
Abstract
The diversity of Lb. rhamnosus and Lb. fermentum strains isolated from feces, saliva, and the vaginal cavity of 18–22-year-old healthy women residing in central regions of the Russian Federation has been characterized. The results obtained using multilocus sequence typing were identical to those obtained with the analysis of genetic and genomic polymorphism in TA systems. Different as well as identical Lb. rhamnosus and Lb. fermentum sequence types (ST) were isolated from various parts of the body of the same person. Identical ST were also isolated from different women, suggesting that such strains belong to a common pool of strains circulating among the population members. Our results demonstrate that TAs are suitable for characterizing intra-specific diversity of Lb. rhamnosus and Lb. fermentum strains. The advantage of using polymorphisms in TA systems for genotyping is based on the weak number of genes used, and consequently, less time is required for the analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Many cavities of the human body are inhabited (to various extent) by bacteria (Faust et al. 2012). The most densely populated and well studied are the gut, oral, and vaginal microbiota. The number of microbial cells living on and inside the human body is approximately 1014–1015 belonging to thousands of bacterial species; the overwhelming majority of bacteria cannot be cultivated (Qin et al. 2010). Lactobacilli are cultured bacteria that play a crucial role in human health. Despite the fact that they constitute only a small proportion of the overall human microbiota, they are found in almost all parts of the body and are often the dominant species in the vagina (Turroni et al. 2013; Kassaa et al. 2015). More than 50 out of 217 known species of lactobacilli were isolated from the human body (Wall et al. 2007; Rossi et al. 2016). The diversity of strains colonizing different biotopes of the same individual is poorly studied. For species identification of lactobacilli, nucleotide sequence of 16S rRNA gene is usually analyzed (Janda and Abbott 2007). A number of methods are used for determining the intra-species diversity of bacteria (Gosiewski et al. 2012; Herbel et al. 2013); nevertheless, no universal approach is employed today.
Multilocus sequence typing (MLST) based on sequencing of a small number (≥7) of housekeeping genes that are under control of stabilizing selection is the most standardized method for intra-species strains typing. The method is easy and inexpensive, and provides the ability to compare and exchange data between laboratories (Maiden 2006). MLST was used in many studies for typing lactic acid bacteria, including lactobacilli (De las Rivas et al. 2006; Cai et al. 2007; Dan et al. 2015).
Type II toxin–antitoxin (TA) systems are found in the genomes of almost all bacteria and constitute a part of bacterial cells’ regulatory networks. They are typically organized in two-component operons encoding a stable toxin and an unstable antitoxin. When transcription or translation is inhibited as a result of stress, the toxin (usually a ribonuclease) degrades the antitoxin leading to bacterial persistence or cell death. TA systems are involved in essential cellular processes like replication, gene expression, cell wall synthesis, maintenance of chromosomal stability, programmed cell death, biofilm formation, quorum sensing, pathogenicity, tolerance, and persistence (Wen et al. 2014). We have previously shown that polymorphism in TA systems is strain-specific and can be used for genotyping various microorganisms (Alekseeva et al. 2011; Zaychikova et al. 2015).
The aim of this study was to characterize the diversity of Lactobacillus strains isolated from feces, saliva, and vaginal cavity of 18–22-year-old healthy women, residents of the central regions of the Russian Federation. A combination of MLST and a method based on the genetic and genomic polymorphism in TA systems was used for strain differentiation.
Materials and methods
Bacterial strains and growth conditions
Lactobacillus strains were isolated at the Department of Microbiology, Virology and Immunology of Tver State Medical Academy (Supplementary Table S1) from feces, saliva, and vaginal mucus of 13 healthy women residing in the central region of the Russian Federation; a written informed consent of volunteers was obtained prior to study. The volunteers took no probiotic treatment or antibiotic treatment for at least 3 months before examination. Bacteria were cultured in Man–Rogosa–Sharp broth and agar (MRS, HiMedia, India) (De Man et al. 1960) at 37 °С in anaerobic conditions (HiAnaerobic System Mark II, HiMedia) for 24–48 h.
DNA techniques
Genomic DNA was extracted from Lactobacillus cells with DNeasy Blood and Tissue Kit (Qiagen, Germany). DNA amplification was performed with Tersus PCR kit (Evrogen, Russia). Information about primers used for MLST and TA analyses is listed in Tables 2, 3, 4, and 6. 16S rRNA genes were amplified and sequenced using universal primers 27F and 1492R (Lane 1991). PCR products were resolved by electrophoresis in 1% agarose gel in TBE buffer, and purified using QIAquick PCR kit (Qiagen). Both DNA strands were Sanger-sequenced on a 3730xl DNA analyzer (Applied Biosystems, USA) at the Scientific Research Institute of Physical–Chemical Medicine (Moscow, Russian Federation). Species identification of strains was determined by aligning 16S rRNA gene sequence of a particular strain using BLASTn with corresponding sequences from GenBank.
Multilocus sequence typing
Nucleotide sequences of housekeeping genes parB, ychF, leuS, ileS, recG, pyrG, and recA were used for typing Lb. rhamnosus strains; nucleotide sequences of genes parB, ychF, pyrG, atpF, recA, ileS, recG, and leuS were used for typing Lb. fermentum strains. Housekeeping genes were selected according to the following criteria: they code for proteins involved in basic metabolic processes, the genes were represented as a single copy in all genomes, and their nucleotide sequence was not less than 1 kb long (Cai et al. 2007). Primers were designed using sequenced genomes of Lb. rhamnosus and Lb. fermentum (http://www.ncbi.nlm.nih.gov/genbank/) (Tables 2, 3), and used to amplify the DNA of the analyzed strains. PCR fragments were detected by agarose gel electrophoresis, purified, Sanger-sequenced, and aligned using ClustalW (Thompson et al. 1994). The length of analyzed fragments was 307–716 bp. Each unique nucleotide sequence was considered an allele of a particular gene and assigned a number. The combination of alleles for each strain determined its sequence type (ST). The total nucleotide sequence of gene fragments arranged in the above-mentioned order (4151 bp for Lb. rhamnosus and 3560 bp for Lb. fermentum), was used for the construction of phylogenetic trees of strains using MEGA v.5.2 (Tamura et al. 2011) program. Nucleotide sequences of MLST alleles are listed in Supplementary material S4–S5.
TA systems analysis
Primers (Table 4, 6) were designed to bind the flanking regions of TA systems RelBE and MazEF (four in Lb. rhamnosus and two in Lb. fermentum). PCR products were electrophoresed, purified, and Sanger-sequenced. Alleles of genes and combinations of alleles (TA types) were identified for each strain. Nucleotide sequences of TA alleles are listed in Supplementary material S6–S7.
Results and discussion
Lactobacillus strains’ identification
Thirty-one Lactobacillus strains were isolated from feces, saliva, and vaginal cavities of 13 women, and their bacterial species were identified (Table 1). The dominant species in all three biotopes were Lb. rhamnosus and Lb. fermentum. Moreover, Lb. salivarius and Lb. crispatus were present in the vagina, Lb. casei, Lb. buchneri, Lb. johnsonii and Lb. gasseri in feces, and Lb. casei and Lb. plantarum in the saliva. Lb. rhamnosus and Lb. fermentum strains were often isolated from feces, saliva, and vagina upon cultivation on appropriate growth media (Pavlova et al. 2002; Álvarez-Olmos et al. 2004; Walter 2008). In a review on the prevalence of fecal Lactobacillus populations in humans using 16S RNA method, the authors concluded that these strains are allochthonous members of the human microbiota (Walter 2008). However, in a different study, where whole-genome sequencing method of fecal samples was used, Lb. rhamnosus and Lb. fermentum strains were classified as a stable community within the gut (Rossi et al. 2016).
MLST of Lactobacillus rhamnosus and Lactobacillus fermentum
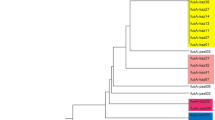
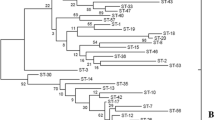
MLST was performed to reveal intra-specific differences for both dominant species—Lb. rhamnosus and Lb. fermentum. Seven and eight housekeeping genes were selected for Lb. rhamnosus and Lb. fermentum, respectively. The number of alleles was 3–4 and 3–6. The total number of polymorphic sites was 156 (Lb. rhamnosus) and 75 (Lb. fermentum). The characteristics of genes are shown in Tables 2 and 3. Lb. rhamnosus strains were divided into 7 ST and Lb. fermentum strains were divided into 5 ST (Supplementary Tables S2–S3). The phylogenetic relationship of strains based on MLST data is shown in Fig. 1.

Phylogenetic trees of Lb. rhamnosus (A) and Lb. fermentum (B) strains generated on the basis of whole nucleotide sequence of MLST loci. The letter F indicates the number of the volunteer. Strains isolated from the same organism are highlighted with same pattern
Strains isolated from the same woman belonged, in most cases, to different species. However, in six women, the same Lactobacillus species were isolated from different biotopes (Lb. rhamnosus in four women and Lb. fermentum in two women). In five cases, strains belonged to the same ST (Lb. rhamnosus strains 76–77, 236–237, 316–360; Lb. fermentum strains 277–279, 309–311). In four cases, the strains of the same ST were isolated from feces and saliva, and in one case, two strains were isolated from saliva and the vagina (Lb. rhamnosus 316–360). Consequently, Lb. rhamnosus and Lb. fermentum strains of the same ST can be isolated from different biotopes of one organism. Strains of the same ST were also isolated from different individuals. Identification of identical STs suggests that a pool of closely related strains is interchangeable between local residents. Similar results were obtained by a group that used whole-genome sequencing to differentiate clinical isolates (Roach et al. 2015) as well as in another study where the authors studied the diversity of Staphylococcus haemolyticus strains using MLST (Kornienko et al. 2016).
Strains isolated from saliva and feces in most cases belonged to the same ST. Only one Lb. rhamnosus and three Lb. fermentum strains were vaginal; MLST of Lb. fermentum vaginal strains revealed differences in ST compared with strains isolated from the gut and the oral cavity. Due to a low number of strains used, the obtained results show preliminary and approximate data concerning the distribution of strains across different biotopes of the human body.
Differentiation of Lactobacillus strains based on TA system polymorphism
Previously, we have demonstrated that polymorphism in TA systems can be used for genotyping lactobacilli (Alekseeva et al. 2011; Klimina et al. 2013). In this study, we used polymorphisms in RelBE and MazEF chromosomal TA systems for typing Lb. rhamnosus and Lb. fermentum strains.
Most Lb. fermentum strains contained the TA system RelBE Lf and a single toxin gene mazF Lf . Both of these loci were present in all investigated Lb. fermentum strains and had four alleles containing three and five polymorphic sites, respectively (Table 4). Differentiation of Lb. fermentum strains based on TA system polymorphism classified them in five TA types, which confirms the MLST data (Table 5).
We identified six TA systems belonging to RelBE and MazEF families in the genome of Lb. rhamnosus (Klimina et al. 2013). Three TA systems consisting of a toxin and an antitioxin (PemK1-A1 Lrh , PemK2-A2 Lrh , and YefM-YoeB Lrh ) and a single toxin gene relE1 Lrh were selected for determining intra-species polymorphism. Only two TA systems were present in all strains, whereas the other two were present only in some strains. The strains demonstrated a high level of genetic polymorphism that was manifested in single-nucleotide polymorphisms, deletions, and insertions, including a mobile element of the IS3 family (Table 6). The latter has been identified earlier in certain Lb. rhamnosus strains (Klimina et al. 2013). Genomic polymorphism (presence or absence of genes) was also considered. The differentiation of strains based on polymorphism in TA systems is shown in Table 7. Both methods allowed to classify Lb. rhamnosus strains into the same seven STs.
MLST allows assessing the diversity of strains on the genetic rather than genomic level. This method is relatively inexpensive and simple; its main advantage is that it provides the ability to compare and exchange data between laboratories. MLST is used for studying genetic diversity, population structure of species, and phylogeny of strains; it was widely employed in studies of pathogenic bacteria (Maiden 2006); databases contain information on tens of thousands of strains (for example, EnteroBase http://enterobase.warwick.ac.uk/). It was also used for typing lactobacilli, such as Lb. casei (Cai et al. 2007; Diancourt et al. 2007), Lb. delbrueckii (Tanigawa and Watanabe 2011), Lb. sanfranciscensis (Picozzi et al. 2010), Lb. fermentum (Dan et al. 2015), Lb. plantarum (De las Rivas et al. 2006; Xu et al. 2015), Lb. salivarius (Raftis et al. 2011), Lb. helveticus (Sun et al. 2015), Lb. ruminis (O’ Donnell et al. 2015), Lb. sakei (Chaillou et al. 2013), and Lb. reuteri (Su et al. 2012). Unfortunately, when it comes to lactobacilli, MLST has not become a universal method yet. Available data are restricted to a small number of papers, and the main websites (http://www.pasteur.fr/mlst and http://pubmlst.org) are not updated. Different genes are used in different laboratories to analyze the same species (De las Rivas et al. 2006; Xu et al. 2015), which makes it impossible to compare the data. Another drawback of this method compared with various amplified-fragment length polymorphism methods is its duration due to the necessity of DNA sequencing.
TA systems were only recently considered as intra-species markers. In Escherichia coli, TA systems have been used to characterize intra-species phylogenetic differences (Fiedoruk et al. 2015). A strong correlation was found between TA types, clonal complexes, and sequence types in 48 Streptococcus pneumoniae strains (Chan et al. 2014). Polymorphism in TA systems was used for typing Mycobacterium tuberculosis strains, allowing their clusterization into basic genotypes and subtypes (Zaychikova et al. 2015). We demonstrated that TAs are suitable for characterizing intra-specific diversity of Lb. rhamnosus and Lb. fermentum strains. The advantage of using polymorphisms in genes of TA systems for genotyping lies in the fewer number of genes, and as a consequence, less time required for the analysis. Since TA systems are involved in many cellular processes in bacteria, we believe that TA markers can also help perform functional intra-specific differentiation. However, since ТА systems are not a stable component of all genomes, it would be wrong to say, at this point, that they are suitable for typing any species. Perhaps, it makes sense combining both MLST and TA methods, using several housekeeping genes and TA systerm(s), like in some pathogenic bacteria where housekeeping genes and virulence factors were used together for typing (Achtman et al. 1999).
References
Achtman M, Azuma T, Berg DE, Ito Y, Morelli G et al. (1999) Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Molec Microbiol 32(3):459–470
Аlekseeva M, Кlimina K, Danilenko V (2011) Use of toxin-antitoxin genes relBE and mazEF for species and strain identification of lactobacilli. Russian patent 2526576, 23.12.2011
Álvarez-Olmos MI, Barousse MM, Rajan L, Van Der Pol BJ, Fortenberry D, Orr D, Fidel PL Jr (2004) Vaginal lactobacilli in adolescents: presence and relationship to local and systemic immunity, and to bacterial vaginosis. Sex Transm Dis 31:393–400
Cai H, Rodríguez BT, Zhang W, Broadbent JR, Steele JL (2007) Genotypic and phenotypic characterization of Lactobacillus casei strains isolated from different ecological niches suggests frequent recombination and niche specificity. Microbiology 153:2655–2665. doi:10.1099/mic.0.2007/006452-0
Chaillou S, Lucquin I, Najjari A, Zagorec M, Champomier-Vergès MC (2013) Population genetics of Lactobacillus sakei reveals three lineages with distinct evolutionary histories. PLoS One 8(9):e73253. doi:10.1371/journal.pone.0073253
Chan WT, Yeo CC, Sadowy E, Espinosa M (2014) Functional validation of putative toxin-antitoxin genes from the Gram-positive pathogen Streptococcus pneumoniae: phd-doc is the fourth bona-fide operon. Front Microbiol 5:677. doi:10.3389/fmicb.2014.00677.
Dan T, Liu W, SongY, Xu H, Menghe B, Zhang H, Sun Z (2015) The evolution and population structure of Lactobacillus fermentum from different naturally fermented products as determined by multilocus sequence typing (MLST). BMC Microbiol 15:107. doi:10.1186/s12866-015-0447-z
De las Rivas B, Marcobal A, Muñoz R (2006) Development of a multilocus sequence typing method for analysis of Lactobacillus plantarum strains. Microbiology 152:85–93. doi:10.1099/mic.0.28482-0
Diancourt L, Passet V, Chervaux C, Garault P, Smokvina T, Brisse S (2007) Multilocus sequence typing of Lactobacillus casei reveals a clonal population structure with low levels of homologous recombination. Appl Environ Microbiol 73(20):6601–6611. doi:10.1128/AEM.01095-07
Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C (2012) Microbial cooccurrence relationships in the human microbiome. PLoS Comput Biol 8:e1002606. doi:10.1371/journal.pcbi.1002606
Fiedoruk K, Daniluk T, Swiecicka I, Sciepuk M, Leszczynska K (2015) Type II toxin-antitoxin systems are unevenly distributed among Escherichia coli phylogroups. Microbiology 161:158–167. doi:10.1099/mic.0.082883-0
Gosiewski T, Chmielarczyk A, Strus M, Brzychczy-Woch M, Heczko P (2012) The application of genetics methods to differentiation of three Lactobacillus species of human origin. Ann Microbiol 62(2):1437–1445. doi:10.1007/s13213-011-0395-2
Herbel SR, Vahjen W, Wieler LH, Guenther S (2013) Timely approaches to identify probiotic species of the genus Lactobacillus. Gut Pathog 5:27. doi:10.1186/1757-4749-5-27
Janda JM, Abbott SL (2007) 16 S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol 45(9):2761–2764. doi:10.1128/JCM.01228-07
Kassaa IA, Hober D, Hamze M, Caloone D, Dewilde A, Chihib NE, Drider D (2015) Vaginal Lactobacillus gasseri CMUL57 can inhibit herpes simplex type 2 but not Coxsackievirus B4E2. Arch Microbiol 197(5):657–664. doi:10.1007/s00203-015-1101-8
Klimina KM, Kjasova DK, Poluektova EU, Krügel H, Leuschner Y, Saluz HP, Danilenko VN (2013) Identification and characterization of toxin–antitoxin systems in strains of Lactobacillus rhamnosus isolated from humans. Anaerobe 22:82–89. doi:10.1016/j.anaerobe.2013.05.007
Kornienko M, Ilina E, Lubasovskaya L, Priputnevich T, Falova O, Sukhikh G, Govorun V (2016) Analysis of nosocomial Staphylococcus haemolyticus by MLST and MALDI-TOF mass spectrometry. Infect Genet Evol 39:99–105. doi:10.1016/j.meegid.2015.12.01
Lane D (1991) 16 S/23 S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, NY, pp 115–175
Maiden MC (2006) Multilocus sequence typing of bacteria. Annu Rev Microbiol 60:561–588. doi:10.1146/annurev.micro.59.030804.121325
De Man JC, Rogosa M, Sharpe M (1960) A medium for the cultivation of Lactobacilli. J Appl Bacteriol 23:130–135. doi:10.1111/j.1365-2672.1960.tb00188.x
O’ Donnell MM, Harris HM, Lynch DB, Ross RP, O’Toole PW (2015) Lactobacillus ruminis strains cluster according to their mammalian gut source. BMC Microbiol 15:80. doi:10.1186/s12866-015-0403-y
Pavlova SI, Kilic AO, Kilic SS, So JS, Nader-Macias ME, Simoes JA, Tao L (2002) Genetic diversity of vaginal lactobacilli from women in different countries based on 16 S rRNA gene sequences. J Appl Microbiol 92:451–459
Picozzi C, Bonacina G, Vigentini I, Foschino R (2010) Genetic diversity in Italian Lactobacillus sanfranciscensis strains assessed by multilocus sequence typing and pulsed-field gel electrophoresis analyses. Microbiology 156(7):2035–2045
Qin J, Li R, Raes J, Arumugam M et al (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65. doi:10.1038/nature08821
Raftis EJ, Salvetti E, Torriani S, Felis GE, O’Toole PW (2011) Genomic diversity of Lactobacillus salivarius. Appl Environ Microbiol 77(3):954–965. doi:10.1128/AEM.01687-10
Roach DJ, Burton JN, Lee C, Stackhouse B, Butler-Wu SM, Cookson BT, Shendure J, Salipante SJ (2015) A year of infection in the intensive care unit: prospective whole genome sequencing of bacterial clinical isolates reveals cryptic yransmissions and novel microbiota. PLoS Genet 11:e1005413. doi:10.1371/journal.pgen.1005413
Rossi M, Martínez-Martínez D, Amarett A, Ulrici A, Raimondi S, Moya A (2016) Mining metagenomic whole genome sequences revealed subdominant but constant Lactobacillus population in the human gut microbiota. Environ Microbiol. doi:10.1111/1462-2920.13295
Su M, Oh PL, Walter J, Gänzle MG (2012) Intestinal origin of sourdough Lactobacillus reuteri isolates as revealed by phylogenetic, genetic, and physiological analysis. Appl Environ Microbiol 78(18):6777–6780. doi:10.1128/AEM.01678-12
Sun Z, Liu W, Song Y, Xu H, Yu J, Bilige M, Zhang H, Chen Y (2015) Population structure of Lactobacillus helveticus isolates from naturally fermented dairy products based on multilocus sequence typing. J Dairy Sci 98(5):2962–2972. doi:10.3168/jds.2014-9133
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. doi:10.1093/molbev/msr121
Tanigawa K, Watanabe K (2011) Multilocus sequence typing reveals a novel subspeciation of Lactobacillus delbrueckii. Microbiology 157:727–738. doi:10.1099/mic.0.043240-0
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680. doi:10.1093/nar/22.22.4673
Turroni F, Ventura M, Buttó LF, Duranti S, O’Toole PW, Motherway MO, van Sinderen D (2013) Molecular dialogue between the human gut microbiota and the host: a Lactobacillus and Bifidobacterium perspective. Cell Mol Life Sci 71:183–203. doi:10.1007/s00018-013-1318-0
Wall R, Fitzgerald G, Hussey S, Ryan T, Murphy B, Ross P, Stanton C (2007) Genomic diversity of cultivable Lactobacillus populations residing in the neonatal and adult gastrointestinal tract. FEMS Microbiol Ecol 59:127–137. doi:10.1111/j.1574-6941.2006.00202.x
Walter J (2008) Implications for ecological role of Lactobacilli in the gastrointestinal tract: fundamental and biomedical research. Appl Environ Microbiol 74:4985–4996. doi:10.1128/AEM.00753-08
Wen Y, Behiels E, Devreese B (2014) Toxin–antitoxin systems: their role in persistence, biofilm formation, and pathogenicity. Pathog Dis 70:240–249. doi:10.1111/2049-632X.12145
Xu H, Liu W, Zhang W, Yu J, Song Y, Menhe B, Zhang H, Sun Z (2015) Use of multilocus sequence typing to infer genetic diversity and population structure of Lactobacillus plantarum isolates from different sources. BMC Microbiol 15:241. doi:10.1186/s12866-015-0584-4
Zaychikova MV, Zakharevich NV, Sagaidak MO, Bogolubova NA, Smirnova TG, Andreevskaya SN, Larionova EE, Alekseeva MG, Chernousova LN, Danilenko VN (2015) Mycobacterium tuberculosis type II toxin-antitoxin systems: genetic polymorphisms and functional properties and the possibility of their use for genotyping. PLoS One 10:e0143682. doi:10.1371/journal.pone.0143682
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Djamel Drider.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Poluektova, E.U., Yunes, R.A., Epiphanova, M.V. et al. The Lactobacillus rhamnosus and Lactobacillus fermentum strains from human biotopes characterized with MLST and toxin-antitoxin gene polymorphism. Arch Microbiol 199, 683–690 (2017). https://doi.org/10.1007/s00203-017-1346-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-017-1346-5