
Abstract
Mechanical loading is thought to provoke a cellular response via loading-induced flow of interstitial fluid through the lacuno-canalicular network of osteocytes. This response supposedly leads to an adaptation of local bone mass and architecture. It has been suggested that loss of estrogen during menopause alters the sensitivity of bone tissue to mechanical load, thereby contributing to the rapid loss of bone. The present study aimed to determine whether estrogen modulates the mechanoresponsiveness of bone cells from osteoporotic women. Bone cell cultures from nine osteoporotic women (aged 62–90 years) were pre-cultured for 24 h with 10−11 mol/l 17β-estradiol (E2) or vehicle, and subjected to 1 h of pulsating fluid flow (PFF) or static culture. E2 alone enhanced prostaglandin E2 (PGE2) and nitric oxide (NO) production by 2.8-fold and 2.0-fold, respectively, and stimulated endothelial nitric oxide synthase protein expression by 2.5-fold. PFF, in the absence of E2, stimulated PGE2 production by 3.1-fold and NO production by 3.9-fold. Combined treatment with E2 and PFF increased PGE2 and NO production in an additive manner. When expressed as PFF-treatment-over-control ratio, the response to fluid shear stress was similar in the absence or presence of E2. These results suggest that E2 does not affect the early response to stress in bone cells. Rather, E2 and shear stress both promote the production of paracrine factors such as NO and PGE2 in an additive manner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteoporosis is a condition characterized by a compromised resistance of the skeleton to mechanical loads as a result of reduced bone mass and an impaired mechanical competence of the trabecular architecture. The cause of bone loss in patients with osteoporosis is likely to be multifactorial, as bone mass and the skeleton’s mechanical performance are affected by many factors. Systemic factors that affect bone mass are calcium-regulating hormones, such as parathyroid hormone, calcitonin and vitamin D, growth hormone, and the sex hormones [1, 2, 3] Of the latter, estrogen is probably the best studied, due to its role in bone loss after menopause.
While all of these hormones are capable of affecting overall bone mass, local bone mass and architecture are primarily determined by mechanical signals. Several studies have shown that mechanical loading determines bone mass, shape, and trabecular architecture [4, 5, 6]. The purpose of this process of mechanical adaptation is to obtain bone that combines a proper resistance against mechanical failure with a minimum use of material. It is currently believed that mechanotransduction, the process whereby mechanical signals are converted into chemical messages, proceeds as follows: when bones are loaded, the hard matrix will undergo a very small deformation (strain). This deformation drives the thin layer of interstitial fluid surrounding the osteocyte network to flow from regions under high pressure to regions under low pressure [7, 8, 9]. Although osteoblasts are sensitive to strain, and might thus sense mechanical loading directly through the deformation of their supporting substratum, it has been found by several investigators that the amount of strain does not correlate with bone formation in vivo [10, 11, 12]. Rather, the strain rate of the applied stimulus, which is directly related to the flow of interstitial fluid within the bone, is related to bone formation [10, 11, 12]. Loading-induced flow of fluid exerts a shear stress on the osteocyte cell membrane, which provokes a cellular response [13, 14]. It is thought that the osteocytes subsequently signal the osteoblasts and osteoclasts to change their bone remodeling activities [15]. In this concept the osteocytes act as the mechanosensors of bone, while the osteoblasts and osteoclasts are the effector cells of bone adaptation.
Bone mass diminishes with increasing age, in part due to declining levels of physical activity and changed mobility patterns. Decreased estrogen levels following menopause also have a strong negative effect on bone mass [16]. Administration of exogenous estrogens has been shown to increase bone mineral density in humans [17, 18], and large doses of estrogens have been shown to induce new bone formation in mice [19, 20]. These effects of estrogen on bone can be explained by its opposite effects on osteoclasts and osteoblasts. Both osteoclasts and osteoblasts express estrogen receptors, and estrogen is able to suppress bone resorption and stimulate bone formation [16, 21]. In addition, it has been suggested that estrogen alters the response of the bone mechanosensing cells to mechanical loading, thereby modulating the anabolic response of bone tissue to mechanical loading [22].
This hypothesis is supported by studies which show that estrogen and mechanical loading can synergistically affect bone formation [18, 23] On the other hand, there are also several in vivo studies that were unable to detect a synergistic effect of estrogen and mechanical loading on bone formation [17, 24, 25, 26]. Estrogen application was shown to affect mechanically induced bone formation whether applied before or after loading [24], and estrogen did not affect the number of osteocytes expressing c-fos and IGF-I mRNA in response to mechanical loading of rat vertebrae [24]. These results do not support the idea of an interaction between mechanical loading and estrogen at the level of mechanosensation in osteocytes.
To study direct effects of E2 on mechanoresponsiveness in an early phase of mechanotransduction we investigated whether E2 modifies the response to shear stress in cultured bone cells from postmenopausal osteoporotic women. Women with bone cells that are highly sensitive to the effects of estrogen are likely to experience severe bone loss during the menopause, which results in an osteoporotic phenotype. Therefore, we specifically chose to investigate bone cells obtained from postmenopausal osteoporotic women. Nitric oxide (NO) and prostaglandin E2 (PGE2) release were determined as parameters of bone cell responsiveness, since both signaling molecules have been shown to be essential for the anabolic response of bone to mechanical loading [27, 28]. We applied mechanical stress by submitting the cells to a pulsating laminar fluid flow (PFF) [13, 14].
We hypothesized that estrogen would affect the magnitude of the fluid flow-induced NO and PGE2 production of the bone cells in an additive manner.
Materials and methods
Donors
Trabecular bone samples (surgical waste) were taken from the femoral neck during hip surgery after osteoporotic, subcapital hip fracture. Nine female osteoporotic donors (mean age 80; range 62–90 years), without other metabolic bone disease, were included in this study. Osteoporotic fracture was defined as a fracture without preceding trauma or in response to minimal trauma. People suffering from hip fracture after severe trauma were excluded from the study. The protocol was approved by the Ethical Review Board of the Vrije University Medical Center, and all subjects gave informed consent.
Culture of primary human bone cells
Trabecular bone biopsies were placed in sterile phosphate-buffered saline (PBS), chopped into small fragments and washed extensively with PBS. The bone fragments were incubated with 2 mg/ml collagenase II (Sigma, St. Louis, Mo., USA) for 2 h at 37°C in a shaking water bath to remove all soft tissue from the bone chip’s surface, after which the denuded bone fragments were transferred to 25 ml flasks (Nunc, Roskilde, Denmark). The fragments were cultured in Dulbecco’s Modified Eagle’s medium (DMEM; Gibco, Paisley, UK) supplemented with 100 U/ml penicillin (Sigma), 50 μg/ml streptomycin sulfate (Sigma), 50 μg/ml gentamicin (Gibco), 1.25 μg/ml fungizone (Gibco), 100 μg/ml ascorbate (Merck, Darmstadt, Germany), and 10% fetal bovine serum (FBS; Hyclone, Logan, Utah, USA). Culture medium was replaced three times per week.
When the cell monolayer growing from the bone fragments reached confluency, the cells were harvested with 0.25% trypsin (Difco Laboratories, Detroit, Mich., USA) and 0.1% EDTA (Sigma) in PBS, plated at 25×103 cells per well in six-well culture dishes (Costar, Cambridge, Mass., USA) and cultured in 3 ml medium as described above, until the cell layer reached confluency again. Then the cells were characterized as described below, or used for pulsating fluid flow and E2 experiments.
Cell characterization: cbfa1 mRNA expression and 1,25-dihydroxyvitamin D3 challenge
To test the osteoblastic phenotype, we isolated RNA isolated from the bone-derived cell cultures, using TRIzol reagent (Gibco), in accordance with the manufacturers instructions. cDNA synthesis was performed with 1 μg of total RNA in 50 μl reaction mix consisting of 1x first-strand buffer, 500 μmol dNTPs, 10 units RNAse inhibitor, 8 mmol dithiothreithol, 50 units Superscript RT (Gibco) and 2 pmol primer p(dT)15 (Boehringer, Mannheim, Germany). For the amplification of the cbfa1 product, 4 μl of cDNA was added to the PCR reaction mixture consisting of 1× Thermal Ace buffer (Invitrogen, Carlsbad, Calif., USA), 0.2 mmol dNTP, 6 mmol sense primer, 6 mmol antisense primer and 1 U Thermal Ace polymerase (Invitrogen), in a final volume of 50 μl. The cbfa1 upstream and downstream primer sequences were 5′ atg ctt cat tcg cct cac aaa c 3′ for the forward primer, and 5′ ttt gat gcc ata gtc cct cct t 3′ for the reverse primer, respectively. The samples were pre-heated for 10 min at 95°C, followed by a three-step PCR procedure consisting of 45 s at 95°C, 15 s at 53°C, and 15 s at 74°C, for 45 cycles. The PCR products were subjected to electrophoresis on a 1.5% agarose gel containing 0.5 μg/ml ethidium bromide.
Alternatively, bone cell cultures were incubated for 3 days in the presence or absence of 10−8 mol/l 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] in medium supplemented with 0.2% bovine serum albumin (BSA). After 3 days’ incubation, the cells were harvested for determination of alkaline phosphatase (ALP) activity and total protein content of the cell layer. ALP activity was determined in the cell lysate by use of p-nitrophenyl phosphate (Merck) as a substrate at pH 10.3, in accordance with the method described by Lowry [29]. The absorbance was read at 410 nm with a Dynatech MR7000 microplate reader (Dynatech, Billinghurst, UK). The amount of protein in the cell layer was measured with a BSA protein assay reagent kit (Pierce, Rockford, Ill., USA); the absorbance was read at 570 nm. ALP values were expressed per amount of protein in the cell layer.
Pulsating fluid flow in the presence or absence of estrogen
Two days before PFF treatment, the cells were harvested from the six-well plates and seeded onto polylysine-coated (50 μg/ml; poly-l-lysine hydrobromide, molecular weight 15–30×104; Sigma) glass slides (size 2.5 cm×6.5 cm). The cells were plated at 5×105 cells/glass slide and cultured overnight in Petri dishes with 13 ml culture medium as described above in “Culture of primary human bone cells”. The next morning the culture medium was replaced by DMEM without phenol red (Gibco) containing 10−11 mol/l 17β-estradiol (E2; Sigma) or vehicle, and supplemented with 0.2% BSA. The medium also contained antibiotics and ascorbate as described above. Similar medium, with or without E2, was used the next day during the PFF experiments.
The cells were subjected to 1 h of PFF as described previously [30]. Briefly, we generated PFF by pumping 13 ml of culture medium through a parallel-plate flow chamber containing the bone cells. The cells were subjected to a 5 Hz pulse with a mean shear stress of 0.6 Pa, a pulse amplitude of 0.3 Pa and a peak shear stress rate of 8.4 Pa/s. Stationary control cultures were kept in a Petri dish under similar conditions as the experimental cultures, i.e., at 37°C in a humidified atmosphere of 5% CO2 in air. After 5 min of PFF or static culture, the medium was collected and assayed for NO production. After 1 h the medium was collected for determination of PGE2 production. After 1 h PFF treatment was terminated, and the cells were either directly harvested in TRIzol reagent for determination of total DNA content or harvested in ice-cold cell lysis buffer (R&D systems, Minneapolis, Minn., USA) for determination of human endothelial nitric oxide synthase (eNOS) protein expression.
Nitric oxide and prostaglandin E2
We measured NO as nitrite (NO2−) accumulation in the conditioned medium, using Griess reagent consisting of 1% sulfanilamide, 0.1% naphthylethylene-diamine-dihydrochloride and 2.5 mol H3PO4. Serial dilutions of NaNO2 in non-conditioned medium were used as the standard curve. The absorbance was measured at 540 nm.
PGE2 release in the medium was measured by an enzyme immunoassay (EIA) system (Amersham, Buckinghamshire, UK) that used an antibody raised against mouse PGE2. The detection limit was 16 pg/ml, and the absorbance was measured at 450 nm.
Isolation of total DNA
DNA was isolated from the bone cell cultures with TRIzol reagent in accordance with the manufacturer’s instructions. DNA content was determined by measurement of the absorbance in water at 260 nm with an Ultrospec III spectrophotometer (Amersham).
Determination of endothelial nitric oxide synthase protein
Human eNOS protein expression was quantified with an eNOS Quantikine immunoassay kit for determination of eNOS in endothelial cells (R&D systems), in accordance with the manufacturer’s protocol. This kit employs the quantitative sandwich enzyme immunoassay [enzyme-linked immunosorbent assay (ELISA)] technique and uses antibodies raised against E. coli-expressed recombinant human eNOS.
Statistical analysis
Data of absolute NO and PGE2 production were not normally distributed and were, therefore, analyzed with the Wilcoxon signed-rank test. When we were comparing treated-over-control (T/C) values, the data were analyzed by a paired t-test. Differences were considered significant when P<0.05.
Results
Cells started to grow out of the collagenase-stripped bone chips along the bottom of the culture flask after approximately 6 days in culture and reached confluency within 19 days, when they were passaged. Expression levels of cbfa1 mRNA by the osteoporotic (OP) bone cell cultures were comparable to that of a MC3T3-E1 mouse osteoblastic cell line (Fig. 1), while treatment of the passaged cells with 10−8 mol/l 1,25(OH)2D3 resulted in a significant 1.6-fold (range 1.2-fold to 2.2-fold) mean increase in alkaline phosphatase activity.

Cbfa1 mRNA expression by untreated primary human bone cell cultures from osteoporotic donors (numbers 1–9). Reaction mixes were subjected to electrophoresis on a 1.5% agarose gel and visualized by ethidium bromide staining. MC3T3-E1, a mouse osteoblast cell line, served as a positive control for cbfa1 expression. Negative negative control, Ladder 100 bp ladder.
Treatment of the cells with PFF and/or E2 did not affect the total amount of DNA per culture (static, 0.60±0.13 μg; static+E2, 0.59±0.12 μg; PFF, 0.57±0.14 μg; PFF+E2, 0.64±0.12 μg. Values are mean ± SEM of nine cultures), which demonstrated that the cells were not removed by the application of fluid flow and that 24 h of E2 treatment did not affect cell numbers.
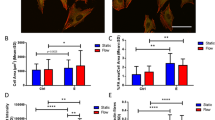
Treatment of the cells with 10−11 mol/l E2 for 24 h stimulated NO and PGE2 production by 2.0-fold and 2.8-fold, respectively (Fig. 2a, b) and increased eNOS protein expression by 2.5-fold (Fig. 2c). One hour’s treatment with PFF slightly, but not significantly, increased eNOS expression (data not shown). Application of PFF rapidly stimulated NO production, in both the vehicle and the E2-treated cells (Fig. 3a). PFF also stimulated the production of PGE2, both in the absence and presence of E2 (Fig. 3b). For both NO and PGE2 the combined treatment with PFF and E2 seemed to have an additive, but not synergistic, effect when mean group values (Fig. 3a, b) were considered.

Effect of 24 h treatment with E2 on NO and PGE2 production, and eNOS protein expression by OP bone cells. a, b Each symbol represents the mean NO and PGE2 production, normalized for the amount of DNA, by duplicate cultures from one donor. Lines connect data of vehicle-treated cultures (circles) with data of E2-treated cultures (triangles) from the same donor. Horizontal bars express mean NO or PGE2 production. Note that the y-axis is logarithmic. c Treatment over control (T/C) ratio of eNOS expression. E2 stimulated NO (a) and PGE2 (b) production and eNOS protein expression (c). *Significant effect of E2, P<0.05.

Effect of E2 and/or PFF on NO and PGE2 production by OP bone cells. Bars represent mean ± SEM. Both PFF and E2 treatment significantly enhanced NO (a) and PGE2 (b) production by the bone cell cultures. Combined treatment with PFF and E2 further increased NO and PGE2 production in an additive, but not synergistic, manner. *Significantly different from static (stat) culture + vehicle; # significantly different from stat + E2, P<0.05.
When the same data were expressed as PFF-treated over static culture (PFF/static) ratios this observation became even clearer (Fig. 4a, b). Five minutes of PFF treatment stimulated the NO production by 3.9-fold in the vehicle-treated cells and by 3.5-fold in the E2-treated cultures (Fig. 4a). After 1 h, PFF increased PGE2 production by 3.1-fold in the vehicle-treated cells and by 2.7-fold in E2-treated cultures (Fig. 4b). Thus, PFF had a similar stimulatory effect on the production of NO and PGE2 in the presence and absence of E2.

Effect of E2 on the magnitude of the NO and PGE2 response to PFF by OP bone cells. Data are expressed as PFF-treated over static culture (PFF/static) ratios. Bars are mean ± SEM of nine donors. Application of PFF for 5 min enhanced NO production both in vehicle and E2-treated cultures in a similar manner (a). There was no difference in magnitude of the PGE2 response to PFF between E2 and vehicle-treated cells (b). *Significant effect of PFF, P<0.05.
Discussion
In the present study we tested whether estrogen modulates the early response of bone cells from post menopausal osteoporotic donors to a mechanical stimulus, applied in the form of a pulsating fluid flow. Bone cells were grown from surgical waste trabecular bone, obtained during hip surgery for osteoporotic, subcapital femur fracture. Our population did not include any pre-menopausal donors. It is, however, unlikely that factors such as hormonal status of the donor would have affected the mechanosensitivity of the bone cells in vitro, since the cells were cultured for an average of 30 days, in the presence of 10% fetal calf serum containing low concentrations of estrogen, before they were used for the PFF experiments. The bone fragments were treated with collagenase to remove all soft tissue from the bone surface before being put into culture, and it took 6 days before cells started to grow out of the denuded bone chips. This suggests that they were derived from a cell source within the bone matrix rather than from its surface.
The “osteocytic osteoblast”, a cell stage between osteoblast and osteocyte [31] seems a good candidate. Treatment of the cell cultures with 1,25(OH)2D3 stimulated ALP activity, and the cells expressed cbfa1 mRNA, indicating that they were of the osteoblast lineage. We have previously shown that human cells isolated as outgrowth from denuded bone chips respond to treatment with 1,25(OH)2D3 with increased osteocalcin expression [32] and do not express the endothelial cell-specific factor VIII, von Willebrand factor [33]. Osteocytes, the terminally differentiated cells from the osteoblastic lineage, are highly responsive to mechanical loading in the form of a fluid flow [13]. The strong response to PFF of the cells isolated as outgrowth from denuded bone chips supports our contention that, to a certain extent, they mimic osteocytes.
We measured NO and PGE2 production as parameters of the bone cell response to mechanical stimulation because animal studies have shown that NO as well as PGE2 are essential for the induction of new bone formation in reaction to mechanical loading [27, 28]. In addition, both NO and PGE2 production are influenced by estrogen [34, 35, 36, 37}, and the anabolic effect of estrogen on bone seems to be mediated by prostaglandins and NO [20, 34, 38]. We used the physiological concentration of 10−11 mol/l E2, because higher concentrations did not affect NO production in human and mouse osteoblastic cells or in bovine endothelial cells [36, 39]. Moreover, this E2 concentration has previously been shown to increase PGE2 production by primary human bone cells [40]. The limited amount of donor bone precluded an extensive dose/effect study. Mechanical loading was applied by a pulsating fluid flow of 0.6±0.3 Pa at 5 Hz because this regime has been shown to cause a submaximal stimulation of NO and PGE2 production [14].
The question whether estrogen and mechanical loading act synergistically to increase bone mass and bone strength is still unresolved. An in vitro study found a synergistic effect of estrogen and mechanical loading on 3H-thymidine and 3H-proline incorporation in cultured ulnae of female rats [23], and a clinical study reported a synergistic effect of weight-bearing exercise and hormone replacement therapy (HRT) on whole-body bone mineral density (BMD) in elderly women [18]. However, that study found no synergistic effect of exercise and HRT on BMD of the spine and hip, two sites prone to osteoporotic fracture [18].
In another clinical study HRT and exercise both increased BMD of the proximal tibia of postmenopausal women, but no synergistic effect was observed at this site either [17]. In two separate studies with rats, the anabolic response of bone to treadmill exercise was not negatively affected by ovariectomy, which suggests that estrogen status does not affect the mechanoresponsiveness of bone tissue [25, 26]. In rat bone, estrogen did not significantly alter the proportion of osteocytes that expressed c-fos and IGF-1 mRNA in response to mechanical loading, and affected mechanically induced osteogenesis similarly when administered before or after application of loading [24]. This latter observation suggests that even if estrogen alters the mechano-adaptive response of bones as an organ, estrogen does not act on the strain-sensing mechanism itself.
Our present in vitro data support that conclusion. We found that E2 and fluid flow-induced shear stress both promote the rapid production of NO and PGE2 and that the combined application of E2 and PFF had an additive effect on NO and PGE2 production, rather than a synergistic one. Our results are in agreement with an earlier study that showed that estrogen enhanced fluid shear stress-induced prostaglandin production by bone cells from elderly women [40]. When the results of that study are expressed as PFF-treated over static culture (T/C) ratios, it becomes clear that the response to shear stress was similar in the absence or presence of E2 (PGE2 production, mean ± SEM, vehicle treated 3.6±1.3; E2 treated 3.5±1.4). In the latter study, cells were used from osteoarthritic rather than osteoporotic donors. The lack of a synergistic effect does not, therefore, appear to be related to the osteoporotic status of the donor.
We found that treatment for 24 h with E2 more than doubled eNOS expression, in agreement with other studies [35, 36, 37]. The enhanced eNOS protein expression after 24 h E2 might explain the enhanced NO production under static culture conditions. The finding that E2 stimulated eNOS expression in the cells, but that PFF and E2 nonetheless acted in an additive manner, suggests that eNOS protein expression in the cells was not rate limiting for PFF-induced NO production. We had found earlier that mRNA levels for eNOShad doubled after 1 h of static post-incubation following 1 h of PFF treatment [33]. In the present study eNOS protein expression was not significantly enhanced immediately after 1 h exposure to PFF, likely because the time period was too short to allow for a significant increase in eNOS protein expression.
Our results are also consistent with the finding by Damien et al. that estrogen and mechanical strain had an additive, rather than synergistic, effect on osteoblast proliferation in vitro [41]. That study by Damien et al., as well as subsequent papers, found evidence for an important role of the estrogen receptor α (ERα) in the proliferative response of bone cells to mechanical stress [31, 42, 43, 44, 45]. In addition, in ERα knockout mice the adaptive bone formation in response to mechanical loading was blunted [46]. Thus, mechanical loading and estrogen do seem to share a common signaling pathway in the regulation of osteoblast proliferation and bone formation. Of course, the responses of osteoblasts, which are the effector cells of bone adaptation, are downstream of the response of the mechanosensors to a mechanical stimulus [15]. It is the latter response that we have studied in the present paper.
Although we found no synergistic effect of E2 and PFF on NO and PGE2 production by OP bone cells, we did find additive effects, both on NO and PGE2 production. The production of both factors was highest when shear stress was applied in the presence of estrogen and lowest when neither estrogen nor shear stress was applied. If we assume that both factors are related to anabolic events further downstream in the adaptive response to mechanical loading [27, 28], this suggests that estrogen and mechanical usage both promote bone mass and strength. As such, these in vitro results are in agreement with clinical studies [17, 18]. Our results do not rule out the possibility that estrogen modulates the adaptive response of whole bones to mechanical load but do contradict the concept that estrogen, by itself, promotes mechanotransduction in osteocytes, the principal bone mechanosensing cells in the adaptive cascade.
References
Parfitt AM (1976) The actions of parathyroid hormone on bone: relation to bone remodeling and turnover, calcium homeostasis, and metabolic bone diseases II. PTH and bone cells: bone turnover and plasma calcium regulation. Metabolism 25:909–955
Pondel M (2000) Calcitonin and calcitonin receptors: bone and beyond. Int J Exp Pathol 81:405–422
Lips P (2001) Vitamin D deficiency and secondary hyperparathyroidism in the elderly: consequences for bone loss and fractures and therapeutic implications. Endocr Rev 22:477–501
Rubin CT, Lanyon LE (1984) Regulation of bone formation by applied dynamic loads. J Bone Joint Surg Am 66:397–402
Mosley JR, Lanyon LE (1998) Strain rate as a controlling influence on adaptive modeling in response to dynamic loading of the ulna in growing male rats. Bone 23:313–318
Turner CH, Forwood MR, Otter MW (1994) Mechanotransduction in bone: do bone cells act as sensors of fluid flow? FASEB J 8:875–878
Piekarski K, Munro M (1977) Transport mechanism operating between blood supply and osteocytes in long bones. Nature 269:80–82
Cowin SC, Weinbaum S (1998) Strain amplification in the bone mechanosensory system. Am J Med Sci 316:184–188
Knothe Tate ML, Knothe U (2000) An ex vivo model to study transport processes and fluid flow in loaded bone. J Biomech 33:247–254
Luo G, Cowin SC, Sadegh AM, Arramon YP (1995) Implementation of strain rate as a bone remodeling stimulus. J Biomech Eng 117:329–338
Lanyon LE, Rubin CT (1984) Static versus dynamic loads as an influence on bone remodelling. J Biomech 17:897–905
Turner CH, Owan I, Takano Y (1995) Mechanotransduction in bone: role of strain rate. Am J Physiol 269:E438–E442
Klein-Nulend J, van der Plas A, Semeins CM, Ajubi NE, Frangos JA, Nijweide PJ, Burger EH (1995) Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J 9:441–445
Bakker AD, Soejima K, Klein-Nulend J, Burger EH (2001) The production of nitric oxide and prostaglandin E2 by primary bone cells is shear stress dependent. J Biomech 34:671–677
Burger EH, Klein-Nulend J (1999) Mechanotransduction in bone—role of the lacunocanalicular network. FASEB J 13:S101–S112
Turner RT, Riggs BL, Spelsberg TC (1994) Skeletal effects of estrogen. Endocr Rev 15:275–300
Cheng S, Sipila S, Taaffe DR, Puolakka J, Suominen H (2002) Change in bone mass distribution induced by hormone replacement therapy and high-impact physical exercise in post-menopausal women. Bone 31:126–135
Kohrt WM, Snead DB, Slatopolsky E, Birge SJ Jr (1995) Additive effects of weight-bearing exercise and estrogen on bone mineral density in older women. J Bone Miner Res 10:1303–1311
Samuels A, Perry MJ, Tobias JH (1999) High-dose estrogen induces de novo medullary bone formation in female mice. J Bone Miner Res 14:178–186
Samuels A, Perry MJ, Gibson RL, Colley S, Tobias JH (2001) Role of endothelial nitric oxide synthase in estrogen-induced osteogenesis. Bone 29:24–29
Chow J, Tobias JH, Colston KW, Chambers TJ (1992) Estrogen maintains trabecular bone volume in rats not only by suppression of bone resorption but also by stimulation of bone formation. J Clin Invest 89:74–78
Frost HM (1987) The mechanostat: A proposed pathogenic mechanism of osteoporoses and the bone mass effects of mechanical and nonmechanical agents. Bone Miner 2:73–85
Cheng MZ, Zaman G, Rawlinson SC, Suswillo RF, Lanyon LE (1996) Mechanical loading and sex hormone interactions in organ cultures of rat ulna. J Bone Miner Res 11:502–511
Jagger CJ, Chow JW, Chambers TJ (1996) Estrogen suppresses activation but enhances formation phase of osteogenic response to mechanical stimulation in rat bone. J Clin Invest 98:2351–2357
Tromp AM, Bravenboer N, Tanck E, Kostense PJ, Lips P (2002) The effects of additional weight bearing during exercise and estrogen on bone mass and structure in female rats. Acta Bioeng Biomech 4 [Suppl 1]:399
Järvinen TLN, Kannus P, Pajamäki I, Vuohelainen T, Tuukkanen J, Järvinen M, Sievänen H (2003) Estrogen deposits extra mineral into bones of female rats in puberty, but simultaneously seems to suppress the responsiveness of female skeleton to mechanical loading. Bone 32:642–651
Forwood MR (1996) Inducible cyclo-oxygenase (COX-2) mediates the induction of bone formation by mechanical loading in vivo. J Bone Miner Res 11:1688–1693
Turner CH, Takano Y, Owan I, Murrell GA (1996) Nitric oxide inhibitor L-NAME suppresses mechanically induced bone formation in rats. Am J Physiol 270:E634–E639
Lowry OH (1955) Micromethods for the assay of enzyme. II Specific procedure. Alkaline phosphatase. Methods Enzymol 4:371
Klein-Nulend J, Semeins CM, Ajubi NE, Nijweide PJ, Burger EH (1995) Pulsating fluid flow increases nitric oxide (NO) synthesis by osteocytes but not periosteal fibroblasts: correlation with prostaglandin upregulation. Biochem Biophys Res Commun 217:640–648
Nijweide PJ, Van der Plas A, Scherft JP (1981) Biochemical and histological studies on various bone cell preparations. Calcif Tissue Int 33:529–540
Sterck JG, Klein-Nulend J, Lips P, Burger EH (1998) Response of normal and osteoporotic human bone cells to mechanical stress in vitro. Am J Physiol 274:E1113–E1120
Klein-Nulend J, Helfrich MH, Sterck JG, MacPherson H, Joldersma M, Ralston SH, Semeins CM, Burger EH (1998) Nitric oxide response to shear stress by human bone cell cultures is endothelial nitric oxide synthase dependent. Biochem Biophys Res Commun 250:108–114
Samuels A, Perry MJ, Tobias JH (1999) High-dose estrogen-induced osteogenesis in the mouse is partially suppressed by indomethacin. Bone 25:675–680
Armour KE, Ralston SH (1998) Estrogen upregulates endothelial constitutive nitric oxide synthase expression in human osteoblast-like cells. Endocrinology 139:799–802
Riancho JA, Zarrabeitia MT, Fernandez-Luna J, Gonzales-Macias J (1995) Mechanisms controlling nitric oxide synthesis in osteoblasts. Mol Cell Endocrinol 107:87–92
Chambliss KL, Shaul PW (2002) Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev 23:665–686
Armour KE, Armour KJ, Gallagher ME, Godecke A, Helfrich MH, Ralston SH (2001) Defective bone formation and anabolic response to exogenous estrogen in mice with targeted disruption of endothelial nitric oxide synthase. Endocrinology 142:760–766
Stewart KG, Zhang Y, Davidge ST (1999) Estrogen decreases prostaglandin H synthase products from endothelial cells. J Soc Gynecol Investig 6:322–327
Joldersma M, Klein-Nulend J, Oleksik AM, Heyligers IC, Burger EH (2001) Estrogen enhances mechanical stress-induced prostaglandin production by bone cells from elderly women. Am J Physiol 280:E436–E442
Damien E, Price JS, Lanyon LE (1998) The estrogen receptor’s involvement in osteoblasts’ adaptive response to mechanical strain. J Bone Miner Res 13:1275–1282
Jessop HL, Sjoberg M, Cheng MZ, Zaman G, Wheeler-Jones CP, Lanyon LE (2001) Mechanical strain and estrogen activate estrogen receptor alpha in bone cells. J Bone Miner Res 16:1045–1055
Zaman G, Cheng MZ, Jessop HL, White R, Lanyon LE (2000) Mechanical strain activates estrogen response elements in bone cells. Bone 27:233–239
Cheng MZ, Rawlinson SCF, Pitsillides AA, Zaman G, Mohan S, Baylink DJ, Lanyon LE (2002) Human osteoblasts’ proliferative responses to strain and 17β-estradiol are mediated by the estrogen receptor and the receptor for insulin-like growth factor I. J Bone Miner Res 17:593–602
Jessop HL, Suswillo RFL, Rawlinson SCF, Zaman G, Lee K, Das-Gupta V, Pitsillides AA, Lanyon LE (2004) Osteoblast-like cells from estrogen receptor α knockout mice have deficient responses to mechanical strain. J Bone Miner Res 19:938–946
Lee K, Jessop HL, Suswillo RFL, Zaman G, Lanyon LE (2003) Bone adaptation requires estrogen receptor-α. Nature 424:389
Acknowledgments
The authors thank C.M. Semeins and H.F. Teshale for their expert technical assistance. We thank Drs P. Patka, A. van Kampen, E. Hoebink and T. Patt for their assistance in obtaining osteoporotic bone material, M.N. Helder for kindly providing PCR-primers for cbfa1, and M. Joldersma for providing the raw data of her study on osteoarthritic women. The Netherlands Organization for Scientific Research supported the work of A.D. Bakker (NWO Grant 903-41-193) who also received a financial contribution from the Stichting Anna Fonds. The European Community supported the work of J. Klein-Nulend (fifth framework grant QLK3-1999-00559).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bakker, A.D., Klein-Nulend, J., Tanck, E. et al. Additive effects of estrogen and mechanical stress on nitric oxide and prostaglandin E2 production by bone cells from osteoporotic donors. Osteoporos Int 16, 983–989 (2005). https://doi.org/10.1007/s00198-004-1785-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-004-1785-0