAP of the host.
With the vacuolar damage caused by Salmonella, vacuolar (H+)-ATPase (V-ATPase) recruits ATG16L1 to SCVs and initiates autophagy; nevertheless, this process is inhibited by the T3SS effector SopF, which targets Gln124 of ATP6V0C (the main component of the C ring) in V-ATPase for ADP-ribosylation (Xu et al. 2019). In both epithelial cells and macrophages, SpvB (a protein encoded by the pSLT plasmid) also inhibits the autophagy of host cells during autophagosome formation, possibly by depolymerizing the actin cytoskeleton (Chu et al. 2016). As SpvB acts as an intracellular toxin, it covalently modifies monomeric actin, resulting in F-actin filament loss in human macrophages infected with Salmonella (Browne et al. 2008). Two other effectors of Salmonella, i.e., SseF and SseG, inhibit autophagosome formation by interfering with Rab1A signaling (Feng et al. 2018). The binding of these two effectors to Rab1A (a GTPase) disrupts the interaction of Rab1A and transport protein particle III complex, a guanine nucleotide exchange factor, and blocks the activation of Rab1A. The interruption of Rab1A signaling blocks the recruitment and activation of Unc-51-like autophagy-activating kinase-1 and reduces the production of phosphatidylinositol 3-phosphate, ultimately preventing autophagosome formation. Additionally, SpvC inhibits autophagosome formation via phosphothreonine lyase activity (Zhou et al. 2021). In conclusion, Salmonella inhibits autophagy initiation and autophagosome formation via many effector proteins.
Focal adhesion kinase (FAK) recruited to the SCV surface in Salmonella-infected cells amplifies signaling through the Akt-mTOR pathway and inhibits autophagy, possibly because FAK negatively regulates autophagy associated with Beclin-1 (Cheng et al. 2017). AvrA, an effector secreted by both T3SS1 and T3SS2, which can reduce Beclin-1 levels through the c-Jun N-terminal kinase (JNK) pathway, also inhibits autophagy. Increased cholesterol levels in the plasma membrane at the site of Salmonella entry into host cells reportedly leads to SCV formation and decreased autophagy (Huang 2014). These strategies used by Salmonella to inhibit autophagy wait for further exploration and explanation.
Polarization of M1 macrophages to M2 macrophages
The M1 macrophages are characterized by the expression of high levels of proinflammatory cytokines, CXCL9, CXCL10, inducible nitric oxide synthase, and reactive oxygen intermediates and exhibit strong antigen presentation abilities. Activation of M1 macrophages occurs through pathogen-associated molecular patterns, such as lipopolysaccharides (LPSs) and inflammatory cytokines, i.e., interferon-γ or tumor necrosis factor (TNF). While M1 macrophages play crucial roles in bactericidal activities, clearance of intracellular pathogens, and antitumor responses, they can also inadvertently inhibit the proliferation of neighboring cells and cause damage to adjacent tissues.
In contrast, atypical M2 macrophages highly express interleukin (IL)-4Rα and CD301 and are activated by cytokines produced by T helper-2 cells, including IL-4 or IL-13. Notably, M2 macrophages contribute to the proliferation of neighboring cells and participate in tissue repair and combating multicellular eukaryotic parasites. However, they also play a role in promoting pathological fibrosis and tumor growth (Brodsky 2020). They produce higher levels of anti-inflammatory cytokines, such as IL-10, and higher levels of ornithine and polyamine via the arginase pathway. Salmonella preferentially replicates in M2 rather than M1 bone marrow-derived macrophages (Lathrop et al. 2015). Notably, S. Typhimurium infection increases the production of proinflammatory cytokines TNF-α, IL-1β, IL-6, and CXCL8 in M1 macrophages, and the levels of these cytokines are lower in anti-inflammatory M2 macrophages than in M1 macrophages (Lathrop et al. 2015).
Salmonella can polarize macrophages to the M2 state by utilizing the effector SteE, which enhances the survival of Salmonella in macrophages. The serine/ threonine kinase glycogen synthase kinase-3 (GSK3) promotes SteE phosphorylation. The protein complex containing phosphorylated SteE and GSK3 binds to signal transduction and transcription activator 3 (STAT3), leading to the phosphorylation of STAT3 at tyrosine-705 and activation of this transcription regulator. The activated STAT3 (pY705) drives macrophages towards polarization into the M2 state and promotes the production of the anti-inflammatory cytokine IL-10 (Panagi et al. 2020).
Modulation of inflammatory pathways
Nuclear factor (NF)-κB belongs to a family of crucial transcriptional regulation factors involved in immune homeostasis in intestinal cells and early pathogen detection, including Salmonella. It consists of homo- and heterodimeric complexes formed by various Rel/NF-κB proteins. In mammals, five known Rel/NF-κB proteins are RelA (p65), RelB, c-Rel, p50 (NF-κB1), and p52 (NF-κB2). Under normal conditions, NF-κB is bound to the inhibitory protein IκB and remains inactive in the cytoplasm. Upon stimulation by membrane-bound or cytoplasmic PRRs, IκB kinase is activated, leading to IκB phosphorylation and dissociation from NF-κB. The phosphorylated IκB is then ubiquitinated by a specific E3 ubiquitin ligase SCFβ−TrCP for degradation, enabling NF-κB to enter the nucleus and activate the expression of target genes (Pilar et al. 2013). Activated NF-κB upregulates the expression of proinflammatory cytokines and enhances inflammatory response. In the early infection phase, a few Salmonella effectors and other components are needed for invasion and the induction of inflammation. Once bacteria are inside the host cells, Salmonella secretes other effectors to down-regulate inflammatory responses. Salmonella effectors, such as SopE, SopE2, and SopB, play a role in activating GTPases of the Rho family (Cdc42, Rac1, and RhoG), disrupting tight junctions, stimulating innate immune responses, and promoting inflammation in intestinal epithelial cells. These processes are critical for Salmonella to invade organs, such as the liver and spleen, during systemic infections (Huang et al. 2004).
Cdc42 and Rac1 activation ultimately triggers the NF-κB pathway, in addition to mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated protein kinases (ERKs), JNKs, and p38 MAPKs. This activation leads to significant transcriptional reprogramming in the host cell, resembling the response of innate immune receptors to stimulation (Du and Galan 2009). Salmonella LPS and TNF-α also activate the NF-κB pathway. The Salmonella effector SipA contains the central region of the SipA–SipA interaction (F2) required for T3SS1 translocation, which is critical for NF-κB activation. Additionally, NOD1, NOD2, and their downstream adapter RIP2 are required for SipA to activate the NF-κB pathway; however, its underlying mechanism remains unknown (Yang et al. 2021a).
Salmonella employs various strategies to ensure survival upon entering host cells by downregulating the immune response in the host. One such approach is the downregulation of NF-κB signaling (Table 1). Salmonella inhibits NF-κB and ERK signaling by hampering the phosphorylation of IκBα and ERK in macrophages, respectively. This anti-inflammatory effect of Salmonella is mediated by Nlrp12, as Nlrp12-deficient mice display higher tolerance to S. Typhimurium infection, with significantly upregulated NF-κB and ERK signaling pathways compared with that in wild-type mice (Zaki et al. 2014). AvrA, an effector, acetylates MAPK kinase 4 (MKK4) and MAPK kinase 7 (MKK7), inhibiting NF-κB signaling. Additionally, AvrA can deubiquitinate IκBα, further preventing the activation of NF-κB. AvrA-induced acetylation of MKK4 and MKK7 effectively inhibits the JNK signaling pathway, thus suppressing JNK-mediated apoptosis and rapid bacterial spread. Inhibition of apoptosis through the JNK pathway may represent a conservative survival strategy for intracellular Salmonella (Wu et al. 2012). SseL, another effector, deubiquitinates IκBα and ribosomal protein S3 (RPS3), inhibiting its degradation and thereby suppressing the activation of NF-κB in macrophages (Wu et al. 2018).
The zinc metalloprotease GtgA, GogA, and PipA secreted by Salmonella can recognize the relatively conserved P1′ sites in both subunits RelA (p65) and RelB of the NF-κB transcription factor, leading to the cleavage of both RelA and RelB transcription factors, ultimately inhibiting the NF-κB pathway (Jennings et al. 2018). The GogB effector interacts with Skp1 and F-box only 22 proteins to inhibit the poly-ubiquitination of IκB, thereby preventing IκB degradation and blocking the NF-κB pathway (Pilar et al. 2013). SpvB inhibits the activation of NF-κB, which is associated with the downregulated expression and phosphorylation of IκB kinase-β, as well as the upregulation of E3 ligase Kelch-like ECH-associated protein 1 (KEAP1). The underlying mechanism may be intricate, as KEAP1 downregulates Nrf2 expression, an inhibitor of the NF-κB pathway, through several axes (Yang et al. 2021b). The effector SpvD, located on the pSLT plasmid and secreted by T3SS2, interacts with exportin Xpo2, disrupting normal importin-α circulation from nucleus to cytoplasm, leading to the accumulation of importin-α in the nucleus and the subsequent failure of p65 nuclear translocation, inhibiting the NF-κB pathway (Rolhion et al. 2016).
A group of effectors encoded by SPI-2, including SseK1, SseK2, and SseK3, inhibits the TNF-α-activated NF-κB pathway. SseK1, an effector secreted by T3SS2 with protein-arginine N-acetylglucosaminyltransferase activity, modifies TNF-α receptor 1 (TNFR1), TNFR1-associated death domain protein (TRADD), and FAS-associated death domain protein (FADD). Similarly, SseK3 modifies TNFR1 and TRADD and is a member of the TNFR superfamily 10B. Consequently, SseK1 and SseK3 jointly prevent TNF-α-induced NF-κB pathway activation and inhibit macrophage apoptosis. Although SseK2 translocates during Salmonella infection in macrophages, its effect on the NF-κB pathway is limited and appears confined to the arginine N-acetylglucosaminylation of FADD (Xue et al. 2020). The E3 ubiquitin ligase SspH1 can inhibit NF-κB signaling, but its precise mechanism of action remains unclear, as it does not affect the activity of NF-κB despite its interaction with PKN1 through the LLR domain (Keszei et al. 2014).
SptP has GTPase-activated protein (GAP) activity, which can lead to the inactivation of Cdc42 and Rac1. Thus, SptP downregulates the MAPK signaling pathway. Additionally, the tyrosine phosphatase activity of SptP inhibits MAPK activation. Both GAP and tyrosine phosphatase activities of SptP inhibit Raf activation, ERK activation, and IL-8 production (Zaki et al. 2014), thereby reducing the inflammatory response of host cells and enhancing the intracellular replication of Salmonella (Lin et al. 2003). The Salmonella phosphothreonine lyase SpvC inactivates ERK1/2, p38, and JNKs by β-elimination, thereby inhibiting their downstream signaling pathways (Haneda et al. 2012).
Salmonella interferes with antigen presentation in the host
During Salmonella infection, both innate and adaptive immune responses of the host are initiated; however, Salmonella utilizes strategies to dampen these immune pathways (Schleker et al. 2012). Salmonella actively hinders the migration of dendritic cells and interferes with antigen presentation via MHC-I and MHC-II molecules. Several studies have identified key effectors, such as SifA, SspH2, SlrP, PipB2, and SopD, that play crucial roles in inhibiting antigen presentation, whereas SseF and SseG exhibit a milder impact on this process than that of other effectors (Halici et al. 2008). Additionally, an unidentified Salmonella effector disrupts MHC-I antigen presentation and impedes the transportation of antigen-carrying vesicles to MHC-II compartments. Interaction between SlrP and ERdj3, an endoplasmic reticulum DnaJ homolog, leads to the inhibition of antigen presentation (Bernal-Bayard et al. 2010). Certain T3SS2-secreted effectors are involved in curtailing dendritic cell migration along chemokine gradients. For instance, SseI reduces the migration of Salmonella-infected dendritic cells from the mouse gut to mesenteric lymph nodes, potentially causing delays in T-cell responses and enhancing Salmonella virulence. Structural and biochemical analyses have revealed the ability of SseI to deamidate heterotrimeric G proteins of the Gαi family, including Gαi2, leading to persistent G-protein activation (Cerny and Holden 2019).
Antigen presentation by dendritic cells to CD4+ T cells is crucial for controlling intracellular bacterial replication during systemic Salmonella infection, and specific effectors secreted by T3SS2 of Salmonella disrupt this process. In dendritic cells, SteD interacts with mature MHC-II (mMHC-II) and host E3 ubiquitin ligase membrane-associated RING-CH (MARCH) 8 in vesicles, promoting the ubiquitination of mMHC-II β chains. This ubiquitination of mMHC-II β chains results in mMHC-II degradation, directly obstructing antigen presentation mediated by MHC-II. Additionally, SteD reduces the surface levels of the costimulatory molecule CD86, which is necessary for full T-cell activation and is regulated by MARCH 1. Therefore, SteD inhibits T cell activation during Salmonella infection in mice (Alix et al. 2020). SteD also enhances CD97 ubiquitination for degradation. The removal of CD97 by SteD hinders interactions between dendritic cells and T cells, ultimately reducing T cell activation, independent of its effect on MHC-II (Cerny et al. 2021). However, Salmonella indirectly impairs CD4+ T cell function via asparaginase, which converts exogenous L-asparagine to aspartic acid and ammonia, leading to downregulation of the T cell receptor and inhibition of T cell proliferation (Kullas et al. 2012). Salmonella utilizes T3SS2 to clear CD4+ T cells, and this process correlates with an increase in programmed death ligand 1 (PD-L1) expression on antigen-specific CD4+ T cells. The binding of PD-L1 to its programmed cell death protein 1 receptor present on dendritic cells restricts the proliferation of antigen-specific CD4+ T cells (Cerny and Holden 2019).
Inhibition of antimicrobial peptides and the use of hepcidin by Salmonella
Hitherto, extensively studied antimicrobial peptides include lysozymes, hepcidin, defensins, and cathelicidins. Antimicrobial peptides can disrupt cell walls and outer membranes in Salmonella, followed by host-derived proteases hydrolyzing periplasmic proteins; however, this process is often hindered by SCVs (Slauch 2011). Bacteria that establish symbiotic relationships with hosts or cause diseases have evolved diverse strategies to evade these antimicrobial peptides. Lysozymes represent a critical element of the ancient innate immune system in animals. They are secreted extracellularly and function to hydrolyze peptidoglycan, a constituent of the bacterial cell wall. Salmonella has developed multiple mechanisms to counteract lysozymes, including the production of lysozyme inhibitors. Salmonella’s lysozyme inhibitors, MliC (membrane-bound) and PliC (periplasmic), exhibit inhibitory effects on C-type lysozymes (Callewaert et al. 2008).
Hepcidin produced by hepatocytes can decrease iron release from macrophages. Notably, S. Typhimurium exploits this metabolic pathway for its benefit, growth, or survival, as S. Typhimurium infection induces the expression of estrogen-related receptor γ, which, in turn, stimulates the expression of hepcidin. Thus, S. Typhimurium ensures sufficient intracellular iron concentrations while infecting macrophages (Kim et al. 2014). Additionally, Salmonella promotes hepcidin synthesis in a STAT3-dependent manner through the effector SpvB. The liver contains a high content of hepcidin, which may be a primary reason why the liver is a major site for the growth and replication of Salmonella in systemic infection. Hepcidin binds to ferroportin in an outward-open conformation to promote its degradation, thereby increasing the iron content in the cytoplasm of host cells (Deng et al. 2021). As iron is an essential element for Salmonella growth, increased iron levels are associated with increased hepcidin levels to facilitate Salmonella replication (Fig. 4) (Kim et al. 2014).

Salmonella infection upregulates hepcidin production by host cells to inhibit iron export and promote intracellular replication
Resistance against reactive oxygen species and reactive nitrogen species
Salmonella possesses several superoxide dismutases, namely SodA, SodB, and SodC, which utilize Mn, Fe, or Cu–Zn as cofactors. These superoxide dismutases protect cells against superoxide anion radicals produced within them. Notably, a Salmonella sodC mutant exhibits significantly reduced intracellular survival and virulence in mice and shows heightened sensitivity to superoxide and nitric oxide (NO) (De Groote et al. 1997). While reactive nitrogen species (RNS) have traditionally been regarded as critical mediators of host defense against pathogens, Salmonella can exploit the RNS produced during infection to enhance its virulence (Henard and Vazquez-Torres 2011). Notably, NO, a type of RNS, can inhibit bacterial growth by modifying various intracellular targets, including protein thiols, heme-containing proteins, thiol-coordinated metals, lipid bilayers, and DNA. Salmonella has evolved multiple mechanisms to limit its exposure to NO during infection. For instance, the flavin protein Hmp of Salmonella converts NO to nitrate, attenuating its inhibitory effects on Salmonella growth. In addition, NO can be oxidized to dinitrogen trioxide (N2O3) or peroxynitrite, mediating the deamination and oxidation of DNA bases, respectively. For example, N2O3 deaminates cytosine to form uracil, which can mutate DNA; however, the uracil DNA glycosylase of Salmonella can remove uracil (dU) that is mistakenly inserted into DNA, resulting in apurinic/apyrimidinic (AP) sites, thus avoiding mutations. Peroxynitrates oxidize adenine, guanine, and xanthine nucleosides, resulting in genetic mutations. Formamidopyrimidine-DNA glycosylase (Fpg) in Salmonella can remove oxidized guanine (dG) and limit peroxynitrate-mediated supermutations. In addition, adenine DNA glycosylase (MutY) blocks genetic mutations in Fpg-deficient cells. Salmonella can avoid NO-induced mutations, such as endonuclease V (Nfi) mutations, by not producing AP sites. In summary, the base excision repair system of Salmonella can prevent NO damage to DNA (Richardson et al. 2009).
Inhibition of neutrophil function
Host neutrophils defend against pathogens in various ways, such as via phagocytosis, degranulation, cytokine production, and neutrophil extracellular traps (Delgado-Rizo et al. 2017); however, despite their potent antibacterial activity, their ability to clear pathogens is partly impaired by Salmonella. In the early stages of infection, neutrophils contain live Salmonella that can escape into more permissive cell types. D-alanine exists in peptidoglycan stem peptides of Salmonella and is a substrate of D-amino acid oxidase (DAO) produced by host cells. Salmonella prevents DAO-induced oxidative damage in neutrophils by expressing a D-alanine ABC importer (DalS). DalS-mediated neutrophil DAO subversion is a novel host–pathogen interaction that enhances Salmonella survival during systemic infections (Tuinema et al. 2014). SptP inhibits Cdc42 and Rac1 activities after Salmonella invasion into epithelial cells, thus inhibiting IL-8 production and reducing neutrophil recruitment (Lin et al. 2017).
Disruption of host metabolism and looting of nutrition
Carbon sources
Salmonella infection upregulates the glycolytic pathway of macrophages and downregulates the tricarboxylic acid cycle, mainly manifested by the accumulation of glycolytic intermediates and is related to a few effectors, such as SopE2 and SseK3 (Yu et al. 2020; Jiang et al. 2021). The upregulation of host glycolysis contributes to the intracellular replication of S. Typhimurium and systemic infection in mice, as glucose is the primary carbon source required for intracellular replication of S. Typhimurium (Bowden et al. 2009). Other glycolytic products such as 2-phosphoglycerate, 3-phosphoglycerate, and phosphoenolpyruvate can also be used as carbon sources by intracellular S. Typhimurium (Jiang et al. 2021).
Recently, we demonstrated that S. Typhi—which causes systemic infection in humans—increases glucose content, glucose uptake, and glycolysis rates in human primary macrophages and reduces oxidative phosphorylation levels (Wang et al. 2021). Additionally, Salmonella in macrophages can directly obtain fatty acids from SCVs using FadL transporters and degrade them via β-oxidation (Taylor and Winter 2020). Different types of macrophages differ significantly regarding carbon metabolism during Salmonella infection. Glycolysis is significantly upregulated in M1 macrophages compared with that in M2 macrophages. In M2 macrophages, Salmonella activates transcription factor peroxisome proliferator-activated receptor δ, which drives fat oxidation in host cells, supplying additional available carbon sources to Salmonella (Eisele et al. 2013).
Nitrogen sources
Previous studies have shown that Salmonella uses various amino acids in host cells—such as arginine, glutamine, aspartic acid, asparagine, alanine, and proline—for survival and replication (Popp et al. 2015). Salmonella infection increases arginine uptake and expression of two cationic amino acid transporters, mCAT1 and mCAT2B, in macrophages and dendritic cells. To access the cytoplasmic arginine pool of macrophages, live Salmonella recruits mCAT1 transporter to SCVs. Salmonella present in SCVs acquires host-derived arginine via arginine transporter ArgT, as the knockout of argT attenuates the growth of Salmonella in mouse infection models and macrophages (Das et al. 2010). Additionally, polyamines—the degradation products of arginine—contribute to the translation of HilA, the main positive regulator of SPI-1 genes (Guerra et al. 2020). We recently reported that Salmonella utilizes inflammation-induced nitrate to replicate in macrophages and cause systemic infection (Li et al. 2022). How intracellular Salmonella obtains other amino acids and whether these amino acids promote the virulence of Salmonella are thus worth exploring in the future.
Metal ions
Salmonella requires metal ions (such as iron, magnesium, and zinc) from host cells for normal metabolism, growth, and virulence. Iron promotes the growth and adhesion of Salmonella, as well as its invasion and translocation across the epithelial monolayer (Kortman et al. 2012). Salmonella can directly acquire free ferrous iron using a ferrous transporter, Feo. Salmonella secretes two catecholate-type siderophores to obtain iron, including enterobactin and its glycosylated derivative, salmochelin (Crouch et al. 2008). Intracellular Salmonella maintain intracellular magnesium balance through regulatory systems, such as PhoQ/PhoP and magnesium ion transport systems, thus contributing to virulence gene expression and intracellular survival (Choi and Groisman 2016). The sequestration of zinc by macrophages is considered a vital host defense strategy against intracellular Salmonella infection; however, Salmonella has evolved several strategies to obtain zinc, such as using a high-affinity zinc transport system. Salmonella infection increases zinc levels in macrophages, helping to inhibit the transcriptional activation of p65 and thus inhibiting the production of ROS and RNS mediated by NF-κB (Wu et al. 2017). Further research is needed to elucidate how intracellular Salmonella acquire other essential nutrients for ensuring their survival and growth.
Host intracellular environment promotes the virulence of Salmonella
During infection, macrophages shift their metabolism to aerobic glycolysis, resulting in the intracellular accumulation of glycolytic products like succinate. The increased succinate can be sensed by intracellular S. Typhimurium, promoting expression of the T3SS, its secreted effectors, and the factors involved in resistance to antimicrobial peptides (Rosenberg et al. 2021). Additionally, the accumulation of pyruvate and lactate in macrophages increases the expression of a Salmonella two-component system called CreBC. CreB, in turn, enhances VrpB expression by binding to the vrpB promoter. VrpB further binds to the promoter of ssrA/B, directly activating the expression of T3SS2-related genes (Jiang et al. 2021). The acidic pH (< 5–5.5) of SCVs is crucial for the expression of genes encoding T3SS2 and its secreted effectors, which are required for the replication of Salmonella in host cells (Yu et al. 2010). In S. Typhimurium, a weakly acidic cytoplasmic pH contributes to the activation of at least three two-component systems, i.e., PhoQ/PhoP, EnvZ/OmpR, and SsrA/B, thereby promoting the expression of T3SS2-related genes (Kenney 2019). In addition, intracellular signals inside host cells contribute to increased kinase activity of PhoQ, enhancing PhoP phosphorylation and the expression of T3SS2-related genes. The periplasmic domain of PhoQ senses low levels of Mg2+ ions, certain antimicrobial peptides (e.g., antimicrobial peptide C18G), and polymyxin B, whereas its cytoplasmic domain senses a mildly acidic pH, collectively activating the expression of T3SS2-related genes (Choi and Groisman 2016). The transcriptional regulatory protein AsiR positively regulates flagellar gene expression by directly binding to the flhDC promoter. However, the acidic pH in macrophages downregulates the expression of AsiR, leading to the downregulation of flagellar gene expression, ultimately promoting the intracellular survival and systemic infection of Salmonella (Ma et al. 2021). Additionally, the acidic pH in SCVs promotes yaeB expression (encoding an N6 methyltransferase) through inhibiting global regulator histone-like nucleoid structuring protein (H-NS), directly promoting the expression of genes associated with phosphate acquisition and the virulence of S. Typhimurium (Zhang et al. 2019).
Effects of Salmonella infection on host cell survival
A few serovars of S. enterica cause chronic inflammation and mucosal damage and produce toxins such as cytolethal distending toxins (CDTs) that cause DNA damage and cell cycle inhibition. In a few salmonellae, such as S. enterica serovars Javiana and Typhi, CDTs are hetero-trimer toxins composed of PltA, CdtB, and PltB subunits. The CdtB subunit exhibits DNAse I activity, leading to cell cycle arrest in the G2/M phase, whereas PltA and PltB facilitate CdtB entry into host cells (Zha et al. 2019). The Salmonella effector PheA can be translocated into the nucleus of RAW 264.7 macrophages to mimic the E2F7 transcription factor of host cells, promoting cell cycle arrest in the G1/S phase, thereby reducing the proportion of cells in the G2/M phase (Na et al. 2015). The effector AvrA acetylates P53, retarding the cycle of intestinal epithelial cells, resulting in an increase in the number of cells in the G0/G1 phase and a decrease in the number of cells in the G2/M phase (Wu et al. 2010).
Salmonella infection can lead to cell death through apoptosis, necroptosis, and pyroptosis. Apoptosis is related to caspases 3/7/8/9, pyroptosis to caspases 1/4/5/11, and necroptosis to mixed lineage kinase domain-like protein and other unknown factors (Wemyss and Pearson 2019). Moreover, Salmonella-derived LPS and effectors (SlrP, SpvB, and SpvC) promote host cell apoptosis in Salmonella infection (Man et al. 2013). Pyroptosis is a vital host defense mechanism against Salmonella, and many virulence factors participate in its activation, including flagellin proteins (FliC and FljB), LPS, rod-shaped proteins (PrgJ), and effectors (SopE and SipB). Moreover, Salmonella-induced ROS and dsDNA can accelerate pyroptosis (Wemyss and Pearson 2019). Certain Salmonella effectors, such as SseK1 and SseK3, inhibit apoptosis and necroptosis in macrophages through arginine glycosylation of FADD and TRADD. Salmonella effector protein SopB delays the apoptosis of epithelial cells by sustaining activation of Akt, allowing sufficient time for its intracellular replication (Knodler et al. 2005; Chu et al. 2021). Salmonella can survive in B cells but the mechanisms are not very clear. Two studies found SopB activates PI3K/Akt pathway to downregulate of NLRC4 transcription and IL-1β secretion of B cells, which delaying pyroptosis of infected B cells and therefore providing a stable niche for Salmonella survival (García-Gil et al. 2018; Luis et al. 2022). Additionally, the effector AvrA inhibits apoptosis, whereas SpvC inhibits pyroptosis (Haneda et al. 2012). Hence, Salmonella exploits various effectors to regulate the host cell death response, which might be a conservative strategy employed by Salmonella to increase its populations and prepare for its next invasion, finally spreading to other cells and organs.
Conclusions and future perspectives
Salmonella establishes infection in hosts by overwhelming host cell defenses. Following invasion into epithelial or M cells in the intestine, the pathogen is taken up by phagocytes that transport the bacteria to organs, such as the liver, spleen, kidney, and bone marrow, through the blood or lymph circulation. If hosts fail to quickly eliminate Salmonella in the early infection/invasion processes because of the slowness of immune signaling activation and shortage of immune cells/substances/molecules, the rapid replication of Salmonella inside host cells destroys the host cells and damages host organs, further promoting the spread of Salmonella and weakening the host immunity.
Salmonella exploits various strategies to combat epithelial and immune cells, including macrophages, dendritic cells, and neutrophils. It uses many secreted effectors/ proteins to survive in the host, and resists immune responses of the host through the production of inhibitors, blockage of immune signaling pathways, disruption of host metabolism, cell cycle arrest, or modulation of host cell death. The exact functions of many proteins and effectors of Salmonella and their interactions with host cells remain unknown. Therefore, there is much room to explore the mechanisms underlying Salmonella pathogenicity regarding its survival strategies in host cells, tissues, and organs.
Data availability
Data sharing does not apply to this article as no new data were created or analyzed in this review.
References
Al-Shehri SS (2021) Reactive oxygen and nitrogen species and innate immune response. Biochimie 181:52–64. https://doi.org/10.1016/j.biochi.2020.11.022
Alix E et al (2020) The tumour suppressor TMEM127 Is a Nedd4-family e3 ligase adaptor required by Salmonella SteD to ubiquitinate and degrade MHC class II molecules. Cell Host Microbe 28:54-68.e57. https://doi.org/10.1016/j.chom.2020.04.024
Amaya FA et al (2022) Identification of type VI secretion systems effector proteins that contribute to interbacterial competition in Salmonella Dublin. Front Microbiol 13:811932. https://doi.org/10.3389/fmicb.2022.811932
Bakowski MA et al (2010) The phosphoinositide phosphatase SopB manipulates membrane surface charge and trafficking of the Salmonella-containing vacuole. Cell Host Microbe 7:453–462. https://doi.org/10.1016/j.chom.2010.05.011
Bao H, Wang S, Zhao JH, Liu SL (2020) Salmonella secretion systems: Differential roles in pathogen-host interactions. Microbiol Res 241:126591. https://doi.org/10.1016/j.micres.2020.126591
Behnsen J, Perez-Lopez A, Nuccio SP, Raffatellu M (2015) Exploiting host immunity: the Salmonella paradigm. Trends Immunol 36:112–120. https://doi.org/10.1016/j.it.2014.12.003
Benjamin JL, Sumpter R, Levine B, Hooper LV (2013) Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe 13:723–734
Bernal-Bayard J, Cardenal-Munoz E, Ramos-Morales F (2010) The Salmonella type III secretion effector, Salmonella leucine-rich repeat protein (SIrP), targets the human chaperone ERdj3. J Biol Chem 285:16360–16368. https://doi.org/10.1074/jbc.M110.100669
Boddy KC et al (2021) Salmonella effector SopD promotes plasma membrane scission by inhibiting Rab10. Nat Commun 12:4707. https://doi.org/10.1038/s41467-021-24983-z
Bowden SD, Rowley G, Hinton JC, Thompson A (2009) Glucose and glycolysis are required for the successful infection of macrophages and mice by Salmonella enterica serovar Typhimurium. Infect Immun 77:3117–3126. https://doi.org/10.1128/IAI.00093-09
Brodsky IE (2020) JAK-ing into M1/M2 polarization SteErs Salmonella-containing macrophages away from immune attack to promote bacterial persistence. Cell Host Microbe 27:3–5. https://doi.org/10.1016/j.chom.2019.12.007
Browne SH, Hasegawa P, Okamoto S, Fierer J, Guiney DG (2008) Identification of Salmonella SPI-2 secretion system components required for SpvB-mediated cytotoxicity in macrophages and virulence in mice. FEMS Immunol Med Microbiol 52:194–201. https://doi.org/10.1111/j.1574-695X.2007.00364.x
Callewaert L et al (2008) A new family of lysozyme inhibitors contributing to lysozyme tolerance in gram-negative bacteria. PLoS Pathog 4:e1000019. https://doi.org/10.1371/journal.ppat.1000019
Cerny O, Holden DW (2019) Salmonella SPI-2 type III secretion system-dependent inhibition of antigen presentation and T cell function. Immunol Lett 215:35–39. https://doi.org/10.1016/j.imlet.2019.01.006
Cerny O et al (2021) CD97 stabilises the immunological synapse between dendritic cells and T cells and is targeted for degradation by the Salmonella effector SteD. PLoS Pathog 17:e1009771. https://doi.org/10.1371/journal.ppat.1009771
Cheng Z et al (2017) Focal adhesion kinase-mediated phosphorylation of Beclin1 protein suppresses cardiomyocyte autophagy and initiates hypertrophic growth. J Biol Chem 292:2065–2079. https://doi.org/10.1074/jbc.M116.758268
Choi J, Groisman EA (2016) Acidic pH sensing in the bacterial cytoplasm is required for Salmonella virulence. Mol Microbiol 101:1024–1038. https://doi.org/10.1111/mmi.13439
Chu Y et al (2016) A novel contribution of spvB to pathogenesis of Salmonella Typhimurium by inhibiting autophagy in host cells. Oncotarget 7:8295–8309. https://doi.org/10.18632/oncotarget.6989
Chu B-X et al (2021) Salmonella infantis delays the death of infected epithelial cells to aggravate bacterial load by intermittent phosphorylation of Akt with SopB. Front Immunol. https://doi.org/10.3389/fimmu.2021.757909
Clark L et al (2011) Differences in Salmonella enterica serovar Typhimurium strain invasiveness are associated with heterogeneity in SPI-1 gene expression. Microbiology 157:2072–2083. https://doi.org/10.1099/mic.0.048496-0
Crouch ML, Castor M, Karlinsey JE, Kalhorn T, Fang FC (2008) Biosynthesis and IroC-dependent export of the siderophore salmochelin are essential for virulence of Salmonella enterica serovar Typhimurium. Mol Microbiol 67:971–983. https://doi.org/10.1111/j.1365-2958.2007.06089.x
Dai S, Sarmiere PD, Wiggan ON, Bamburg JR, Zhou D (2004) Efficient Salmonella entry requires activity cycles of host ADF and cofilin. Cell Microbiol 6:459–471. https://doi.org/10.1111/j.1462-5822.2004.00375.x
Das P et al (2010) Cationic amino acid transporters and Salmonella Typhimurium ArgT collectively regulate arginine availability towards intracellular Salmonella growth. PLoS ONE 5:e15466. https://doi.org/10.1371/journal.pone.0015466
De Groote MA et al (1997) Periplasmic superoxide dismutase protects Salmonella from products of phagocyte NADPH-oxidase and nitric oxide synthase. Proc Natl Acad Sci USA 94:13997–14001. https://doi.org/10.1073/pnas.94.25.13997
Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A, Fafutis-Morris M (2017) Neutrophil Extracellular traps and its implications in inflammation: an overview. Front Immunol 8:1–20. https://doi.org/10.3389/fimmu.2017.00081
Deng Q et al (2021) Salmonella effector SpvB aggravates dysregulation of systemic iron metabolism via modulating the hepcidin-ferroportin axis. Gut Microbes 13:1–18. https://doi.org/10.1080/19490976.2020.1849996
Di Martino ML, Ek V, Hardt W-D, Eriksson J, Sellin ME (2019) Barcoded consortium infections resolve cell type-dependent Salmonella enterica Serovar Typhimurium entry mechanisms. Mbio 10:1–15. https://doi.org/10.1128/mBio.00603-19
Drecktrah D, Knodler LA, Ireland R, Steele-Mortimer O (2006) The mechanism of Salmonella entry determines the vacuolar environment and intracellular gene expression. Traffic 7:39–51. https://doi.org/10.1111/j.1600-0854.2005.00360.x
Du F, Galan JE (2009) Selective inhibition of type III secretion activated signaling by the Salmonella effector AvrA. PLoS Pathog 5:e1000595. https://doi.org/10.1371/journal.ppat.1000595
Eisele NA et al (2013) Salmonella require the fatty acid regulator PPARδ for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe 14:171–182. https://doi.org/10.1016/j.chom.2013.07.010
Eswarappa SM, Negi VD, Chakraborty S, Sagar BKC, Chakravortty D (2010) Division of the Salmonella-containing vacuole and depletion of acidic lysosomes in Salmonella-infected host cells are novel strategies of Salmonella enterica to avoid lysosomes. Infect Immun 78:68–79. https://doi.org/10.1128/iai.00668-09
Feng Z-Z et al (2018) The Salmonella effectors SseF and SseG inhibit Rab1A-mediated autophagy to facilitate intracellular bacterial survival and replication. J Biol Chem 293:9662–9673. https://doi.org/10.1074/jbc.M117.811737
Fookes M et al (2011) Salmonella bongori provides insights into the evolution of the Salmonellae. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1002191
Fuentes JA, Villagra N, Castillo-Ruiz M, Mora GC (2008) The Salmonella Typhi hlyE gene plays a role in invasion of cultured epithelial cells and its functional transfer to S. typhimurium promotes deep organ infection in mice. Res Microbiol 159:279–287. https://doi.org/10.1016/j.resmic.2008.02.006
Gal-Mor O (2019) Persistent infection and long-term carriage of typhoidal and nontyphoidal Salmonellae. Clin Microbiol Rev. https://doi.org/10.1128/cmr.00088-18
García-Gil A, Galán-Enríquez CS, Pérez-López A, Nava P, Alpuche-Aranda C, Ortiz-Navarrete V (2018) SopB activates the Akt-YAP pathway to promote Salmonella survival within B cells. Virulence 9:1390–1402. https://doi.org/10.1080/21505594.2018.1509664
Ghosh S et al (2011) An adhesion protein of Salmonella enterica serovar Typhi is required for pathogenesis and potential target for vaccine development. Proc Natl Acad Sci USA 108:3348–3353. https://doi.org/10.1073/pnas.1016180108
Gillis CC et al (2018) Dysbiosis-associated change in host metabolism generates lactate to support Salmonella growth. Cell Host Microbe 23:54–64. https://doi.org/10.1016/j.chom.2017.11.006
Griffin AJ, McSorley SJ (2011) Development of protective immunity to Salmonella, a mucosal pathogen with a systemic agenda. Mucosal Immunol 4:371–382. https://doi.org/10.1038/mi.2011.2
Guerra PR et al (2020) Polyamine depletion has global effects on stress and virulence gene expression and affects HilA translation in Salmonella enterica serovar Typhimurium. Res Microbiol 171:143–152. https://doi.org/10.1016/j.resmic.2019.12.001
Günster RA, Matthews SA, Holden DW, Thurston TLM (2017) SseK1 and SseK3 type III secretion system effectors inhibit NF-κB signaling and necroptotic cell death in Salmonella-infected macrophages. Infect Immun 85:e00010-00017. https://doi.org/10.1128/IAI.00010-17
Hahn MM, González JF, Gunn JS (2021) Salmonella Biofilms Tolerate Hydrogen Peroxide by a Combination of Extracellular Polymeric Substance Barrier Function and Catalase Enzymes. Front Cell Infect Microbiol 11:683081. https://doi.org/10.3389/fcimb.2021.683081
Halici S, Zenk SF, Jantsch J, Hensel M (2008) Functional analysis of the Salmonella pathogenicity island 2-mediated inhibition of antigen presentation in dendritic cells. Infect Immun 76:4924–4933. https://doi.org/10.1128/IAI.00531-08
Haneda T et al (2012) Salmonella type III effector SpvC, a phosphothreonine lyase, contributes to reduction in inflammatory response during intestinal phase of infection. Cell Microbiol 14:485–499. https://doi.org/10.1111/j.1462-5822.2011.01733.x
Hassuna NA, Monk PN, Ali F, Read RC, Partridge LJ (2017) A role for the tetraspanin proteins in Salmonella infection of human macrophages. J Infect 75:115–124. https://doi.org/10.1016/j.jinf.2017.06.003
Hébrard M, Viala JP, Méresse S, Barras F, Aussel L (2009) Redundant hydrogen peroxide scavengers contribute to Salmonella virulence and oxidative stress resistance. J Bacteriol 191:4605–4614. https://doi.org/10.1128/jb.00144-09
Heffernan EJ, Reed S, Hackett J, Fierer J, Roudier C, Guiney D (1992) Mechanism of resistance to complement-mediated killing of bacteria encoded by the Salmonella typhimurium virulence plasmid gene rck. J Clin Invest 90:953–964. https://doi.org/10.1172/jci115972
Henard CA, Vazquez-Torres A (2011) Nitric oxide and Salmonella pathogenesis. Front Microbiol 2:1–11. https://doi.org/10.3389/fmicb.2011.00084
Herb M, Schramm M (2021) Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants (Basel) 10. https://doi.org/10.3390/antiox10020313
Hernandez LD, Hueffer K, Wenk MR, Galan JE (2004) Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304:1805–1807. https://doi.org/10.1126/science.1098188
Hernandez RE, Gallegos-Monterrosa R, Coulthurst SJ (2020) Type VI secretion system effector proteins: effective weapons for bacterial competitiveness. Cell Microbiol 22:e13241. https://doi.org/10.1111/cmi.13241
Huang F-C (2014) The critical role of membrane cholesterol in Salmonella-induced autophagy in intestinal epithelial cells. Int J Mol Sci 15:12558–12572
Huang FC, Werne A, Li Q, Galyov EE, Walker WA, Cherayil BJ (2004) Cooperative interactions between flagellin and SopE2 in the epithelial interleukin-8 response to Salmonella enterica serovar Typhimurium infection. Infect Immun 72:5052–5062. https://doi.org/10.1128/iai.72.9.5052-5062.2004
Jennings E, Esposito D, Rittinger K, Thurston TLM (2018) Structure–function analyses of the bacterial zinc metalloprotease effector protein GtgA uncover key residues required for deactivating NF-κB. J Biol Chem 293:15316–15329. https://doi.org/10.1074/jbc.RA118.004255
Jiang L et al (2021) Salmonella Typhimurium reprograms macrophage metabolism via T3SS effector SopE2 to promote intracellular replication and virulence. Nat Commun. https://doi.org/10.1038/s41467-021-21186-4
Kenney LJ (2019) The role of acid stress in Salmonella pathogenesis. Curr Opin Microbiol 47:45–51. https://doi.org/10.1016/j.mib.2018.11.006
Keszei AFA et al (2014) Structure of an SspH1-PKN1 complex reveals the basis for host substrate recognition and mechanism of activation for a bacterial E3 ubiquitin ligase. Mol Cell Biol 34:362–373. https://doi.org/10.1128/mcb.01360-13
Kim DK et al (2014) Inverse agonist of estrogen-related receptor γ controls Salmonella typhimurium infection by modulating host iron homeostasis. Nat Med 20:419–424. https://doi.org/10.1038/nm.3483
Knodler LA, Finlay BB, Steele-Mortimer O (2005) The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J Biol Chem 280:9058–9064. https://doi.org/10.1074/jbc.M412588200
Kortman GA, Boleij A, Swinkels DW, Tjalsma H (2012) Iron availability increases the pathogenic potential of Salmonella Typhimurium and other enteric pathogens at the intestinal epithelial interface. PLoS ONE 7:e29968. https://doi.org/10.1371/journal.pone.0029968
Kullas Amy L et al (2012) L-Asparaginase II produced by Salmonella Typhimurium inhibits T cell responses and mediates virulence. Cell Host Microbe 12:791–798. https://doi.org/10.1016/j.chom.2012.10.018
Lahiri A, Eswarappa SM, Das P, Chakravortty D (2010) Division of the Salmonella-containing vacuole and depletion of acidic lysosomes in Salmonella-infected host cells are novel strategies of Salmonella enterica to avoid lysosomes. Virulence 1:325–329. https://doi.org/10.4161/viru.1.4.12361
Lambert MA, Smith SGJ (2008) The PagN protein of Salmonella enterica serovar Typhimurium is an adhesin and invasin. BMC Microbiol 8:142
Lambert MA, Smith SGJ (2009) The PagN protein mediates invasion via interaction with proteoglycan. FEMS Microbiol Lett 297:209–216. https://doi.org/10.1111/j.1574-6968.2009.01666.x
LaRock DL, Chaudhary A, Miller SI (2015) Salmonellae interactions with host processes. Nat Rev Microbiol 13:191–205. https://doi.org/10.1038/nrmicro3420
Lathrop SK et al (2015) Replication of Salmonella enterica serovar Typhimurium in human monocyte-derived macrophages. Infect Immun 83:2661–2671. https://doi.org/10.1128/IAI.00033-15
Li W et al (2022) Nitrate utilization promotes systemic infection of Salmonella Typhimurium in mice. Int J Mol Sci. https://doi.org/10.3390/ijms23137220
Lin SL, Le TX, Cowen DS (2003) SptP, a Salmonella Typhimurium type III-secreted protein, inhibits the mitogen-activated protein kinase pathway by inhibiting Raf activation. Cell Microbiol 5:267–275. https://doi.org/10.1046/j.1462-5822.2003.t01-1-00274.x
Lin Z et al (2017) Immunogenicity and protective efficacy of a Salmonella Enteritidis sptP mutant as a live attenuated vaccine candidate. BMC Vet Res 13:1–9. https://doi.org/10.1186/s12917-017-1115-3
Luis L-B et al (2022) Salmonella promotes its own survival in B cells by inhibiting autophagy. Cells. https://doi.org/10.3390/cells11132061
Ma S et al (2021) Downregulation of a novel flagellar synthesis regulator AsiR promotes intracellular replication and systemic pathogenicity of Salmonella Typhimurium. Virulence 12:298–311. https://doi.org/10.1080/21505594.2020.1870331
Mallo GV et al (2008) SopB promotes phosphatidylinositol 3-phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J Cell Biol 182:741–752. https://doi.org/10.1083/jcb.200804131
Mambu J et al (2020) Rck of Salmonella Typhimurium delays the host cell cycle to facilitate bacterial invasion. Front Cell Infect Microbiol 10:1–15. https://doi.org/10.3389/fcimb.2020.586934
Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE (2013) Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1 production. J Immunol 191:5239–5246. https://doi.org/10.4049/jimmunol.1301581
Masud S et al (2019a) Macrophages target Salmonella by Lc3-associated phagocytosis in a systemic infection model. Autophagy 15:796–812. https://doi.org/10.1080/15548627.2019.1569297
Masud S, van der Burg L, Storm L, Prajsnar TK, Meijer AH (2019b) Rubicon-dependent Lc3 recruitment to Salmonella-containing phagosomes is a host defense mechanism triggered independently from major bacterial virulence factors. Front Cell Infect Microbiol 9:1–13. https://doi.org/10.3389/fcimb.2019.00279
McGhie EJ, Hayward RD, Koronakis V (2004) Control of actin turnover by a salmonella invasion protein. Mol Cell 13:497–510. https://doi.org/10.1016/s1097-2765(04)00053-x
McGourty K, Thurston TL, Matthews SA, Pinaud L, Mota LJ, Holden DW (2012) Salmonella inhibits retrograde trafficking of mannose-6-phosphate receptors and lysosome function. Science 338:963–967. https://doi.org/10.1126/science.1227037
Mesquita FS, Thomas M, Sachse M, Santos AJM, Figueira R, Holden DW (2012) The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog 8:1–14. https://doi.org/10.1371/journal.ppat.1002743
Na HN, Yoo YH, Yoon CN, Lee JS (2015) Unbiased proteomic profiling strategy for discovery of bacterial effector proteins reveals that Salmonella protein PheA is a host cell cycle regulator. Chem Biol 22:453–459. https://doi.org/10.1016/j.chembiol.2015.03.008
Orf K, Cunnington AJ (2015) Infection-related hemolysis and susceptibility to Gram-negative bacterial co-infection. Front Microbiol. https://doi.org/10.3389/fmicb.2015.00666
Panagi I et al (2020) Salmonella effector SteE converts the mammalian serine/threonine kinase GSK3 into a tyrosine kinase to direct macrophage polarization. Cell Host Microbe 27:41–53. https://doi.org/10.1016/j.chom.2019.11.002
Pilar AV, Reid-Yu SA, Cooper CA, Mulder DT, Coombes BK (2012) GogB is an anti-inflammatory effector that limits tissue damage during Salmonella infection through interaction with human FBXO22 and Skp1. PLoS Pathog 8:e1002773. https://doi.org/10.1371/journal.ppat.1002773
Pilar AV, Reid-Yu SA, Cooper CA, Mulder DT, Coombes BK (2013) Active modification of host inflammation by Salmonella. Gut Microbes 4:140–145. https://doi.org/10.4161/gmic.23361
Popp J, Noster J, Busch K, Kehl A, Zur Hellen G, Hensel M (2015) Role of host cell-derived amino acids in nutrition of intracellular Salmonella enterica. Infect Immun 83:4466–4475. https://doi.org/10.1128/IAI.00624-15
Richardson AR, Soliven KC, Castor ME, Barnes PD, Libby SJ, Fang FC (2009) The base excision repair system of Salmonella enterica serovar Typhimurium counteracts DNA damage by host nitric oxide. PLoS Pathog 5:e1000451. https://doi.org/10.1371/journal.ppat.1000451
Rolhion N et al (2016) Inhibition of nuclear transport of NF-kB p65 by the Salmonella type III secretion system effector SpvD. PLoS Pathog 12:e1005653. https://doi.org/10.1371/journal.ppat.1005653
Rosenberg G et al (2021) Host succinate is an activation signal for Salmonella virulence during intracellular infection. Science 371:400–405. https://doi.org/10.1126/science.aba8026
Ruby T, McLaughlin L, Gopinath S, Monack D (2012) Salmonella’s long-term relationship with its host. FEMS Microbiol Rev 36:600–615. https://doi.org/10.1111/j.1574-6976.2012.00332.x
Sana TG et al (2016) Salmonella Typhimurium utilizes a T6SS-mediated antibacterial weapon to establish in the host gut. Proc Natl Acad Sci USA 113:E5044-5051. https://doi.org/10.1073/pnas.1608858113
Schleker S et al (2012) The current Salmonella-host interactome. Proteom Clin Appl 6:117–133. https://doi.org/10.1002/prca.201100083
Shelton CD et al (2022) Salmonella enterica serovar Typhimurium uses anaerobic respiration to overcome propionate-mediated colonization resistance. Cell Rep 38:110180. https://doi.org/10.1016/j.celrep.2021.110180
Sibinelli-Sousa S et al (2020) A family of T6SS antibacterial effectors related to l, d-transpeptidases targets the peptidoglycan. Cell Rep 31:107813. https://doi.org/10.1016/j.celrep.2020.107813
Slauch JM (2011) How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol 80:580–583. https://doi.org/10.1111/j.1365-2958.2011.07612.x
Stebbins CE, Galan JE (2000) Modulation of host signaling by a bacterial mimic: structure of the Salmonella effector SptP bound to Rac1. Mol Cell 6:1449–1460. https://doi.org/10.1016/s1097-2765(00)00141-6
Sun H, Kamanova J, Lara-Tejero M, Galan JE (2016) A family of Salmonella type III secretion effector proteins selectively targets the NF-kappaB signaling pathway to preserve host homeostasis. PLoS Pathog 12:1–19. https://doi.org/10.1371/journal.ppat.1005484
Sun YH et al (2019) Infection-generated electric field in gut epithelium drives bidirectional migration of macrophages. PLoS Biol 17:29. https://doi.org/10.1371/journal.pbio.3000044
Tattoli I et al (2012) Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11:563–575
Taylor SJ, Winter SE (2020) Salmonella finds a way: metabolic versatility of Salmonella enterica serovar Typhimurium in diverse host environments. PLoS Pathog 16:e1008540. https://doi.org/10.1371/journal.ppat.1008540
Terebiznik MR et al (2002) Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat Cell Biol 4:766–773. https://doi.org/10.1038/ncb854
Tuinema BR, Reid-Yu SA, Coombes BK (2014) Salmonella evades D-amino acid oxidase to promote infection in neutrophils. Mbio 5:1–9. https://doi.org/10.1128/mBio.01886-14
Tyrkalska SD et al (2016) Neutrophils mediate Salmonella Typhimurium clearance through the GBP4 inflammasome-dependent production of prostaglandins. Nat Commun 7:12077. https://doi.org/10.1038/ncomms12077
van der Heijden J, Bosman ES, Reynolds LA, Finlay BB (2015) Direct measurement of oxidative and nitrosative stress dynamics in Salmonella inside macrophages. Proc Natl Acad Sci U S A 112:560–565. https://doi.org/10.1073/pnas.1414569112
Wan S et al (2018) Overexpression of toll-like receptor 4 contributes to phagocytosis of Salmonella Enterica Serovar typhimurium via phosphoinositide 3-kinase signaling in sheep. Cell Physiol Biochem 49:662–677. https://doi.org/10.1159/000493032
Wang J et al (2021) Salmonella enterica serovar Typhi induces host metabolic reprogramming to increase glucose availability for intracellular replication. Int J Mol Sci 22:1–14. https://doi.org/10.3390/ijms221810003
Wang H et al (2022) Salmonella enterica serovar Typhi influences inflammation and autophagy in macrophages. Braz J Microbiol 53:525–534. https://doi.org/10.1007/s42770-022-00719-z
Wemyss MA, Pearson JS (2019) Host cell death responses to non-typhoidal Salmonella infection. Front Immunol 10:1–10. https://doi.org/10.3389/fimmu.2019.01758
Wiedemann A et al (2016) Identification of the epidermal growth factor receptor as the receptor for Salmonella Rck-dependent invasion. Faseb J 30:4180–4191. https://doi.org/10.1096/fj.201600701R
Winter SE, Baumler AJ (2011) A breathtaking feat: to compete with the gut microbiota, Salmonella drives its host to provide a respiratory electron acceptor. Gut Microbes 2:58–60. https://doi.org/10.4161/gmic.2.1.14911
Wu S et al (2010) Salmonella Typhimurium infection increases p acetylation in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 298:784–794. https://doi.org/10.1152/ajpgi.00526.2009
Wu H, Jones RM, Neish AS (2012) The Salmonella effector AvrA mediates bacterial intracellular survival during infection in vivo. Cell Microbiol 14:28–39. https://doi.org/10.1111/j.1462-5822.2011.01694.x
Wu A et al (2017) Salmonella utilizes zinc to subvert antimicrobial host defense of macrophages via modulation of NF-κB signaling. Infect Immun. https://doi.org/10.1128/iai.00418-17
Wu M, El Qaidi S, Hardwidge PR (2018) SseL deubiquitinates RPS3 to inhibit its nuclear translocation. Pathogens. https://doi.org/10.3390/pathogens7040086
Xu Y et al (2019) A bacterial effector reveals the V-ATPase-ATG16L1 axis that initiates xenophagy. Cell 178:552-566.e520. https://doi.org/10.1016/j.cell.2019.06.007
Xue J et al (2020) Arg-GlcNAcylation on TRADD by NleB and SseK1 Is crucial for bacterial pathogenesis. Front Cell Dev Biol 8:1–10. https://doi.org/10.3389/fcell.2020.00641
Yang F, Sheng X, Huang X, Zhang Y (2021a) Interactions between Salmonella and host macrophages - dissecting NF-κB signaling pathway responses. Microb Pathog 154:104846. https://doi.org/10.1016/j.micpath.2021.104846
Yang S et al (2021b) Salmonella effector SpvB inhibits NF-κB activity via KEAP1-mediated downregulation of IKKβ. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2021.641412
Yin C et al (2020) AvrA exerts inhibition of NF-kappaB pathway in its naive Salmonella serotype through suppression of p-JNK and beclin-1 molecules. Int J Mol Sci 21:6063. https://doi.org/10.3390/ijms21176063
Yu X-J, McGourty K, Liu M, Unsworth KE, Holden DW (2010) pH sensing by intracellular Salmonella induces effector translocation. Science 328:1040–1043. https://doi.org/10.1126/science.1189000
Yu C et al (2020) Salmonella enterica serovar Typhimurium sseK3 induces apoptosis and enhances glycolysis in macrophages. BMC Microbiol 20:1–9. https://doi.org/10.1186/s12866-020-01838-z
Zaki MH, Man SM, Vogel P, Lamkanfi M, Kanneganti TD (2014) Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc Natl Acad Sci U S A 111:385–390. https://doi.org/10.1073/pnas.1317643111
Zha L, Garrett S, Sun J (2019) Salmonella Infection in chronic inflammation and gastrointestinal cancer. Diseases 7:28. https://doi.org/10.3390/diseases7010028
Zhang H, Song X, Wang P, Lv R, Ma S, Jiang L (2019) YaeB, expressed in response to the acidic ph in macrophages, promotes intracellular replication and virulence of Salmonella Typhimurium. Int J Mol Sci 20:4339. https://doi.org/10.3390/ijms20184339
Zhou L et al (2021) Salmonella spvC gene inhibits autophagy of host cells and suppresses NLRP3 as well as NLRC4. Front Immunol 12:639019. https://doi.org/10.3389/fimmu.2021.639019
Acknowledgements
The authors are thankful for the suggestions provided by Dr. Lingyan Jiang from Nankai University for the revision of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 82170606] and the Basic Research Project of Key Scientific Research Projects of Universities in Henan Province [grant number 23ZX006].
Author information
Authors and Affiliations
Contributions
W.L. wrote and revised the manuscript. Q.R. drew the figures and made revisions for the manuscript. T.N., Y.Z., Z.S., and R.L. collected the references and verified the points presented in the manuscript. Z.L. and S.L. conceived and revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Communicated by Yusuf Akhter.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, W., Ren, Q., Ni, T. et al. Strategies adopted by Salmonella to survive in host: a review. Arch Microbiol 205, 362 (2023). https://doi.org/10.1007/s00203-023-03702-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-023-03702-w