
Abstract
Objectives
To determine the effects of passive leg raising (PLR) on hemodynamics and on cardiac function according to the preload dependency defined by the superior vena cava collapsibility index (ΔSVC).
Results
Forty patients with shock, sedated and mechanically ventilated, were included. Transesophageal echocardiography was performed. At baseline (T1), two groups were defined according to ΔSVC. Eighteen patients presenting a ΔSVC > 36%, an indicator of preload dependency, formed group 1, whereas 22 patients (group 2) exhibited a ΔSVC < 30% (not preload-dependent). Measurements were then performed during PLR (T2), back to baseline (T3), and after volume expansion (T4) in group 1 only. At T1, ΔSVC was significantly higher in group 1 than in group 2, 50 ± 9% and 7 ± 6%, respectively. In group 1, we found a decrease in ΔSVC at T2 (24 ± 9%) and T4 (17 ± 7%), associated with increased systolic, diastolic and arterial pulse pressures. Cardiac index also increased, from 1.92 ± 0.74 (T1) to 2.35 ± 0.92 (T2) and 2.85 ± 1.2 l/min/m2 (T4) and left ventricular end-diastolic volume from 51 ± 41 to 61 ± 51 and 73 ± 51 ml/m2. None of these variations was found in group 2. No change in heart rate was observed.
Conclusion
Hemodynamic changes related to PLR were only induced by increased cardiac preload.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Passive leg raising (PLR), a reversible fluid-loading maneuver [1], may potentially increase intrathoracic blood volume (ITBV), cardiac preload, and then cardiac output [2], by shifting venous blood from the legs [3] towards the thorax [4]. Studies performed in normovolemic healthy volunteers [1], in anesthetized patients [4], and in hypovolemic patients [5] linked the cardiovascular response to PLR to preload dependence of the heart. Recently, some studies [6–8] have suggested that PLR could be used in ventilated and spontaneously breathing patients with hemodynamic instability to assess fluid responsiveness.
However, hemodynamic changes induced by PLR could be difficult to interpret, as several mechanisms may simultaneously contribute to the observed cardiovascular changes: not only an increase in cardiac preload, but also a sympathetic effect resulting from nociceptive stimulation, an activation of the baroreflex [9], and finally a possible activation of the cardiopulmonary reflex mediated by the low-pressure baroreceptors [10, 11]. The cardiopulmonary reflex may serve to shift blood out of the congested venous system into the arterial system, as first reported by Bainbridge [12]. Tachycardia, vasoplegia and so an increase in cardiac index are expected. The baroreflex may serve to regulate blood pressure in the case of increased arterial pressure. Bradycardia and vasoplegia are expected.
Respiratory variations in superior vena cava (SVC) diameter, measured by transesophageal echocardiography, provide an indirect and minimally invasive parameter of fluid responsiveness in ventilated patients [13]. Collapse of the vessel during tidal ventilation is well correlated with “delta down”, a strong indicator of low ITBV [14]. A threshold SVC collapsibility of 36% also discriminates between responders and nonresponders to blood volume expansion [15].
The aim of our study was to test the hypothesis that increased cardiac index during PLR was related only to a shift of venous blood from the legs toward the thorax. We report the various effects of PLR on hemodynamics and on cardiac function, according to the preload dependency defined by the SVC collapsibility index.
Material and methods
We performed a prospective clinical study over a period of 7 months, between February and September 2006, in a medical intensive care unit of a university hospital. The study was approved by the Ethics Committee of the Société de Réanimation de Langue Française (SRLF) and no informed consent was required from the patients' next of kin.
We included all consecutive patients exhibiting acute circulatory failure, defined as systolic arterial pressure (SAP) lower than 90 mmHg, or persistent lactic acidosis, or the need for catecholamine infusion. All patients were mechanically ventilated and adapted to their ventilator. They were all sedated with midazolam and sufentanil. Heart rate (HR) and systolic, diastolic, mean and arterial pulse pressures (respectively SAP, DAP, MAP and PP) were recorded at each step of the study, using an indwelling radial artery catheter.
We also collected the Simplified Acute Physiologic Score II (SAPS II), a severity score [16] calculated within the first 24 h of admission to the ICU, and clinical data at inclusion, e.g., height, body surface area, ventilatory parameters, blood gas analysis, and catecholamine dose.
Transesophageal echocardiography
Transesophageal echocardiography (TEE) was performed with a Siemens Sequoia C-256 equipped with a multiplane Acuson TE-V5Ms 5-MHz transducer (Acuson, Mountain View, CA). Using the signal from the respirator, airway pressure was displayed on the screen of the ultrasound machine, permitting accurate timing of cardiac events during the respiratory cycle [17, 18].
The SVC was examined by a long-axis view, using the two-dimensional view to direct the M-mode beam across the maximal diameter. As previously described, we measured the SVC collapsibility index, as the maximal diameter during expiration minus the minimal diameter during insufflation, divided by the maximal diameter [13]. We also reported the maximal SVC diameter, a surrogate of central venous pressure.
Left ventricular (LV) end-diastolic volume (EDV) was measured at end-expiration according to the Simpson method [19] on a long-axis view of the left ventricle and indexed to body area.
Pulmonary artery (PA) flow velocity was also recorded in a long-axis view at the level of right ventricular (RV) outflow tract. From the pulsed Doppler velocity profile, recorded at a speed of 25 cm/s, we measured PA velocity–time integrals (VTIPA). PA systolic diameter (D) was also measured and we calculated PA cross-sectional area as ΠD2/4. RV stroke volume was calculated by multiplying averaged VTIPA during the respiratory cycle by the PA cross-sectional area [20], and was expressed as RV stroke index (RVSI) after dividing by body surface area. Cardiac index (CI) was calculated by multiplying RVSI by HR.
Finally, RV size was assessed at baseline on a long-axis view of the heart by the ratio of the end-diastolic area of the right ventricle to the end-diastolic area of the left ventricle (RV/LV EDA). A right ventricle was considered dilated if this ratio was above 0.6 [21]. LV function was also assessed at baseline on a short-axis view of the left ventricle by calculating the fractional area change (FAC) as the end-diastolic area minus the end-systolic area, divided by the end-diastolic area.
All views collected were directly stored numerically, allowing measurement of most parameters, with the exception of SVC collapsibility index, by an expert unaware of the study.
Study protocol
Patients were prospectively assigned to two groups according to their SVC collapsibility index. At baseline, patients presenting an SVC collapsibility index strictly higher than 36% formed group 1. We previously demonstrated that all of these patients had significantly increased cardiac output after blood volume expansion [15]. Group 2 comprised patients presenting a SVC collapsibility index lower than 30%, a threshold value predicting unresponsiveness to fluid loading in 100% of cases [15].
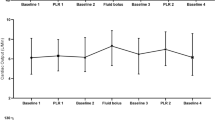
A first set of measurements (baseline, T1) was obtained in the semirecumbent position (25°). Using an automatic bed-elevation technique, the lower limbs were then raised to a 20° angle while the patient's trunk was lowered into the supine position, using Trendelenburg's maneuver. A second set of measurements (PLR, T2) was then obtained after 2 min of stabilization. Patients were then returned to baseline and a third set of measurements was recorded (T3). Finally, for group 1 only, a fourth set of measurements (T4) was obtained after fluid loading in less than 15 min with 500 ml of hydroxylethyl starch (Voluven®; Fresenius Kabi, Sèvres, France), and patients were considered as responders to fluids if their cardiac index increased by more than 10%.
The ventilator settings and vasoactive therapy were kept constant throughout the study period.
Statistical analysis
Continuous variables were reported as mean ± SD and between-group comparison was performed using a Mann–Whitney U test. Qualitative variables were reported as number and percentage and compared between groups using a chi-square test (or Fisher's exact test).
Hemodynamic and echocardiographic changes over time were analyzed using a Friedman test, followed, if appropriate, by two-by-two post-hoc comparisons using the paired Wilcoxon rank sum test. An alpha risk below 0.05 (p ≤ 0.05) was considered as significant. After Bonferroni's adjustment, p values < 0.008 and < 0.017 for the Wilcoxon rank sum test in groups 1 and 2, respectively, were regarded as significant. Statistical analysis was performed using Statview 5.0 (Abacus Concepts, Berkeley, CA) for all tests.
Intra-observer variability for maximum and minimum SVC diameters was previously reported as between 2 ± 3% and 3 ± 3% and between 5 ± 6% and 4 ± 4%, respectively [15]. Inter-observer variability was reported as 7 ± 7% [15].
Results
Forty patients were included, 18 in group 1 and 22 in group 2. Four patients were excluded because of SVC collapsibility between 30% and 36%. Demographic and clinical data at inclusion are reported in Table 1. Body height and body area surface were the only variables that differed significantly between the two groups. No patient received beta-blockers or digoxin.
At baseline (T1), the SVC collapsibility index was 50 ± 9% (38–67%) in group 1 and 7 ± 6% (0–24%) in group 2. HR, SAP, DAP, PP, RVSI, and CI did not differ between the two groups, and neither did LVEDV. There was a slight, nonsignificant increase in RV/LV EDA ratio in group 2 (0.61 ± 0.36 vs. 0.47 ± 0.15), whereas maximal SVC diameter was significantly higher. Finally, LVFAC was not different at baseline between groups 1 and 2 (44 ± 12% vs. 44 ± 14%, respectively).
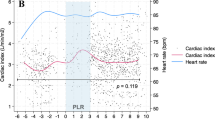
All but one of the 18 patients in group 1 were responders to fluids. Group 1 patients had a significant decrease in SVC collapsibility index during PLR (T2) and after fluids (T4), respectively 24 ± 9 and 17 ± 7%, compared with T1 (p < 0.008) (Table 2, Fig. 1). LVEDV and maximal SVC diameter were also significantly increased at T2 and T4 (Table 2). Finally, we noted significant increases in SAP, DAP, PP, RVSI, and CI (Table 2, Fig. 2). No change in HR was observed.

Examination of superior vena cava (SVC) by transesophageal echocardiography using M-mode. a Group 1 patients at baseline (T1), during passive leg raising (T2), and after 500 ml rapid fluid loading (T4). b Group 2 patients at T1 and T2. Arrow, airway pressure

Comparison of superior vena cava (SVC) collapsibility index and cardiac index between the two groups (group 1, left panel; group 2, right panel) at baseline (T1), during passive leg raising (T2), after return to baseline (T3), and after rapid fluid loading (T4) for group 1 only. The central box represents the values from the lower to the upper quartile (25th to 75th percentile). The middle line represents the median. The vertical line extends from the minimum to the maximum value. The circle represents the mean value. *p < 0.008 vs. T1, ‡ p < 0.008 vs. T2, † p < 0.017 vs. T2
Patients in group 2 did not exhibit any significant changes in HR, SAP, DAP, PP, RVSI, and CI between T1, T2, and T3 (Table 3, Fig. 2). LVEDV and maximal SVC diameter remained unchanged.
Discussion
It has been suggested that PLR be used in ventilated and spontaneously breathing patients with hemodynamic instability to assess fluid responsiveness [6–8] and so to indicate fluid requirements. However, different mechanisms can contribute to the hemodynamic changes observed during PLR.
The first mechanism is an increase in systemic venous return. This induces an increase in cardiac output in preload-dependent patients [22], i.e. those in whom the ventricles are operating on the steep part of the Frank–Starling curve, by increasing ITBV and cardiac preload. This is actually the effect one aims to test when performing PLR to assess fluid responsiveness. Patients in group 1 had a mean SVC collapsibility index of 50%, and their arterial pressures, RVSI and CI significantly increased during PLR. This is attributed to an increase in ITBV and in cardiac preload. Indeed, the partial collapse of SVC observed at baseline was corrected during PLR. Moreover, maximal SVC diameter and LVEDV significantly increased after PLR. The same effects were observed in these patients after fluid loading, confirming a posteriori the effect of PLR. Hemodynamic changes induced by PLR were less pronounced than those induced by fluid administration. Indeed, whereas PLR should mobilize less than 300 ml of blood, we actually administered 500 ml of fluids. Interestingly, the fact that not only hemodynamics but also SVC collapsibility index and cardiac preload returned to baseline after PLR (T3) demonstrates the temporary and reversible effect of PLR.
However, other mechanisms, such as baroreflex activation [23], stimulation of the cardiopulmonary reflex, mediated by the low-pressure baroreceptors [10, 11], and sympathetic stimulation through partial wakening of the patient can contribute to the hemodynamic changes observed during PLR. Interestingly, patients in group 1 had no significant changes in HR despite a significant increase in systolic arterial pressure. This could suggest a desensitization of the baroreflex, previously observed during heart failure [24], sedation [25], or prolonged bed rest [26, 27]. The two latter conditions were present in our patients.
As expected in patients in group 2, those with a mean SVC collapsibility index of less than 30% at baseline, PLR did not induce any significant increase in LVEDV or in RVSI and CI. Arterial pressures and HR remained unchanged, which does not indicate sympathetic stimulation induced by PLR, resulting from a hypothetical and partial wakening of the patient. The degree of sedation of our patients, and the method used for PLR, with an automatic bed elevation technique instead of direct leg raising by an operator, may in part explain these results, which could be different in less sedated patients or using another PLR technique [28]. In these patients, whose ventricles operated on the horizontal part of the Frank–Starling curve, PLR should induce an increase in cardiac filling pressure, but not in cardiac volume, thus stimulating the low-pressure baroreceptors and then the cardiopulmonary reflex [11]. By inducing arterial vasodilation and tachycardia, the cardiopulmonary reflex would serve to shift blood out of the congested venous system into the arterial system [12, 29, 30]. As a matter of fact, PLR did not induce any increase in LVEDV and in HR. Moreover, we did not observe any decrease in diastolic and pulse pressures, whereas RVSI remained unchanged. This may argue against the role of such a reflex in our population. Different results were previously observed in awake and spontaneously breathing patients [5]. Briefly, the only significant change in group 2 induced by PLR was in SVC collapsibility index, which decreased from 7 ± 6% to 4 ± 4%. This could reflect a slight increase in venous return, insufficient to induce significant hemodynamic changes. However, in our experience, such value has no clinical meaning [15].
Interestingly, at baseline, HR, blood pressures, CI and LV fractional area changes were not significantly different between group 1 and group 2. The only significantly different parameter was the maximal SVC diameter, which was higher in group 2 patients. This was associated with a trend towards a higher RV/LV end-diastolic area ratio and a lower LV end-diastolic volume. This means that RV function could be a major determinant of the response to fluids, as previously suggested [31]. In other words, patients with RV dysfunction (suggested by higher SVC diameter, increased RV size, and decreased LV size) were unresponsive to PLR, whereas patients with preserved RV function (suggested by lower SVC diameter, normal RV size, and normal LV size) were responsive to PLR and fluids. In the first situation the heart is expected to be on the flat part of the Frank–Starling curve, whereas in the second situation it is expected to be on the steep part.
Our study has some limitations. First, in this clinical investigation baroreflex and cardiopulmonary reflex were not directly studied at the bedside, but the absence of changes in HR in groups 1 and 2 and in blood pressures in group 2 suggests the absence of a significant impact or opposite and offsetting effects of such reflexes on HR [32]. However, one can say that HR and pulse pressure alone are insufficient to evaluate it. Second, our results are applicable only to sedated patients adapted to their ventilator. How sympathetic stimulation or cardiac reflexes may alter the hemodynamic response to PLR is unknown in awake and spontaneously breathing patients. Finally, inter- and intraobserver variability for SVC diameters were calculated not in the current study but in a previous analysis [15].
Nevertheless, our results are of importance as they demonstrate in our population that increased CI related to PLR was due only to an increase in cardiac preload.
References
Gaffney FA, Bastian BC, Thal ER, Atkins JM, Blomqvist CG (1982) Passive leg raising does not produce a significant or sustained autotransfusion effect. J Trauma 22:190–193
Thomas M, Shillingford J (1965) The circulatory response to a standard postural change in ischaemic heart disease. Br Heart J 27:17–27
Rutlen DL, Wackers FJ, Zaret BL (1981) Radionuclide assessment of peripheral intravascular capacity: a technique to measure intravascular volume changes in the capacitance circulation in man. Circulation 64:146–152
Reich DL, Konstadt SN, Raissi S, Hubbard M, Thys DM (1989) Trendelenburg position and passive leg raising do not significantly improve cardiopulmonary performance in the anesthetized patient with coronary artery disease. Crit Care Med 17:313–317
Wong DH, O'Connor D, Tremper KK, Zaccari J, Thompson P, Hill D (1989) Changes in cardiac output after acute blood loss and position change in man. Crit Care Med 17:979–983
Boulain T, Achard JM, Teboul JL, Richard C, Perrotin D, Ginies G (2002) Changes in BP induced by passive leg raising predict response to fluid loading in critically ill patients. Chest 121:1245–1252
Lamia B, Ochagavia A, Monnet X, Chemla D, Richard C, Teboul JL (2007) Echocardiographic prediction of volume responsiveness in critically ill patients with spontaneously breathing activity. Intensive Care Med 33:1125–1132
Maizel J, Airapetian N, Lorne E, Tribouilloy C, Massy Z, Slama M (2007) Diagnosis of central hypovolemia by using passive leg raising. Intensive Care Med 33:1133–1138
Roddie IC, Shepherd JT (1957) The effects of carotid artery compression in man with special reference to changes in vascular resistance in the limbs. J Physiol 139:377–384
Barbieri R, Triedman JK, Saul JP (2002) Heart rate control and mechanical cardiopulmonary coupling to assess central volume: a systems analysis. Am J Physiol Regul Integr Comp Physiol 283:R1210–1220
Parati G, Grassi G, Coruzzi P, Musiari L, Ravogli A, Novarini A, Mancia G (1987) Influence of cardiopulmonary receptors on the bradycardic responses to carotid baroreceptor stimulation in man. Clin Sci (Lond) 72:639–645
Bainbridge FA (1915) The influence of venous filling upon the rate of the heart. J Physiol 50:65–84
Vieillard-Baron A, Augarde R, Prin S, Page B, Beauchet A, Jardin F (2001) Influence of superior vena caval zone condition on cyclic changes in right ventricular outflow during respiratory support. Anesthesiology 95:1083–1088
Vieillard-Baron A, Chergui K, Augarde R, Prin S, Page B, Beauchet A, Jardin F (2003) Cyclic changes in arterial pulse during respiratory support revisited by Doppler echocardiography. Am J Respir Crit Care Med 168:671–676
Vieillard-Baron A, Chergui K, Rabiller A, Peyrouset O, Page B, Beauchet A, Jardin F (2004) Superior vena caval collapsibility as a gauge of volume status in ventilated septic patients. Intensive Care Med 30:1734–1739
Le Gall JR, Loirat P, Alperovitch A (1983) Simplified acute physiological score for intensive care patients. Lancet 2:741
Vieillard-Baron A, Prin S, Chergui K, Dubourg O, Jardin F (2003) Hemodynamic instability in sepsis: bedside assessment by Doppler echocardiography. Am J Respir Crit Care Med 168:1270–1276
Vieillard-Baron A, Loubieres Y, Schmitt JM, Page B, Dubourg O, Jardin F (1999) Cyclic changes in right ventricular output impedance during mechanical ventilation. J Appl Physiol 87:1644–1650
Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I (1989) Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr 2:358–367
Maslow A, Comunale ME, Haering JM, Watkins J (1996) Pulsed wave Doppler measurement of cardiac output from the right ventricular outflow tract. Anesth Analg 83:466–471
Weyman A (1982) Cross-Sectional echocardiography. In: Lea & Febiger (ed), Philadelphia PA, pp 501–502
Starling EH (1965) The Linacre Lecture of the Law of the Heart. In: Chapman CB, Mitchell JH (ed) Starling on the heart. Dawsons, London, pp 119–147
Roddie IC, Shepherd JT, Whelan RF (1957) Reflex changes in vasoconstrictor tone in human skeletal muscle in response to stimulation of receptors in a low-pressure area of the intrathoracic vascular bed. J Physiol 139:369–376
White CW (1981) Reversibility of abnormal arterial baroreflex control of heart rate in heart failure. Am J Physiol 241:H778–H782
Marty J, Gauzit R, Lefevre P, Couderc E, Farinotti R, Henzel C, Desmonts JM (1986) Effects of diazepam and midazolam on baroreflex control of heart rate and on sympathetic activity in humans. Anesth Analg 65:113–119
Chobanian AV, Lille RD, Tercyak A, Blevins P (1974) The metabolic and hemodynamic effects of prolonged bed rest in normal subjects. Circulation 49:551–559
Nixon JV, Murray RG, Bryant C, Johnson RL Jr, Mitchell JH, Holland OB, Gomez-Sanchez C, Vergne-Marini P, Blomqvist CG (1979) Early cardiovascular adaptation to simulated zero gravity. J Appl Physiol 46:541–548
Wong DH, Tremper KK, Zaccari J, Hajduczek J, Konchigeri HN, Hufstedler SM (1988) Acute cardiovascular response to passive leg raising. Crit Care Med 16:123–125
Jones JJ (1962) The Bainbridge reflex. J Physiol 160:298–305
Pawelczyck JA, Levine BD (1995) Cardiovascular responses to rapid volume infusion: the human Bainbridge reflex. Circulation 92:3148
Schneider AJ, Teule GJ, Groeneveld AB, Nauta J, Heidendal GA, Thijs LG (1988) Biventricular performance during volume loading in patients with early septic shock, with emphasis on the right ventricle: a combined hemodynamic and radionuclide study. Am Heart J 116:103–112
Barbieri R, Triedman JK, Saul JP (2002) Heart rate control and mechanical cardiopulmonary coupling to assess central volume: a system analysis. Am J Physiol Regul Integr Comp Physiol 283:R1210–R1220
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Caille, V., Jabot, J., Belliard, G. et al. Hemodynamic effects of passive leg raising: an echocardiographic study in patients with shock. Intensive Care Med 34, 1239–1245 (2008). https://doi.org/10.1007/s00134-008-1067-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-008-1067-y