
Abstract
The operation of mitogen-activated protein kinase (MAPK) signal transduction pathways is one of the most important mechanisms for the transfer of extracellular information into the cell. These pathways are highly conserved in eukaryotic organisms. In fungi, MAPK pathways are involved in the regulation of a number of cellular processes such as metabolism, homeostasis, pathogenesis and cell differentiation and morphogenesis. Considering the importance of pathways, in the present work we proceeded to identify all the genes that are regulated by the signal transduction pathway involved in mating, pathogenesis and morphogenesis of Ustilago maydis. Accordingly we made a comparison between the transcriptomes from a wild-type strain and an Ubc2 mutant affected in the interacting protein of this pathway by use of microarrays. By this methodology, we identified 939 genes regulated directly or indirectly by the MAPK pathway. Of them, 432 were positively, and 507 were negatively found regulated. By functional grouping, genes encoding cyclin-dependent kinases, transcription factors, proteins involved in signal transduction, in synthesis of wall and cell membrane, and involved in dimorphism were identified as differentially regulated. These data reveal the importance of these global studies, and the large (and unsuspected) number of functions of the fungus under the control of this MAPK, providing clues to the possible mechanisms involved.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Signal transduction pathways are involved in the transfer of information from the exterior to the nucleus in all eukaryotic cells. Among them, we may cite the Mitogen-activated protein kinase (MAPK) signaling pathway that has important roles in several cellular processes such as transformation, proliferation, differentiation, development and apoptosis (reviewed by Zhang and Liu 2002). In lower eukaryotic organisms such as fungi, MAPK pathways are rather conserved, and among their functions, there are the regulations of gene expression during cellular responses, allowing the adaptation of the organisms to different environmental conditions. In these organisms, up to five different MAPK pathways have been described to be involved in different cellular processes; for example in Saccharomyces cerevisiae, MAPK pathway responding to different cues have been described: pheromone response, filamentous growth, high osmolarity, cell wall integrity and spore wall assembly (Gustin et al. 1998; Pan et al. 2000; Gancedo 2001; Palecek et al. 2002; Chen and Thorner 2007).
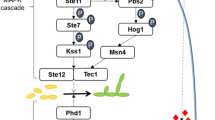
The MAPK pathways involve the operation of three protein kinases in hierarchical order: MAPKKK (MAP kinase kinase kinase), MAPKK (MAP kinase kinase) and MAPK (MAP kinase), which are sequentially activated by phosphorylation at specific sites. Normally these protein kinases become associated through an adaptor protein. The activation process initiates by the perception of a signal by a transmembrane receptor and the action normally of heterotrimeric G proteins that transfer the signal to MAPKKK that starts the cascade of phosphorylation reactions. Finally, the MAPK (MAP kinase) phosphorylates distinct transcription factors that regulate gene expression in response to the sensed stimuli (Brefort et al. 2009; Ruiz-Herrera et al. 2009; Raudaskoski and Kothe 2010; Vollmeister et al. 2011; Ruiz-Herrera and Campos-Góngora 2012).
Contrasting to other fungi, in U. maydis it has been described the existence of only two MAPK pathways. The best one characterized is involved in mating, pathogenesis and dimorphism (Brefort et al. 2009; Vollmeister et al. 2011). A second MAPK pathway involved in cell wall integrity (CWI) was described further on (Carbó and Pérez-Martín 2010), and the possible existence of a third one that would be involved in response to osmotic stress has been entertained (Perez-Nadales et al. 2014).
Previously, we isolated a mutant of U. maydis (CL211) that is unable to carry out the yeast-to-mycelium dimorphic transition, grows constitutively in the yeast-like form, and is non-virulent to maize (Martínez-Espinoza et al. 1997). A genetic analysis of the mutant demonstrated that it is affected in UBC2 (Ustilago bypass of cyclase; Mayorga and Gold, 2001) gene encoding the MAP kinase adaptor protein belonging to the best known MAPK pathway (Martínez-Espinoza et al. 2004) and that for facility, we refer to as PMM (pathogenesis, mating and morphogenesis).
U. maydis is a dimorphic Basidiomycota fungus and an attractive model organism for the analysis of different cellular processes (Bölker 2001; Brefort et al. 2009; Ruiz-Herrera et al. 2009; Vollmeister et al. 2011; Valdés-Santiago et al. 2012; Ruiz-Herrera and Campos-Góngora 2012, Ruiz-Herrera et al. 2013; Valdés-Santiago and Ruiz-Herrera 2014; León-Ramirez et al. 2014). In nature U. maydis infects maize (Zea mays L.) and teozintle (Z. mays subsp. parviglumis), where it completes its sexual life cycle. Under axenic conditions, the fungus is able to infect plant species phylogenetically unrelated to maize (León-Ramírez et al. 2004; Méndez-Morán et al. 2005, Martínez-Soto et al. 2013; Ruiz-Herrera et al. 2013). In addition, when incubated under defined environmental in vitro conditions, U. maydis performs a completely different sexual life cycle with the formation of basidiocarps (Cabrera-Ponce et al. 2012).
Considering the importance of MAPK pathways in the regulation of metabolism in fungi that there is almost no knowledge of the whole set of genes whose transcription is regulated by the MAPK pathways in U. maydis and in other fungi, and that the approaches to understand their operation have relied mostly on reverse genetics and biochemical techniques (Banuett and Herskowitz 1994; Mayorga and Gold 1999; Müller et al. 1999; Andrews et al. 2000; Müller et al. 2003; Martínez-Espinoza et al. 2004), we have proceeded to make an analysis of all the U. maydis genes regulated by the PMM MAPK pathway, making use of the CL211 ubc2 mutant affected in this system.
Materials and methods
Ustilago maydis strains and culture conditions
In this work, we used the wild-type strain FB2 (a 2 b 2 ; Banuett and Herskowitz 1989) and the CL211 strain (a 2 b 2 , ubc2; Martínez-Espinoza et al. 1997, 2004), a constitutive monomorphic yeast mutant deficient in the gene encoding the MAP kinase pathway-interacting protein UBC2 (Martínez-Espinoza et al. 1997, 2004; Mayorga and Gold 2001). The strains were maintained in 50 % glycerol in complete medium (MC; Holliday 1974) at −70 °C, and recovered in MC liquid medium. Growth in minimal medium (MM; Holliday 1974) took place at pH 7. Under these conditions, the wild-type strain and the mutant grow yeast-like (Ruiz-Herrera et al. 1995; Martínez-Espinoza et al. 1997).
Isolation of RNA and microarrays hybridization
Similar conditions to those described by Martínez-Soto and Ruiz-Herrera (2013) were used. Three independent cultures (biological replicates) of U. maydis strains (106 cells/ml) were grown in MM pH 7 at 28 °C under shaking conditions for 16 h. The cells were recovered by centrifugation, and RNA was isolated using Trizol (Invitrogen, Carlsbad, CA, USA) and purified with QIAGEN (Hilden, Germany) columns. RNA concentration was measured by absorbance at 260 nm with a Nanodrop (Termo Scientific, Waltham, MA, USA), and its integrity was determined by electrophoresis in denaturing agarose gels. The RNA samples coming from the three independent cultures were mixed together, and used for synthesis and subsequent labeling of cDNA, and microarray hybridization. These two last procedures were performed by Roche NimbleGen Inc. (Reykjavík, Iceland).
Microarray analysis
The type of microarrays used in this work were the same as previously described (Martínez-Soto and Ruiz-Herrera 2013) from NimbleGen, characterized by having five different oligonucleotides of 60 nt in length designed along the full gene length in duplicate, according to a design from Scott Gold (University of Georgia). It must be stressed that these conditions secure that data for each one of the 6,883 U. maydis genes represents an average of ten determinations. Scan and normalization of the data were done as described previously (Martínez-Soto and Ruiz-Herrera 2013). ArrayStar software from DNAStar was used for microarray analyses, P values were adjusted by the false discovery rate (FDR) method (Benjamini and Hochberg 1995) and P values <0.05 were considered differentially expressed. Genes whose expression values were higher (up-regulated) in the wild-type strain were considered to be positively regulated by the MAPK pathway, and those whose expression was higher in the mutant (down-regulated) were considered to be negatively regulated by the pathway. A value of twofold change up or down was considered the cutoff to determine whether a gene was positively or negatively regulated.
Functional grouping and search for specific genes
The functional annotation of the total regulated genes by the PMM MAPK pathway in U. maydis was performed with the Functional Catalogue (FunCat) online program (Ruepp et al. 2004), and with the aid of R statistical software; and supported by the MIPS Ustilago maydis Database (http://mips.helmholtz-muenchen.de/genre/proj/ustilago/), and the Ustilago maydis Database-Broad Institute (http://www.broadinstitute.org/annotation/genome/ustilago_maydis). The search of genes previously described and related to U. maydis dimorphism was made based on the data previously reported by Heimel et al. (2010) and Martínez-Soto and Ruiz-Herrera (2013). For handling of data sets, Venn diagrams were used (http://www.bioinformatics.lu/venn.php).
Search of domains in genes differentially regulated as clusters
For the identification of possible domains, and the presence of a signal peptide in proteins encoded by genes described as Unclassified, which are present in clusters, their amino acid sequences were analyzed with the Pfam (Punta et al. 2012), SMART (Letunic et al. 2012), and SignalP (Bendtsen et al. 2004) online programs; and supported by NCBI database (http://www.ncbi.nlm.nih.gov/).
Search of consensus sequences and binding sites for transcription factors
To identify consensus sequences for transcription factors in genes encoding cyclin-dependent kinases, protein kinases, serine/threonine protein kinases, and genes grouped in clusters, 1,000 base pairs upstream of the ATG site were analyzed using the JASPAR online program (http://jaspar.binf.ku.dk/) based on the S. cerevisiae genome. BLAST of NCBI page online (http://blast.ncbi.nlm.nih.gov/Blast.cgi), MIPS Ustilago maydis Database and Ustilago maydis Database-Broad Institute were used to identify U. maydis homologue genes for the transcription factors identified.
Results and discussion
To identify the genes regulated by the PMM MAPK pathway in U. maydis, we proceeded to make a comparison of the transcriptomes of the wild-type strain FB2 and the CL211 ubc2 mutant, both grown at pH 7. Under these conditions, the morphology of both strains is yeast-like, and the only variable involved is the mutation in the UBC2 gene. The results obtained revealed that of total 6,883 genes of U. maydis, 939 genes were differentially expressed between the two strains. Of these, 432 (46.0 %) were up-regulated and 507 (54.0 %) were down-regulated in the wild type (see Table S1), indicating that the former are positively regulated and the latter negatively regulated by the PMM MAPK pathway.
Functional grouping of the differentially regulated genes is shown in Fig. 1. The categories with the higher number of differentially expressed genes were Unclassified Proteins with 362 genes (38.6 %), and Metabolism and Energy with 202 genes (21.5 %). Genes grouped in this latter category were mainly related to general metabolism and metabolism of carbohydrates, amino acids and lipids; genes of the second one generally were found repressed, and those from the third one were generally overexpressed. On the other hand, 54 genes (5.7 %) were grouped into the Synthesis and Protein Fate category, and most of them were negatively regulated by PMM MAPK pathway (see Table S1). Six categories with a smaller number of differential genes showed relevance to the metabolic pathway analyzed here. These were the following: (1) Cell cycle and DNA Processing, with 38 differential genes (4.0 %); (2) Transcription, with 42 differential genes (4.5 %); (3) Cellular Transport, with 104 differential genes (11.1 %); (4) Signal Transduction Mechanism, with 41 differential genes (4.4 %); (5) Biogenesis of Cellular Components, with 50 differential genes (5.3 %); and (6) Differentiation and Cell Fate, with 21 differential genes (2.2 %). In the category of Cell Cycle and DNA Processing, a number of genes encoding cyclin-dependent kinases (cdk), cell-division cycle proteins (CDC), and other genes involved in cell cycle were found positively regulated by the PMM MAPK pathway (Table 1). For example, genes um03992 encoding Dip1–Don3 interacting protein, um03234 encoding Cdc5-serine/threonine-protein kinase, um10499 related to Hos4-subunit of the Set3 complex and um00277 encoding an M-phase inducer phosphatase were 2.8-, 2.9-, 3.0- and 4.6-fold up-regulated, respectively. DIP1 gene has been previously mutated in U. maydis, and the mutant strain was not affected in cytokinesis; but the double mutation in DIP1 and DON3 genes affected nuclear separation during mitosis. Moreover, DON3 has been associated with the formation of secondary septa in U. maydis (Sandrock et al. 2006). CDC5 gene has been reported in Schizosaccharomyces pombe as a cell cycle regulator that acts on the G2/M transition (Bernstein and Coughlin 1997). Also this gene has been described as necessary for initiation of DNA replication in S. cerevisiae (Kitada et al. 1993). The Set complex is a multiprotein complex involved in the mitotic cell cycle, as well as in sporulation in yeasts such as S. cerevisiae (Pijnappel et al. 2001). And the last gene, um00277, has been described to encode a CDC protein directly involved in cell cycle regulation (Millar and Russell 1992). Contrary, genes grouped in this same category and related to DNA processing were mostly negatively regulated by the MAPK pathway, for example genes encoding DNA helicases, DNA repair proteins and histone acetyltransferase (Table 1). Differential regulation by the PMM MAPK pathway of genes related to cell cycle supports studies performed on other eukaryotic organisms including fungi, which postulate that this pathway regulates positively or negatively the cell cycle of these organisms when sensing environmental or stress stimuli (Wilkinson and Millar 2000; Clotet and Posas 2007; Carbó and Pérez-Martín 2010).

Functional grouping of the 939 genes regulated by the PMM MAPK pathway in U. maydis. Black bars represent the percentage of differentially regulated genes grouped in each category; white bars represent the percentage of genes up-regulated in each category; and gray bars, the percentage of genes down-regulated in each category in the wild-type strain. Legends under the bars are the designation of each one of the categories (FunCat); and the numbers over the bars are the percentages of differentially expressed genes in each category
Within the Transcription category, different genes encoding transcription factors were positively regulated by the PMM MAPK pathway (Table 2), including the following ones: um10426, encoding the PacC transcription factor; um15103, related to transcription factor AtfA; um03588, related to transcription factor Medusa; and um02052, the homologue of white collar 1 (WC1) gene from Neurospora crassa with 2.0-, 2.0-, 2.5- and 2.7-fold changes, respectively. Regarding PacC, it has been demonstrated that in most fungi, including U. maydis, pH sensing is carried out only by the PAL/RIM pathway, involving PacC (Ramon et al. 1999; Davis et al. 2000; Peñalva and Arst 2002; Lamb and Mitchell 2003; Aréchiga-Carvajal and Ruiz-Herrera 2005; Hua et al. 2010; Cervantes-Chávez et al. 2010; Franco-Frías et al. 2014). This observed increase in expression under the control of the MAPK pathway may be due to a cross talk between both signaling pathways (Fonseca-Garcia et al. 2012). The AtfA transcription factor has been described in A. nidulans and Schizosaccharomyces pombe as a bZIP-type transcription factor involved in response to different types of stress (Balázs et al. 2010). Medusa type transcription factors act in concert with other transcription factors and microRNAs forming a network that regulates the expression of a large number of genes, including some involved in the formation of tumors in animals (Guo et al. 2011). And WC1 gene in Neurospora is involved in the circadian feedback loops and light sensing (Ballario et al. 1996). The overexpression of transcription factors by the PMM MAPK pathway was expected since, as was mentioned above, this pathway activates different transcription factors that in turn regulate the expression of different genes or other transcription factors in response to the sensed stimuli. However, genes directly involved in RNA processing and grouped in this category were down-regulated, and among them, some involved in transcription, protein elongation, splicing factor, aminoacyl-tRNA hydrolases, etc. (Table 2). These latter data are in agreements with the negative effect of the PMM MAPK pathway on processes involved in DNA metabolism (see above).
Interestingly, most of the genes grouped in the Cellular Transport category that encode proteins involved in intracellular transport of different substances including metals such as, zinc, iron, calcium and sodium between organelles, or encode secretion proteins, such as effectors, were generally repressed by the PMM MAPK (Table 3). These data indicate that the cellular transport processes and protein secretion are mostly negatively regulated by the MAPK pathway during saprophytic growth of the fungus.
A category of genes with relevance considering the role of the MAPK pathway was the class of Signal Transduction Mechanism. Interestingly, genes associated within this category, as well as GTPases were grouped here, and most of them were positively regulated by the PMM MAPK pathway (Table 4). Among the genes overexpressed, we may cite the following: (1) um05656, related to Sok1 protein, a suppressor of kinases in S. cerevsiae (Ward and Garrett 1994); (2) um15092, encoding a probable Pbs2-tyrosine protein kinase of the MAP kinase kinase family that specifically phosphorylate threonine and tyrosine residues of the MAPK protein Hog1p, and is essential for the survival of the yeast under conditions of high osmolarity (Wurgler-Murphy et al. 1997); (3) um11007, that encodes a probable GTP-binding protein Rab5c, a member of the family of small GTPases that regulate membrane traffic, and whose role has been suggested in the mitotic cell cycle of eukaryotic cells (Singer-Krüger et al. 1994; Chiariello et al. 1999); and (4) um02382, encoding the Mfa1-a1-specific pheromone mating factor a1 (Bölker et al. 1992) with 3.3-, 3.6-, 5.1- and 13.2-fold change, respectively. Also, we identified genes encoding serine/threonine protein kinases and genes encoding MAPK proteins, for example: um03282, um10855, um04543, um15092 and Crk1 (um11410 with 2.9-fold change), described as a novel MAPK protein involved in the activation of the transcription factor Prf1, and therefore in mating and pathogenesis in U. maydis (Garrido et al. 2004). These results may be related to the recently described additional U. maydis MAPK pathways involved in the integrity of the cell wall, and perhaps the possible pathway responding to osmotic stress (Carbó and Pérez-Martín 2010; Perez-Nadales et al. 2014), and more important, the possible existence of a cross talk among them. In the category of Biogenesis of Cellular Component, different genes related to synthesis of the membrane and cell wall were found to be up-regulated by the PMM MAPK pathway (Table 5). Among these, we may cite genes related to cell wall biogenesis, e.g., those encoding chitin deacetylases, glucan synthases, chitinases and chitin synthases. One example is gene um01640 encoding GAS1, whose homologues have been described in Candida glabrata as of importance for cell wall biosynthesis (Weig et al. 2001). Also up-regulated was gene um05811 encoding KRE6, a protein involved in the synthesis of β 1,6-glucans. These results contrast with the observation that during U. maydis dimorphism induced by pH change, KRE6 was down-regulated (Martínez-Soto and Ruiz-Herrera 2013; Robledo-Briones and Ruiz-Herrera 2013). Finally, we may cite um04364 and um10211 encoding two Exg1-exo-β-1,3-beta-glucanases. Also some genes related to ribosome biogenesis were negatively regulated by the PMM MAPK pathway (see Table S1).
Finally, in the category Differentiation and Cell Fate, several genes related to dimorphism and cell morphology were found to be mostly up-regulated by the PMM MAPK pathway (Table 6). Among these genes, some related to actin may be important, taking into account their role in cell polarity and vesicle trafficking, for example: um11450 encoding Hgl1–Hgl1p is required for dimorphism and teliospore formation in U. maydis (Dürrenberger et al. 2001); um11246 is related to Bzz1, an actin assembly complex component involved in actin polymerization in S. cerevisiae (Soulard et al. 2002); um00896 encodes Kin7a-Kinesin-7a motor protein, probably involved in polar growth of hyphae; and um01671, related to Ysc84-protein, a member of a new class of actin-binding proteins described in yeasts as important for actin polarization and endocitosis (Robertson et al. 2009), which is involved in the organization of the actin cytoskeleton.
Regarding the possible nature of the transcription factors through which the PMM MAPK might control the expression of different genes, we found that the genes described as cyclin-dependent kinases, such as um10705, um02860 and um03234 (Table 1), posses the GGCCAT and TTGGT sequences in their promoter regions. These sequences are known to be binding sites for the Skn7 and Hap2 transcription factors, respectively. These data suggest that their regulation by the PMM MAPK pathway may occur through these two transcription factors, particularly Hap2 (um01597) that has already been described to be activated by the MAPK pathway (Brefort et al. 2009; Vollmeister et al. 2011), and has been described as a regulator of the master transcription factor, Prf1. Deletion of HAP2 affects mating and pathogenesis in U. maydis (Mendoza-Mendoza et al. 2009). The S. cerevisiae gene homologous to U. maydis Skn7 (um03346) have been described as involved in gene expression in response to changes in extracellular osmolarity, oxidative stress, thermal shock and cell wall integrity (Brown et al. 1993; Krems et al. 1996); we postulate that the PMM MAPK pathway through this transcription factor may regulate stress genes identified in this work (Fig. 1). Its homologues in C. albicans and Cryptococcus neoformans are important for virulence (Singh et al. 2004; Wormley et al. 2005); and its regulation in U. maydis has been described during A. thaliana infection (Martínez-Soto et al. 2013). In addition to binding sites for Hap2 and Skn7, we found the GATAA consensus sequence, which is recognized by the S. cerevisiae transcription factor Gln3 (see Tables 1, 4) involved in the expression of various genes in response to limiting nitrogen conditions (Minehart and Magasanik 1991; Xu et al. 1995). Accordingly we searched for potential homologues of this gene in U. maydis. The highest level of homology corresponded to gene um10417.
On the other hand and interestingly, 39 of the total of genes differentially regulated by the PMM MAPK pathway are grouped into twelve clusters; none of them was described as pathogenesis clusters by Kämper et al. (2006); and only one of them (Cluster V) has been described in U. maydis as involved in the biosynthesis of mannosylerythritol lipids (MELs) (Hewald et al. 2006) (Table 7). Moreover, for all genes grouped into clusters, we found in their promoter regions the presence of consensus sequences and binding sites for several transcription factors, and among the most represented are: AGGGG, binding sequence for MNS2, MSN4 and RGM1 transcription factors; AGACGC, binding sequence for ARG80 transcription factor; and also binding sequence for HAP2, GLN3, SKN7 and RIM101 or PacC (CGCCAAG) previously discussed. These data suggest that the regulation of gene clusters by the PMM MAPK pathway is through these transcription factors. All these transcription factors have been described in S. cerevisiae, e.g, MNS2 and MSN4 are transcriptional activators in stress response (Martínez-Pastor et al. 1996); RGM1 induces the expression of metabolism genes (Estruch 1991); and ARG80 is a transcriptional activator of arginine-responsive genes (Dubois et al. 1987). Also, possible homologous genes for MSN2, MSN4, RGM1 and ARG80 were identified in U. maydis (um00946, um12004, um02038 and um01224, respectively).
Agreeing with the fundamental role of the PMM MAPK pathway in the U. maydis dimorphic transition, we found that 60 of the 154 genes reported as differentially regulated during U. maydis dimorphism induced in vitro by a change in pH (Martínez-Soto and Ruiz-Herrera 2013) were regulated by the PMM MAPK pathway. Of these, 40 (67 %) were overexpressed and 20 (33 %) were repressed (see Table S2). In addition, 80 of the 345 genes reported as differential during U. maydis mycelial growth induced by the bE/bW heterodimer (Heimel et al. 2010) were found as differentially regulated by the PMM MAPK pathway; 60 of them (75 %) were up-regulated and 20 (25 %) down-regulated (see Table S3). Differential regulation of these genes suggests a mechanism for the regulation of fungal morphogenesis by this pathway.
In conclusion, these results of the first global analysis of the regulation of expression of the whole genome of a fungus, in this case U. maydis, demonstrate the importance of these studies for comprehension of the complex regulatory networks in these organisms. Accordingly, our data demonstrate that the PMM MAPK pathway positively regulates, directly or through specific transcription factors, genes involved in a large number of physiological processes in U. maydis, including the cell cycle, other signal transduction mechanisms, synthesis of membrane and cell wall, and dimorphism; and that negatively regulates DNA and RNA processing, and ribosome biogenesis, among other processes. Interestingly, these results demonstrate that the regulatory capacities of this pathway exceed what was previously known about its roles, showing the extreme complexity of its possible cross talks with other regulatory networks. No doubt that the data reported here will contribute to the understanding of the operation of the MAPK pathways in this and other fungi, and possibly even in higher organisms, considering that they are conserved in the eukaryotes.
References
Andrews DL, Egan JD, Mayorga ME et al (2000) The Ustilago maydis ubc4 and ubc5 genes encode members of a MAPK kinase cascade required for filamentous growth. Mol Plant Microbe Interact 13:781–786
Aréchiga-Carvajal ET, Ruiz-Herrera J (2005) The RIM101/PacC homologue from the basidiomycete Ustilago maydis is functional in multiple pH-sensitive phenomena. Eukaryot Cell 4:999–1008
Balázs A, Pócsi I, Hamari Z et al (2010) AtfA bZIP-type transcription factor regulates oxidative and osmotic stress responses in Aspergillus nidulans. Mol Genet Genomics 283:289–303
Ballario P, Vittorioso P, Magrelli A et al (1996) White collar-1, a central regulator of blue light responses in Neurospora, is a zinc finger protein. EMBO J 15:1650–1657
Banuett F, Herskowitz I (1989) Different a alleles of Ustilago maydis are necessary for maintenance of filamentous growth but not for meiosis. Proc Natl Acad Sci USA 86:5878–5882
Banuett F, Herskowitz I (1994) Morphological transitions in the life cycle of Ustilago maydis and their genetic control by the a and b loci. Exp Mycol 18:247–266
Bendtsen JD, Nielsen H, von Heijne G et al (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
Bernstein HS, Coughlin SR (1997) Pombe Cdc5-related protein. A putative human transcription factor implicated in mitogen-activated signaling. J Biol Chem 272:5833–5837
Bölker M (2001) Ustilago maydis—a valuble model system for the study of fungal dimorphism and virulence. Microbiology 147:1395–1401
Bölker M, Urban M, Kahmann R (1992) The a mating type locus of U. maydis specifies cell signaling components. Cell 68:441–450
Brefort T, Doehlemann G, Mendoza-Mendoza A et al (2009) Ustilago maydis as a pathogen. Annu Rev Phytopathol 47:423–445
Brown JL, North S, Bussey H (1993) SKN7, a yeast multicopy suppressor of a mutation affecting cell wall beta-glucan assembly, encodes a product with domains homologous to prokaryotic two-component regulators and to heat shock transcription factors. J Bacteriol 175:6908–6915
Cabrera-Ponce JL, León-Ramírez CG, Verver-Vargas A et al (2012) Metamorphosis of the basidiomycota Ustilago maydis: transformation of yeast-like cells into basidiocarps. Fungal Genet Biol 49:765–771
Carbó N, Pérez-Martín J (2010) Activation of the cell wall integrity pathway promotes escape from G2 in the fungus Ustilago maydis. PLoS Genet. doi:10.1371/journal.pgen.1001009
Cervantes-Chávez JA, Ortiz-Castellanos L, Tejada-Santorius M et al (2010) Functional analysis of the pH responsive pathway Pal/Rim in the phytopathogenic basidiomycete Ustilago maydis. Fungal Genet Biol 47:446–457
Chen RE, Thorner J (2007) Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochim Biophys Acta 1773:1311–1340
Chiariello M, Bruni CB, Bucci C (1999) The small GTPases Rab5a, Rab5b and Rab5c are differentially phosphorylated in vitro. FEBS Lett 453:20–24
Clotet J, Posas F (2007) Control of cell cycle in response to osmostress: lessons from yeast. Methods Enzymol 428:63–76
Davis D et al (2000) Candida albicans RIM101 pH response pathway is required for host-pathogen interaction. Infect Immun 68:5953–5959
Dubois E, Bercy J, Messenguy F (1987) Characterization of two genes, ARGRI and ARGRIII required for specific regulation of arginine metabolism in yeast. Mol Gen Genet 207:142–148
Dürrenberger F, Laidlaw RD, Kronstad JW (2001) The hgl1 gene is required for dimorphism and teliospore formation in the fungal pathogen Ustilago maydis. Mol Microbiol 41:337–348
Estruch F (1991) The yeast putative transcriptional repressor RGM1 is a proline-rich zinc finger protein. Nucleic Acids Res 19:4873–4877
Fonseca-Garcia C, León-Ramírez CG, Ruiz-Herrera J (2012) The regulation of different metabolic pathways through the Pal/Rim pathway in Ustilago maydis. FEMS Yeast Res 12:547–556
Franco-Frías E, Ruiz-Herrera J, Aréchiga-Carvajal ET (2014) Transcriptomic analysis of the role of Rim101/PacC in the adaptation of Ustilago maydis to an alkaline environment. Microbiology 160:1985–1998
Gancedo JM (2001) Control of pseudohyphae formation in Saccharomyces cerevisiae. FEMS Microbiol Rev 25:107–123
Garrido E, Voss U, Müller P et al (2004) The induction of sexual development and virulence in the smut fungus Ustilago maydis depends on Crk1, a novel MAPK protein. Genes Dev 18:3117–3130
Guo Y, Feng Y, Trevedi NS et al (2011) Medusa structure of the gene regulatory network: dominance of transcription factors in cancer subtype c1assification. Exp Biol Med (Maywood) 236:628–636
Gustin MC, Albertyn J, Alexander M et al (1998) MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev 62:1264–1300
Heimel K, Scherer M, Vranes M et al (2010) The transcription factor Rbf1 is the master regulator for b-mating type controlled pathogenic development in Ustilago maydis. Plos Pathog. doi:10.1371/journal.ppat.1001035
Hewald S, Linne U, Scherer M et al (2006) Identification a gene cluster for biosynthesis of mannosylerythritol lipids in the basidiomycetous fungus Ustilago maydis. Appl Environ Microbiol 72:5469–5477
Holliday R (1974) Ustilago maydis. In: King RC (ed) The handbook of genetics. Plenum Press, New York, pp 575–595
Hua X, Yuan X, Wilhelmus KR (2010) A fungal pH-responsive signaling pathway regulating Aspergillus adaptation and invasion into the cornea. Investig Ophthalmol Vis Sci 51:1517–1523
Kämper J, Kahmann R, Bölker M et al (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444:97–101
Kitada K, Johnson AL, Johnston LH et al (1993) A multicopy suppressor gene of the Saccharomyces cerevisiae G1 cell cycle mutant gene dbf4 encodes a protein kinase and is identified as CDC5. Mol Cell Biol 13:4445–4457
Krems B, Charizanis C, Entian KD (1996) The response regulator-like protein Pos9/Skn7 of Saccharomyces cerevisiae is involved in oxidative stress resistance. Curr Genet 29:327–334
Lamb TM, Mitchell AP (2003) The transcription factor Rim101p governs ion tolerance and cell differentiation by direct repression of the regulatory genes NRG1 and SMP1 in Saccharomyces cerevisiae. Mol Cell Biol 23:677–686
León-Ramirez CG, Sánchez-Arreguín JA, Ruiz-Herrera J (2014) Ustilago maydis, a delicacy of the aztec cuisine and a model for research. Nat Resour 5:256–267
León-Ramírez CG, Cabrera-Ponce JL, Martínez-Espinoza AD et al (2004) Infection of alternative host plant species by Ustilago maydis. New Phytol 164:337–346
Letunic I, Doerks T, Bork P (2012) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res 40:302–305
Martínez-Espinoza AD, León-Ramírez CG, Elizarraraz G et al (1997) Monomorphic nonpathogenic mutants of Ustilago maydis. Phytopathology 87:259–265
Martínez-Espinoza AD, Ruiz-Herrera J, León-Ramírez CG et al (2004) MAP kinase and cAMP signaling pathways modulate the pH-induced yeast-to-mycelium dimorphic transition in the corn smut fungus Ustilago maydis. Curr Microbiol 49:274–281
Martínez-Pastor MT, Marchler G, Schüller C et al (1996) The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p required for transcriptional induction through the stress response element (STRE). EMBO J 15:2227–2235
Martínez-Soto D, Ruiz-Herrera J (2013) Transcriptomic analysis of the dimorphic transition of Ustilago maydis induced in vitro by a change in pH. Fungal Genet Biol 58–59:116–125
Martínez-Soto D, Briones-Robledo AM, Estrada-Luna AA et al (2013) Transcriptomic analysis of Ustilago maydis infecting Arabidopsis reveals important aspects of the fungus pathogenic mechanisms. Plant Signal Behav. doi:10.4161/psb.25059
Mayorga ME, Gold SE (1999) A MAP kinase encoded by the ubc3 gene of Ustilago maydisis required for filamentous growth and full virulence. Mol Microbiol 34:485–497
Mayorga ME, Gold SE (2001) The Ubc2 gene of Ustilago maydis encodes a putative novel adaptor protein required for filamentous growth, pheromone response and virulence. Mol Microbiol 41:1365–1379
Méndez-Morán L, Reynaga-Peña CG, Springer PS et al (2005) Ustilago maydis infection of the nonnatural host Arabidopsis thaliana. Phytopathology 95:480–488
Mendoza-Mendoza A, Eskova A, Weise C et al (2009) Hap2 regulates the pheromone response transcription factor prf1 in Ustilago maydis. Mol Microbiol 72:683–698
Millar JB, Russell P (1992) The cdc25 M-phase inducer: an unconventional protein phosphatase. Cell 68:407–410
Minehart PL, Magasanik B (1991) Sequence and expression of GLN3, a positive nitrogen regulatory gene of Saccharomyces cerevisiae encoding a protein with a putative zinc finger DNA-binding domain. Mol Cell Biol 11:6216–6228
Müller P, Aichinger C, Feldbrügge M et al (1999) The MAP kinase kpp2 regulates mating and pathogenic development in Ustilago maydis. Mol Microbiol 34:1007–1017
Müller P, Weinzierl G, Brachmann A et al (2003) Mating and pathogenic development of the smut fungus Ustilago maydis are regulated by one mitogen-activated protein kinase cascade. Eukaryot Cell 2:1187–1199
Palecek SP, Parikh AS, Kron SJ (2002) Sensing, signalling and integrating physical processes during Saccharomyces cerevisiae invasive and filamentous growth. Microbiology 148:893–907
Pan X, Harashima T, Heitman J (2000) Signal transduction cascades regulating pseudohyphal differentiation of Saccharomyces cerevisiae. Curr Opin Microbiol 3:567–572
Peñalva MA, Arst HN Jr (2002) Regulation of gene expression by ambient pH in filamentous fungi and yeasts. Microbiol Mol Biol Rev 66:426–446
Perez-Nadales E, Almeida-Nogueira MF, Baldin C et al (2014) Fungal model systems and elucidation of pathogenicity determinants. Fungal Genet Biol 7:42–67
Pijnappel WW, Schaft D, Roquev A et al (2001) The S. cerevisiae SET3 complex two histone deacetylases, Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev 15:2991–3004
Punta M, Coggill PC, Eberhardt RY et al (2012) The Pfam protein families database. Nucleic Acids Res 40:290–301
Ramon AM, Porta A, Fonzi WA (1999) Effect of environmental pH on morphological development of Candida albicans is mediated via the PacC-related transcription factor encoded by PRR2. J Bacteriol 181:7524–7530
Raudaskoski M, Kothe E (2010) Basidiomycete mating type genes and pheromone signaling. Eucaryot Cell 9:847–859
Robertson AS, Allwood EG, Smith AP et al (2009) The WASP homologue Las17 activates novel actin-regulatory activity of Ysc84 to promote endocitosis in yeast. Mol Biol Cell 20:1618–1628
Robledo-Briones AM, Ruiz-Herrera J (2013) Regulation of genes involved in cell wall synthesis and structure during Ustilago maydis dimorphism. FEMS Yeast Res 13:74–84
Ruepp A, Zollner A, Maier D et al (2004) The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res 32:5539–5545
Ruiz-Herrera J, Campos-Góngora E (2012) An introduction to fungal dimorphism. In: Ruiz-Herrera J (ed) Dimorphic fungi: their importance as models for differentiation and fungal pathogenesis. Bentham e Books, eISBN:978-1-60805-364-3
Ruiz-Herrera J, León-Ramírez CG, Guevara-Olvera L et al (1995) Yeast-mycelial dimorphism of haploid end diploid strains of Ustilago maydis. Microbiology 141:695–703
Ruiz-Herrera J, Reynaga-Peña CG, Aréchiga-Carvajal ET (2009) Ustilago maydis as a model for phytopathogenic fungal development. In: Khachatourians GG, Arora DK, Rajendran TP, Srivastava AK (eds) Agriculturally important microorganisms, vol I. Academic World International, Bhopal, pp 107–122
Ruiz-Herrera J, Robledo-Briones M, Martínez-Soto D (2013) Experimental pathosystems as a tool for the identification of virulence factors in pathogenic fungi. In: Deshpande MV, Ruiz-Herrera J (eds) Biotechnology: beyond borders. CSIR-National Chemical Laboratory, Pune, pp 30–38
Sandrock B, Böhmer C, Bölker M (2006) Dual function of the germinal centre kinase Don3 during mitosis and cytokinesis in Ustilago maydis. Mol Micobiol 62:655–666
Singer-Krüger B, Stenmark H, Düsterhöft A et al (1994) Role of three rab5-like GTPases, Ypt51p, Ypt52p, and Ytp53p, in the endocytic and vacuolar protein sorting pathways of yeast. J Cell Biol 125:283–298
Singh P, Chauhan N, Ghosh A et al (2004) SKN7 of Candida albicans: mutant construction and phenotype analysis. Infect Immun 72:2390–2394
Soulard A, Lechler T, Spiridonov V et al (2002) Saccharomyces cerevisiae Bzz1p is implicated with type I myosins in actin patch polarization and is able to recruit actin-polymerizing machinery in vitro. Mol Cell Biol 22:7889–7906
Valdés-Santiago L, Ruiz-Herrera J (2014) Stress and polyamine metabolism in fungi. Front Chem. doi:10.3389/fchem.2013.00042
Valdés-Santiago L, Cervantes-Chávez JA, León Ramírez CG et al (2012) Polyamine metabolism in fungi with emphasis on phytopathogenic species. J Amino Acids. doi:10.1155/2012/837932
Vollmeister E, Schipper K, Baumann S et al (2011) Fungal development of the plant pathogen Ustilago maydis. FEMS Microbiol Rev 36:59–77
Ward MP, Garrett S (1994) Suppression of a yeast cyclic AMP-dependent protein kinase defect by overexpression of SOK1, a yeast gene exhibiting sequence similarity to a developmentally regulated mouse gene. Mol Cell Biol 14:5619–5627
Weig M, Haynes K, Rogers TR et al (2001) A GAS-like gene family in the pathogenic fungus Candida glabrata. Microbiology 147:2007–2019
Wilkinson MG, Millar JB (2000) Control of eukaryotic cell cycle by MAP kinase signaling pathways. FASEB J 14:2147–2157
Wormley FL Jr, Heinrich G, Miller JL et al (2005) Identification and characterization of an SKN7 homolog in Cryptococcus neoformans. Infect Immun 73:5022–5030
Wurgler-Murphy SM, Maeda T, Witten EA et al (1997) Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by PTP2 and PTP3 protein tyrosine phosphatases. Mol Cell Biol 17:1289–1297
Xu S, Falvey DA, Brandriss MC (1995) Roles of URE2 and GLN3 in the proline utilization pathway Saccharomyces cerevisiae. Mol Cell Biol 15:2321–2330
Zhang W, Liu HT (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 12:9–18
Acknowledgments
This work was supported by Consejo Nacional de Ciencia y Tecnología (CONACYT), Mexico. Thanks are given to Dr. Scott Gold for permission to use his microarray design, and QFB Claudia Geraldine León-Ramirez, IBQ Fernando Emilio Pérez-García, Biol. Mayela Fernanda Salazar-Chávez and M.S. Guillermo Antonio Silva-Martínez for assistance in some analyses. DMS is a doctoral student supported by a fellowship from CONACYT (México). JRH is Emeritus National Professor, México.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Martínez-Soto, D., Ruiz-Herrera, J. Regulation of the expression of the whole genome of Ustilago maydis by a MAPK pathway. Arch Microbiol 197, 575–588 (2015). https://doi.org/10.1007/s00203-015-1087-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-015-1087-2