
Abstract
Assimilation by mafic to ultramafic magmas of sulfur-bearing country rocks is considered an important contributing factor to reach sulfide saturation and form magmatic Ni–Cu–platinum group element (PGE) sulfide deposits. Sulfur-bearing sedimentary rocks in the Archean are generally characterized by mass-independent fractionation of sulfur isotopes that is a result of atmospheric photochemical reactions, which produces isotopically distinct pools of sulfur. Likewise, low-temperature processing of iron, through biological and abiotic redox cycling, produces a range of Fe isotope values in Archean sedimentary rocks that is distinct from the range of the mantle and magmatic Fe isotope values. Both of these signals can be used to identify potential country rock assimilants and their contribution to magmatic sulfide deposits. We use multiple S and Fe isotopes to characterize the composition of the potential iron and sulfur sources for the sulfide liquids that formed the Hart deposit in the Shaw Dome area within the Abitibi greenstone belt in Ontario (Canada). The Hart deposit is composed of two zones with komatiite-associated Ni–Cu–PGE mineralization; the main zone consists of a massive sulfide deposit at the base of the basal flow in the komatiite sequence, whereas the eastern extension consists of a semi-massive sulfide zone located 12 to 25 m above the base of the second flow in the komatiite sequence. Low δ56Fe values and non-zero δ34S and Δ33S values of the komatiitic rocks and associated mineralization at the Hart deposit is best explained by mixing and isotope exchange with crustal materials, such as exhalite and graphitic argillite, rather than intrinsic fractionation within the komatiite. This approach allows tracing the extent of crustal contamination away from the deposit and the degree of mixing between the sulfide and komatiite melts. The exhalite and graphitic argillite were the dominant contaminants for the main zone of mineralization and the eastern extension zone of the Hart deposit, respectively. Critically, the extent of contamination, as revealed by multiple S and Fe isotope systematics, is greatest within the deposit and decreases away from it within the komatiite flow. This pattern points to a local source of crustal contamination for the mantle-derived komatiitic melt and a low degree of homogenization between the mineralization and the surrounding lava flow. Coupled S and Fe isotope patterns like those identified at the Hart deposit may provide a useful tool for assessing the potential of a komatiitic sequence to host Ni–Cu–(PGE).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Exploration models for magmatic nickel–copper–platinum group element (PGE) sulfide deposits are relatively well established, and, in komatiite-associated deposits, it is generally accepted that the source of metals is the mantle-derived ultramafic magmas. However, sulfur is generally thought to be derived from an external source (Lesher 1989), typically from the melting of sulfur-bearing country rocks to generate sulfide xenomelts (Lesher and Campbell 1993; Lesher and Burnham 2001). An external sulfur source is necessary due to an increase in sulfur solubility in komatiitic magmas with the decrease in pressure during the magma ascent from the mantle through the crust to near-surface environments (Wendlandt 1982; Lesher and Groves 1986; Mavrogenes and O’Neill 1999). Sulfur sources can be relatively well constrained by field evidence and geological relationships for some komatiite-associated deposits, such as those at Alexo (Houlé et al. 2012), Kambalda (Lesher 1989), or the Agnew-Wiluna belt (Fiorentini et al. 2012a, b). However, for some deposits where more than one possible sulfur source exists, identifying the one responsible for sulfide saturation becomes more difficult.
Early attempts to determine sulfur sources for komatiite-associated mineralization relied on δ34S. However, the range of bulk-rock δ34S values in Archean supracrustal deposits is much smaller than that in their Phanerozoic counterparts, resulting in potential crustal sources with δ34S values that are largely indistinguishable from the mantle range (Ripley 1999), which makes this approach inconclusive in some cases. More recently, multiple sulfur isotopes have been used to link nickel sulfide mineralization to sedimentary sulfur sources in Archean komatiite-associated deposits (Bekker et al. 2009; Fiorentini et al. 2012a, b; Konnunaho et al. 2013; Hofmann et al. 2014).
This study expands on these previous efforts by linking magmatic nickel sulfide mineralization to the sedimentary sulfur source at the Hart deposit and investigates the lateral and vertical variations of the stable isotope signatures, using multiple sulfur isotopes and iron isotopes, with increasing distance away from the mineralization within the host komatiite flows. Stable isotope (δ34S, Δ33S, and δ56Fe) and major and trace element geochemistry are used to identify the most likely sedimentary contaminants for the genesis of the Hart komatiite-associated Ni–Cu–PGE deposit within the Shaw Dome area in the Abitibi greenstone belt (Ontario, Canada).
Background
Under present terrestrial conditions, most sulfur isotope fractionations are controlled by relative isotope mass difference, leading to a close “mass-dependent” correspondence between δ values of δ33S ≈ 0.5 × δ34S. However, as a result of photochemical reactions in the anoxic Archean atmosphere, atmospherically processed Archean sulfur exhibits mass-independent fractionation that can be characterized by the difference between the δ33S value expected from normal mass-dependent fractionation and the measured δ33S value (Farquhar et al. 2000; Farquhar and Wing 2003). It is calculated by
where λ RFL is the slope of the reference mass-dependent fractionation line equal to 0.515.
Photochemically fractionated sulfur was delivered to Archean seawater and ultimately incorporated into sedimentary rocks. Unlike modern oceans, the Archean oceans had relatively low sulfate concentrations, less than 100–200 μmol L−1 (Habicht et al. 2002; Jamieson iet al. 2013), which resulted in the preservation of small isotopic fractionations caused by microbial S cycling in marine sediments (Wing and Halevy 2014). As a result, Archean sediments do not exhibit the large range in δ34S values seen in more recent marine sediments, hampering discrimination between crustal and mantle sulfur. However, Archean sedimentary rocks typically exhibit mass-independent fractionation of S isotopes as shown by their non-zero Δ33S values (Farquhar et al. 2000; Farquhar and Wing 2003). Therefore, multiple sulfur isotope ratios can constrain the sulfur source for nickel–copper–PGE sulfide mineralization in mafic to ultramafic systems and vector toward prospective areas, where crustal sulfur incorporation occurred and triggered sulfide saturation (e.g., Bekker et al. 2009).
Additionally, for several Archean lithologies, such as ferruginous sediments and organic matter-rich shales, the δ56Fe (and δ57Fe) values have been shown to exhibit significant variability (Johnson et al. 2003; Rouxel et al. 2005; Yamaguchi et al. 2005; Archer and Vance 2006; Dauphas and Rouxel 2006) and can be used to provide independent constraints on the mechanisms that triggered sulfur saturation and the extent of country rock assimilation.
Once the signatures of potential sedimentary sources are established, tracing the contamination in the komatiite could provide insights into the flow regime and cooling history of the komatiite (Lesher and Arndt 1995). Utilization of both S and Fe isotopes provides two tracers that are sensitive to different degrees of contamination (Hiebert et al. 2013). Contaminant to komatiite ratios can be quantified with two-component mixing and isotope exchange equations (Campbell and Naldrett 1979; Lesher and Burnham 2001; Ripley and Li 2003); there is more Fe than S in komatiite magmas, which makes Fe isotopes in sulfide xenomelts more sensitive to lower silicate magma–sulfide melt ratios (R factor) than S isotopes.
Geological setting
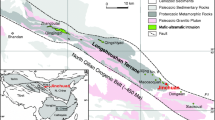
Komatiites and komatiite-associated Ni–Cu–PGE deposits in the Abitibi greenstone belt (Fig. 1) are recognized worldwide for their outstanding preservation and exposure (Barnes and Naldrett 1987; Houlé and Lesher 2011), including well-studied examples at Pyke Hill (Pyke et al. 1973; Houlé et al. 2009), Dundonald Beach (Houlé et al. 2008), and Alexo (Houlé et al. 2012). The Abitibi greenstone belt can be subdivided into seven volcanic episodes with associated lesser sedimentary packages (Thurston 2008). Of these, four episodes contain most of the komatiites, but only two host significant Ni–Cu–PGE mineralization: the 2720–2710 Ma Kidd-Munro and 2710–2704 Ma Tisdale volcanic episodes. Most of the past and ongoing nickel production from this type of deposit in the Abitibi greenstone belt has come from the Shaw Dome area (Fig. 1), which hosts the Hart deposit located approximately 30 km southeast of Timmins (Houlé and Lesher 2011).

The volcano-sedimentary succession in the Shaw Dome comprises from oldest to youngest: (1) massive and pillowed intermediate volcanic rocks, thin, but laterally extensive, iron formations, and subordinate massive to volcaniclastic felsic volcanic rocks of the 2734–2724 Ma Deloro volcanic episode; (2) felsic to intermediate volcaniclastic rocks intercalated with komatiitic dikes, sills, lavas, and less extensive iron formations of the lower part of the 2710–2704 Ma Tisdale volcanic episode; (3) intercalated tholeiitic mafic and komatiitic volcanic rocks of the middle part of the Tisdale volcanic episode; and (4) calc-alkaline felsic to intermediate volcanic rocks in the upper part of the Tisdale volcanic episode (Houlé et al. 2010a, b; Houlé and Lesher 2011).
The main mineralized zone of the Hart deposit is hosted in the basal komatiite flow of the middle Tisdale episode, where several stacked komatiite flows overly and cross-cut the felsic to intermediate volcanic and sedimentary rocks of the lower Tisdale episode (Houlé et al. 2010a, b; Houlé and Lesher 2011). The eastern extension of the Hart deposit is hosted within the second komatiite flow in this succession. Combined, the main zone contains 1.9 Mt with an average grade of 1.38 % Ni (Houlé and Lesher 2011). Following the terminology of Lesher and Keays (2002), the main zone of the Hart deposit is a classic, type I stratiform basal mineralization hosted by thick, olivine orthocumulate to mesocumulate komatiite units in which the mineralization is localized at the base of a wide (>200 m) embayment into its footwall rocks (Figs. 2 and 3). The embayment is interpreted to have been produced by thermomechanical erosion of underlying felsic volcanic rocks and iron formation (Houlé et al. 2010b). In addition to the main zone, a secondary zone of semi-massive, net-textured, and disseminated sulfide, referred to as the eastern extension, is present 12–25 m above the base of the second flow. No significant mineralization is known to exist in komatiites or basalts above the second komatiite flow. Komatiites in the study area have been strongly altered to serpentinite, but pseudomorphs of original olivine cumulates are commonly preserved. All rocks in the study area have been metamorphosed under greenschist facie conditions.

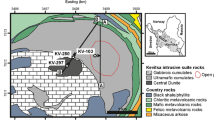
Geologic map of the Hart deposit area (modified from Houlé et al. 2010b). Drill hole collar locations and section lines for composite isotope data traverses through the deposit are indicated. Location of sampled surface trenches are indicated by the green bars

Field photograph of mineralized zone in trench MGH600. From stratigraphic base to top; a felsic volcanic rocks, b exhalite, c felsic volcanic rocks, d massive sulfides, e net-textured sulfides, f barren komatiite, g disseminated sulfides in komatiitic peridotite, h barren komatiitic peridotite
The footwall rocks are dominantly composed of felsic to intermediate volcanic and volcaniclastic rocks with a lesser, but regionally extensive, banded iron formation, having variable content of magnetite-rich layers that are chert-rich locally, and minor graphitic argillite. In the vicinity of the Hart deposit, some of the iron formation has been interpreted to represent an exhalite (Fig. 4) due to the predominance of chert and chert-rich lithologies. These exhalites typically contains minor laminae of Fe oxides or sulfides (e.g., sample H11-16-411.4; 86.2 % SiO2, 10.9 % Fe2O3). Locally, the chert or chert-rich lithologies within the exhalite grade into more typical banded iron formation (e.g., sample H11-13C-387.2; 41.73 % SiO2, 37.44 % Fe2O3), or barren massive sulfides, but these lithologies do not extend laterally for more than a few tens of meters in the area of the Hart deposit (Hiebert et al. 2013), despite the regional extent of iron formation (Houlé et al. 2010b). Sulfides within the exhalite are typically masses of fine-grained pyrrhotite (0.1–0.2 mm in size) that have been locally recrystallized to form larger pyrite grains (0.25–0.6 mm in size; Fig. 4d). Apparent thicknesses of exhalite observed in drill core are typically less than 10 m, but may be up to 25 m thick.

a, b Outcrop photographs taken along strike from the deposit of chert (a) and iron formation (b) stratigraphically equivalent to the exhalite unit in the Hart Deposit area. The coin is 18 mm in diameter on (a) and (b). c, d Reflected light photomicrographs of exhalite in the footwall of the Hart deposit: oxide-rich laminae in chert (c sample H11-13C-363) and coarse pyrite grains surrounded by fine pyrrhotite grains in barren sulfide lens within exhalite unit (d sample H11-08-58.3)
The graphitic argillite is observed as two thin (<5 m) layers in drill core northeast of the main zone of mineralization in the Hart deposit. It is composed predominantly of graphite (45–65 vol.%) and pyrite (10–40 vol.%), with lesser amounts of metamorphic chlorite, epidote, and quartz (10–20 vol.%; Fig. 5a). Sulfide in the graphitic argillite takes two forms: finely disseminated pyrite (Fig. 5b, c; <0.1 mm in size) or large (1–1.5 cm in diameter) pyrite nodules and bands (Fig. 5a, c).

a Core photo of graphitic argillite showing nodules and bands of pyrite. The coin is 18 mm in diameter. b Photomicrograph of graphitic argillite unit in reflected light (sample H11-13C-366.1) showing minor disseminated sulfide (bright grains) and opaque mineral (graphite). c Photomicrograph in reflected light of pyrite nodule in graphitic argillite showing concentric growth bands (sample H11-13C-357.1)
Material analyzed and analytical methods
Sampling methodology
The 93 samples were selected from mechanically stripped trenches and diamond drill cores (Electronic Supplementary Material, ESM 1). Several transects were made within and away from the main mineralized zone at the Hart deposit to create composite traverses that include all footwall lithologies (felsic volcanic rocks, exhalite, and graphitic argillite), the mineralization (massive, semi-massive, net-textured, and disseminated sulfides), and the hosting komatiite flow immediately above the mineralization and upward into barren komatiitic flows. These transects utilized 11 diamond drill holes and 3 trenches along 5 sections on the local mine grid (Fig. 2).
This sampling strategy ensured that the basal komatiitic flow, which hosts the main mineralized zone, was sampled as far as 500 m east and 300 m west of the main mineralized zone. The second komatiitic flow, which hosts the eastern extension zone, was only sampled west of the mineralization due to a lack of drilling east of mineralization at the time of the investigation. Additionally, one sample of the third flow from drill hole H07-33 was taken to represent barren komatiite flow that likely never interacted with the sulfur source rock.
Whole rock geochemistry
Samples were analyzed for major, trace, and rare earth elements in two different laboratories: the Ontario Geological Survey Geoscience Laboratories (GeoLabs; Sudbury, Ontario) and the Acme Laboratories (Acme Labs; Vancouver, British Columbia). All the materials were crushed at the Stable Isotopes for Innovative Research (SIFIR) laboratory at the University of Manitoba to a fine powder (200 mesh) using an agate puck mill before the pulps were sent to these laboratories for geochemical analysis. At GeoLabs, major element analyses were performed by wavelength dispersive X-ray fluorescence (XRF) using a fused disk method. Total sulfur content was determined by oxidation of sulfur through combustion of the sample in an oxygen-rich environment and SO2 detection by infrared absorption (LECO elemental analyzer). Trace elements, including the main refractory elements and rare earth elements (REE), were analyzed by inductively coupled plasma mass spectrometry (ICP-MS) following a closed vessel multiacid digestion. At Acme Labs, concentrations were determined by ICP-MS following a four-acid digestion. One sample (H11-11-407) was analyzed by both labs and showed reproducibility of data between the two laboratories.
Sulfur isotope analysis
Sulfur was extracted from rock powders and converted to Ag2S in both the SIFIR lab (University of Manitoba) and the Stable Isotope Laboratory of the Department of Earth and Planetary Sciences (McGill University) with a Cr(II) reduction procedure that has been already applied to a range of different types of ore metal sulfides from mafic to ultramafic intrusive systems (Fiorentini et al. 2012b; Hiebert et al. 2013), komatiite-associated Fe–Ni–Cu sulfide mineralization (Konnunaho et al. 2013), volcanic massive sulfide deposits (Sharman et al. 2014), and oxidized intrusion-related gold deposits (Helt et al. 2014). All samples were analyzed at McGill University by first fluorinating the Ag2S at 225 °C in a Ni autoclave under ≈20× stoichiometric excess of F2 for >9 h to produce SF6, which was then purified cryogenically and chromatographically and analyzed on a Thermo Electron MAT 253 mass spectrometer for multiple sulfur isotope ratios in a dual-inlet mode. The sulfur isotope compositions are reported with respect to the V-CDT scale, on which the δ34S value of IAEA-S-1 is defined as −0.3 ‰, and the Δ33S value is taken to be 0.094 ‰. Repeat analyses throughout the entire analytical procedure return 2σ uncertainties on δ34S and Δ33S values that are <0.3 and <0.02 ‰, respectively.
Fe isotope analysis
Fe isotope analyses were performed at IFREMER (Brest, France). Aliquots of sample powders were first dissolved in an HF-HNO3-HCl acid mixture on a hot plate. Fe was then purified on a Bio-Rad AG-MP1 anion exchange chromatographic resin. Isotopic ratios were determined with a Thermo Electron Neptune multicollector inductively coupled mass spectrometer (MC-ICP-MS), following methods previously described by Rouxel et al. (2005). Internal precision of data was determined through duplicate analysis of a reference standard, and the long-term external reproducibility is 0.06 ‰ for δ56Fe values (2σ). Fe isotope values are reported relative to the standard IRMM-14.
Results
Sulfur and iron isotopes
Isotope results are described separately for lithologies present in the footwall to the komatiite and those hosted within komatiitic flows (ESM 1), where the latter have been further subdivided based on the visual estimate of the abundance of sulfide mineralization into barren komatiite (<5 % sulfide minerals by volume), disseminated mineralization (5–30 %), semi-massive and net-textured (30–70 %), and massive (>70 %).
Footwall lithologies
The exhalite shows the largest S isotope variability of any footwall lithology in the Hart area with δ34S values ranging from −11.4 to 7.6 ‰ and Δ33S values ranging from −1.4 to −0.3 ‰ (Fig. 6a). The graphitic argillite shows the least variability, with δ34S values ranging from 1.6 to 5.0 ‰ and Δ33S values ranging from −0.9 to −0.2 ‰. Felsic volcanics have δ34S values ranging from −5.7 to 1.9 ‰ and Δ33S values ranging from −0.5 to −0.2 ‰. The δ56Fe values of felsic volcanic rocks range from −0.9 to −0.1 ‰. Exhalite and graphitic argillite have overlapping ranges of δ56Fe values, −2.1 to −0.9 ‰ and −2.0 to −1.7 ‰, respectively, and show systematically lower δ56Fe values than the average bulk silicate Earth (Fig. 7). The δ56Fe values of exhalite also show no relationship to the abundance of sulfides.

Plot of Δ33S versus δ34S values showing the variations in S isotope composition of potential crustal contaminants (a) and komatiite-associated mineralization (b). In (b), exhalite data is represented by the brown field, and graphitic argillite data is represented by the grey field. Orange box represents the range of the mantle values in both (a) and (b) (Bekker et al. 2015)

Fe isotope composition of the lithologies present in the Hart deposit area. Orange rectangle represents the range of the mantle values
Komatiite
The S isotope compositions of komatiite samples can be generally related to sulfide mineralization, with δ34S values of mineralized samples ranging from −3.8 to 1.7 ‰ and Δ33S values ranging from −0.7 to −0.4 ‰ (Fig. 6b). In general, massive sulfides have the lowest δ34S and Δ33S values and samples with disseminated mineralization have negative values close to 0 ‰, although significant overlap exists. Barren komatiites generally have values close to the mantle range, with δ34S values ranging from −4.1 to 3.3 ‰ and Δ33S values ranging from −0.6 to 0.5 ‰.
Barren komatiites have a narrow range of δ56Fe values near 0.0 ‰, similar to near-chondritic Fe isotope composition of the silicate Earth, reflecting minor fractionations during komatiite magma genesis (Dauphas et al. 2010). Mineralized komatiites, however, show significant deviation from mantle values, with δ56Fe values (−1.5 to 0.2 ‰) ranging between those of barren komatiite and footwall lithologies (Fig. 7). The lowest δ56Fe values are generally found in massive and semi-massive sulfides, whereas disseminated sulfides have negative δ56Fe values close to 0 ‰, although, as with S isotopes, significant overlap exists.
Samples from the basal flow have δ34S and Δ33S values that are lowest close to the mineralization and trend toward mantle values (0‰) away from the zone of mineralization, both laterally to the west (2100E; Fig. 8a), and vertically toward the top of the flow (Fig. 9). To the east of the deposit, komatiite δ34S values tend to be more positive than those in the mineralized zone. Δ33S values east of the main zone have a bimodal distribution, with some samples having similar values to the mineralization (approximately −0.5 to −0.6 ‰), and others having positive Δ33S values. Although komatiite above the mineralization shows significant overlap with the range of δ56Fe values observed in mineralization, trends similar to those shown by δ34S and Δ33S values are also observed in δ56Fe data, with values approaching the mantle range both laterally (east and west of the mineralization) and vertically away from the mineralization (Fig. 8b).

Isotope data for the mineralization and the komatiites (above, east, and west of mineralization) from the different komatiite flows in the Hart area. Mineralization and komatiite data for Flow 1 is from sections 2400E and 2450E; Komatiite E is from sections 2700E and 2900E, and Komatiite W is from section 2100E. Mineralization and komatiite data for the Overlying Flows is from section 2900E; Komatiite W is from sections 2450E and 2700E. See Fig. 2 for the location of the sections

δ34S and δ56Fe isotopic profiles through the Main Zone of the basal komatiitic flow along composite sections. a 2400E. b 2450E, c 2900E. Distance from the top of flow is calculated based on the thickness of the flow in sampled drill holes and an approximate dip. See Fig. 2 for the location of the sections
In the second flow, eastern extension mineralization is characterized by positive δ34S values and negative Δ33S values (Fig. 8c). These values trend to mantle values laterally to the west (Fig. 8c), and vertically, both above and below the mineralization (Fig. 9c). Little variability exists in δ56Fe values for samples from komatiite and mineralization in the second flow, with all values within, or near, the mantle range. In the third flow, δ34S and Δ33S values are near 0 ‰, well within the mantle range (Fig. 8d).
Major and trace element geochemistry
The komatiite samples in this study show a wide range of whole-rock compositions that reflects alteration and sulfide accumulation in addition to normal magmatic variability (ESM 1). In order to investigate the latter, komatiite compositions were recalculated on a volatile-free basis and only samples with MgO > 10 %, TiO2 < 1.0 %, 40 % < SiO2 < 58 %, and S < 0.5 % were considered. This procedure eliminates samples with composition strongly influenced by sulfides and high degree of alteration as well as those that do not represent true komatiites (Barnes et al. 2007). On an Al2O3/(2/3–MgO–FeO) versus TiO2/(2/3–MgO–FeO) discrimination diagram utilizing mole proportions and designed to be a projection from olivine (Hanski et al. 2001), the Hart komatiite samples plot in both the Al-depleted (Barberton-type komatiites) and Al-undepleted (Munro-type komatiites) fields (Fig. 10). Note that Sproule et al. (2005) found Tisdale komatiites, such as those at Hart, to be dominantly Munro-type.

Al2O3/(2/3–MgO–FeO) versus TiO2/(2/3–MgO–FeO) discrimination diagram (in mole %) showing chemical affinity of Hart komatiites (after Hanski et al. 2001). Note coexistence of Al-depleted komatiite (Barberton-type) and Al-undepleted komatiite (Munro-type) in the Hart Deposit
In a review of komatiite-associated ores, Barnes and Fiorentini (2012) compared the range of values for a number of trace element ratios normalized to the primitive mantle to constrain which of them displays the largest variability in contaminated rocks. For the Abitibi komatiites, ratios of [Th/Nb]MN and [Zr/Ti]MN most consistently showed the signature of crustal contamination (e.g., values >1), when normalized to the primitive mantle values from McDonough and Sun (1995), even though [Zr/Ti]MN has the smallest range of values and was considered to be the least sensitive of the ratios shown by Barnes and Fiorentini (2012) to indicate contamination. In our study, Th concentrations are commonly below detection limit, so we used La instead, as the [La/Nb]MN ratios have been found to behave similarly to [Th/Nb]MN ratios (Lesher et al. 2001). Most mineralized and barren komatiite samples have [La/Nb]MN and [Zr/Ti]MN ratios above 1, suggesting crustal contamination (Fig. 11), although concentrations of both Zr and Ti are low in our samples, potentially resulting in significant errors.

[La/Nb]MN versus [Zr/Ti]MN plot. Values >1 on both axes are interpreted to represent the signature of contamination by crustal material; see text for further details. Mantle values used for normalization are from McDonough and Sun 1995
Discussion
Since the Δ33S and δ56Fe data are likely to reflect the degree of crustal contamination in the mineralized and barren komatiite samples, these data are compared to the trace element ratios to explore whether any correlation between these two datasets exists (Fig. 12). A weak correlation between the trace element ratios and δ56Fe values (Fig. 12a, b), especially for komatiites, for which Fe budget in the bulk samples is not controlled by sulfide abundances, suggests that non-mantle values for both proxies might be related to crustal contamination. However, when compared to Δ33S values, these trace element ratios exhibit a large range of values, within a limited and consistently negative range of Δ33S values (Fig. 12c, d). These consistently non-mantle Δ33S values formed as a result of crustal contamination suggest a small degree of crustal contamination even in samples that do not have [La/Nb]MN and [Zr/Ti]MN ratios above 1.

Primitive mantle-normalized trace element ratios versus stable isotope values suggesting contamination with crustal materials for mineralization and some komatiite samples. a [La/Nb]MN versus δ56Fe. b [Zr/Ti] MN versus δ56Fe. c [La/Nb]MN versus Δ33S. d [Zr/Ti] MN versus Δ33S. Note that trace element ratios >1 and stable isotope values different from 0 ‰ are considered to indicate crustal contamination. Legend as in Fig. 11
Contaminant composition versus high-temperature fractionations
Prevalent models for the formation of magmatic sulfide deposits suggest that sulfide xenomelt segregates at the base of the magma body during melting and assimilation of the country rock, with an isotopic composition of the original melted material modified by isotope exchange with the magma (Ripley and Li 2003). Consequently, composition of the contaminant material will have some effect on the isotopic values in the sulfide mineralization. However, fractionation processes during melting, crystallization, and isotope exchange with the silicate melt can also account for some variability in δ56Fe values. Experiments conducted by Scheussler et al. (2007) show that pyrrhotite in equilibrium with a peralkaline rhyolite melt have δ56Fe values 0.4 ‰ lower than that of the silicate. However, they suggested that this fractionation factor is dependent on the redox state of the magma and would likely decrease in more reduced ultramafic magmas (Scheussler et al. 2007). Significant range of Fe isotope composition has been also reported in komatiite-associated nickel sulfide deposits and Ni–Cu mineralization in mafic to ultramafic intrusions (δ56Fe = 0.0 ± 0.4 ‰; Bekker et al. 2009; Fiorentini et al. 2012b; Hofmann et al. 2014).
Crystallization of olivine in basalts has been shown to fractionate Fe isotopes with δ56Fe values in olivine 0.1 to 0.3 ‰ lower than in the residual melt (Teng et al. 2008). It is therefore expected that partial melting to produce basaltic magma in the mantle would have a similar effect, with melt δ56Fe values approximately 0.1 ‰ higher than those in the residual mantle materials (Williams et al. 2005; Weyer 2008; Teng et al. 2013). However, a study of Fe isotopes in komatiites has shown that bulk sample δ56Fe values for komatiite are the same as chondritic values (0.044 ‰; Dauphas et al. 2010), or slightly lower (−0.66 ‰; Nebel et al. 2014), suggesting that the high degree of partial melting required to produce komatiite magma minimizes this fractionation effect. Additionally, Dauphas et al. (2010) found no correlation between δ56Fe values and MgO concentrations, indicating that significant fractionation of Fe isotopes did not occur during crystallization of Mg-rich olivine. This implies that high-temperature magmatic fractionation of Fe isotopes in komatiite is minimal, with only small (<1 ‰) fractionation between silicate and sulfide melts in this system. We therefore interpret low δ56Fe values in komatiite (down to −0.9 ‰) and mineralized samples (down to −1.5 ‰) to be due to contamination and isotope exchange with crustal materials, such as exhalite (with δ56Fe values ranging from −2.1 to −0.9 ‰) and graphitic argillite (with δ56Fe values ranging from −2.0 to −1.7 ‰) and unrelated to intrinsic fractionation within the komatiite system between silicate and sulfide melts at high temperatures.
The potential contaminant that could have acted as dominant sulfur source for the Hart deposit has been previously interpreted to be the exhalite unit due to the relative abundance of S in the exhalite compared to the felsic volcanic rocks, and the location of mineralization where the exhalite unit is significantly thinned or entirely removed by thermomechanical erosion (Houlé et al. 2010b). However, the recent discovery of significant concentrations of sulfides in the graphitic argillite unit provides another viable sulfur source for the formation of this deposit. To distinguish between these two potential sulfur sources, we use δ34S, Δ33S, and δ56Fe values to constrain the isotopic signatures of these footwall lithologies for comparison to the signatures observed in the komatiite and associated mineralization.
Two isotopically distinct S pools that formed through photochemical reactions in the Archean oxygen-poor atmosphere have been identified: (1) a reduced pool with positive Δ33S values and (2) an oxidized pool with negative Δ33S values (Farquhar et al. 2002; Ono et al. 2003). The reduced pool is inferred to have reacted with Fe2+ dissolved in the anoxic seawater and precipitated as disseminated Fe sulfide in sediments (Ono et al. 2009; Maynard et al. 2013; Marin-Carbonne et al. 2014). The oxidized pool is thought to have been reduced by bacterial sulfate reduction and incorporated into paleosols on the continents (Maynard et al. 2013), or added to the oceans as dissolved sulfate (Farquhar et al. 2002). Once dissolved in the oceans, sulfate was reduced via bacterial metabolism to form pyrite nodules in organic matter-rich sediments during diagenesis or cycled through submarine hydrothermal systems and was eventually deposited on the ocean floor forming barren massive sulfide lenses distally or volcanogenic massive sulfide deposits proximally to hydrothermal centers (e.g., Bekker et al. 2009). However, the slightly positive δ34S values of the Archean seawater sulfate could be modified by mass-dependent fractionation, for example, via bacterial or thermogenic sulfate reduction. This would produce nodules or layers of sulfides in sediments with consistently negative Δ33S values, but highly variable δ34S values, reflecting S source from dissolved seawater sulfate (cf. Ono et al. 2003, 2009), as seen in the data from the exhalite (Figs. 13 and 14a).

Broadly defined fields of Δ33S and δ34S values for the different volcanic, atmospheric, and seawater S pools in the Archean suggesting that sulfides in exhalite and graphitic argillite originated via bacterial reduction of seawater sulfate (SRB). Purple oval represents composition of mantle-derived volcanic sulfur; blue oval and red circle represent composition of S8 and SO4 2−, respectively, formed by photochemical reactions in the atmosphere and delivered with aerosols to seawater. Orange dashed line represents the most likely range in composition of sulfides formed by sulfate-reducing bacteria that resulted in a horizontal shift in seawater sulfate composition (red oval) to higher δ34S values (fields are after Ono et al. 2003)

Schematic cross-section through the Hart deposit (facing north), showing general trends in isotopic data. Geology is based on drill hole logs and correlation among sampled drill holes
Fe isotope fractionation is thought to be dominantly controlled by redox reactions, with igneous rocks having values between 0.0 and 0.1 ‰, and subsequent weathering and low-temperature, surface reactions resulting in a range of δ56Fe values that depends on the extent, and process of alteration (Rouxel et al. 2003). Circulation of submarine hydrothermal fluids along mid-oceanic ridges results in fluids having negative to 0 ‰ δ56Fe values (as low as −0.9 ‰; Beard et al. 2003; Rouxel et al. 2008; Bennett et al. 2009). As a result, Fe sulfides that precipitate from these hydrothermal fluids have similarly negative δ56Fe values as low as −2 ‰ (Rouxel et al. 2008). Fe oxides and hydroxides that precipitated Fe added to the oceans by hydrothermal fluids form iron formations that tend to have relatively high δ56Fe values (Dauphas et al. 2004; Rouxel et al. 2005; Planavsky et al. 2012; Moeller et al. 2014), although isotopically light compositions have been also reported in oxide-facies BIF (Bekker et al. 2010; Planavsky et al. 2012). The residual Fe in seawater could then precipitate as sulfide with δ56Fe <0 ‰ (Rouxel et al. 2005; Guilbaud et al. 2011).
The negative Δ33S values of both the exhalite and graphitic argillite at the Hart deposit suggest that sulfides in both lithologies formed as a result of bacterial reduction of Archean seawater sulfate (Fig. 13). The Δ33S values of the exhalite and graphitic argillite do not provide an adequate means of distinguishing between these potential sulfur sources for the komatiite sulfides. Additionally, the negative values of δ56Fe in exhalite (average −1.9 ± 0.06 ‰) and graphitic argillite (average −1.8 ± 0.06 ‰) also do not differentiate between these two lithologies. Notably, the average values of δ34S are distinct between exhalite (−2.1 ‰) and graphitic argillite (+3.4 ‰), although the range of values for the exhalite (−11.4 to +7.6 ‰) overlaps with that of the graphitic argillite (+1.6 to +5.0 ‰).
The exhalite is characterized by negative Δ33S values, negative δ56Fe values, and δ34S values that range from positive to negative. The graphitic argillite also has negative Δ33S and δ56Fe values, but always has positive δ34S values. Therefore, consideration of these three isotopic tracers might provide a signature to identify the dominant contaminant that contributed to sulfur saturation in the komatiite at the Hart deposit.
Significance of negative δ56Fe values in exhalite and graphitic argillite
Negative, and highly variable, δ56Fe values are relatively common in sulfides from Archean organic matter-rich sediments (Rouxel et al. 2005). Precipitation of Fe sulfides from Fe2+ dissolved in an aqueous solution has been shown to produce fractionations between −0.3 and −0.9 ‰ in the temperature range of 2 to 40 °C (Butler et al. 2005), although even larger fractionations (−2.5 ‰ between pyrite and Fe2+) have been shown by kinetic experiments (Guilbaud et al. 2011). Under hydrothermal conditions, non-equilibrium Fe isotope fractionation between pyrite in hydrothermal chimneys and hydrothermal fluid has been found to be about −0.9 ‰ (Rouxel et al. 2008). Hence, in order to account for the values observed in this study (−2.0 ‰ in graphitic argillite and −2.1 ‰ in exhalite), an additional pathway is likely required to lower the δ56Fe value of the water prior to precipitation of Fe sulfide. Two mechanisms have been proposed to produce isotopically light Fe2+ in solution, which might be archived in Archean sedimentary rocks: dissimilatory iron reduction (DIR) in pore waters by bacteria (Yamaguchi et al. 2005; Archer and Vance 2006) and reservoir effects (e.g. Rayleigh fractionation) involving the precipitation of isotopically heavy Fe oxides (Rouxel et al. 2005; Planavsky et al. 2012). We infer the latter mechanism for the origin of very low Fe isotope values in the graphitic argillite and exhalite in our study due to the presence of iron formation at the same stratigraphic level as the exhalite in the Hart deposit area.
Isotopic variations within komatiitic flows
Since we have established typical isotopic signature of the two most likely sulfur sources in the vicinity of the Hart deposit, isotope ratios of the mineralization and associated komatiite might identify which of the footwall lithologies was responsible for providing sulfur to form the deposit. The isotope signatures (Δ33S, δ34S, and δ56Fe) observed in komatiites and the associated mineralization at Hart deposit can be clustered into four main groups, each associated with a unique location (Fig. 14).
Barren komatiite of the third flow, which is not associated with significant sulfide mineralization, is characterized by near mantle values of δ34S, δ56Fe, and Δ33S and trace element concentrations showing no evidence of contamination. In contrast, the main zone of mineralization, in the central part of the basal embayment, is characterized by negative Δ33S, δ34S, and δ56Fe values. These values are generally lowest where mineralization is most abundant, gradually increasing toward 0 ‰ above the mineralization and laterally to the west of the main zone (Figs. 8a, b; 9a, b; and 15). This isotopic signature is most consistent with the dominant crustal contaminant for the main zone mineralization being the exhalite.

R factor modeling showing range of R factor between 5 and 100 for massive sulfides, semi-massive sulfides, disseminated sulfides, and barren komatiites based on δ56Fe and Δ33S values (a) and between 5 and 50 based on Ni concentrations and Δ33S values (b)
The eastern extension of sulfide mineralization (Fig. 14) is characterized by negative Δ33S, positive δ34S, and slightly negative to near zero δ56Fe values (Figs. 8c, d; 9c; and 14). This isotopic signature is more characteristic of the graphitic argillite as the dominant crustal contaminant once the effect of isotope exchange is taken into account. As no graphitic argillite has been observed between the first and second komatiite flows, it is likely that the second flow completely assimilated an interflow graphitic argillite unit as a result of thermomechanical erosion. Alternatively, this komatiitic flow may have been in direct contact with a graphitic argillite unit upstream from the present location of the mineralization. Both scenarios are plausible, as thick komatiite flows are known to be capable of extensive thermomechanical erosion, and mineralization within a komatiitic flow could be deposited relatively far from the site of assimilation (Lesher and Campbell 1993; Arndt et al. 2008; Barnes et al. 2013).
An additional isotopic signature is present in the barren komatiite of the basal flow east of the main zone (Fig. 14) where only trace amounts up to ∼3 % of sulfides by volume are present. This pattern is characterized by slightly positive Δ33S and δ34S and δ56Fe values near 0 ‰. Although most of the δ34S values fall within the expected mantle range of approximately 0 ± 2 ‰ (Chaussidon et al. 1989; Labidi et al. 2012, 2013, 2015; Bekker et al. 2015), this represents a definite shift from dominantly negative δ34S values, elsewhere in the basal komatiite flow, to dominantly positive δ34S values in the eastern area. Considering the positive Δ33S values, this may be due to local assimilation of early diagenetic sulfide that had been formed through reduced atmospheric sulfur reacting with Fe present in seawater and precipitating as disseminated Fe sulfide in shale (Marin-Carbonne et al. 2014). This could have resulted in a small amount of assimilated sulfur, insufficient to produce a sulfide xenomelt at the base of the molten komatiite but sufficient to preserve the signature of this local contaminant when sulfur saturation was reached in the late stages of fractional crystallization of the komatiite lava. Alternatively, this could represent the remnant isotopic signature after the majority of komatiite melt and possible sulfide xenomelt have been flushed downstream. Different isotopic signatures of, and correspondingly S sources for, the basal flow mineralization and the zone immediately east of the mineralization emphasize a local S source for minor amounts of mineralization, which might not have exchanged with the rest of the komatiite flow.
Considering the low sulfur content of komatiite in the area to the east of the basal flow mineralization, it seems likely that only assimilation of organic matter-rich shales with pyrite nodules would result in an economic-grade mineralization, unless massive sulfides deposited from submarine hydrothermal fluids were present in the footwall. Both these lithologies would have negative to near zero Δ33S values. The negative Δ33S signature in mineralization and komatiite therefore does indicate presence of favorable environmental conditions and lithologies in the footwall for generation of economic-grade mineralization in komatiites.
R factor and isotope exchange
Silicate to sulfide mass ratio plays important role in controlling the chemical composition and isotopic ratios in sulfide mineralization (Lesher and Burnham 2001; Ripley and Li 2003). Calculations of R factor are based on simple mass-balance equations and can be expressed as mixing models (Lesher and Burnham 2001) or isotope exchange equations (Ripley and Li 2003). As such, we use the following equation from Ripley and Li (2003) for R factor calculations, involving isotopes:
where δm is the isotope ratio of the sulfide product after isotope exchange with the silicate melt, and δ sul and δ sil are the initial isotope ratios of sulfide and silicate melts, respectively, ϵ sul,sil represents the sulfide–silicate fractionation factor at the appropriate temperature, and R 0 = (Csil/Csul) * R, where Csil and Csul are S concentrations in initial silicate and sulfide melts, respectively, and R is the silicate to sulfide mass ratio. For high-temperature magmatic processes, ϵ sul,sil for S isotopes is expected to be close to 0 ‰ (Ripley and Li 2003). For Fe isotopes, however, there may exist a small fractionation between sulfide and silicate melts that might be significant for the range of Fe isotope values observed at the Hart deposit. As already discussed above, Scheussler et al. (2007) found a δ56Fe fractionation factor of 0.38 ‰ between pyrrhotite and silicates for a peralkaline rhyolite melt. They attributed most of the fractionation to redox reactions and suggested that the fractionation may approach 0 ‰ in completely reduced magmas. As such, we use ϵ sul,sil = 0.05 ‰ to allow for a small fractionation at the high magmatic temperatures and reducing conditions thought to exist in komatiite magma. R factors were calculated using Ni concentrations with the magma mixing equation (Lesher and Burnham 2001). In this case, Ni concentrations were recalculated to 100 % sulfide using the method described by Kerr (2003). Since there is a large range of δ34S values in exhalite, only Δ33S and δ56Fe values were used in R factor calculations, as local variations in δ34S values in the footwall would have a large impact on the average value adopted for the sulfide mineralization, complicating R factor calculations. The similar values of Δ33S and δ56Fe for exhalite and graphitic argillite allow the use of a composite value as the initial sulfide value in these calculations. Other values used in our calculations are presented in Table 1.
Many studies of komatiite-associated Ni-sulfide deposits have reported a wide range of R factor values from 10 to >500, calculated mainly through the use of Ni, Cu, and PGE concentrations in the ores (Arndt et al. 2008). Estimates of R factor using δ56Fe and Δ33S tracers produce values that vary from 5 to 250, with the higher R factor values common to disseminated sulfide and weakly mineralized komatiite relative to massive and semi-massive sulfide mineralization (Fig. 15a). Mineralized samples from the eastern extension generally have δ56Fe values close to 0 ‰, compared to the more negative δ56Fe values for the main zone, suggesting a slightly higher R factor for the eastern extension. R factors calculated from Ni concentrations vary from 5 to 50, and overlap with the estimates from the Fe and S isotope systems (Fig. 15b). This study also emphasizes the value of using multiple methods to estimate R factor as proxies such as δ56Fe values and Ni concentrations are more sensitive to small R factor values and show a greater range for the mineralization at the Hart deposit. S isotopes (specifically Δ33S values) are more sensitive to larger R factor values than those observed in mineralization at the Hart deposit. This resulted in a very limited range of Δ33S values observed in the mineralized samples, making R factor estimates based on S isotopes alone very difficult in this case. As an independent estimate, average Pt concentration was used to calculate an R factor based on data from Barnes and Naldrett (1987). This calculation yielded an estimate of 27 for the R factor (Table 1), which is within the range of values generated in this study (5 to 50).
These R factor values are somewhat lower than those for most comparable komatiite-associated Ni-sulfide deposits, although still within the known range for these deposits (10–1100; Arndt et al. 2008). Magmatic systems with low R factors could still produce significant mineralization in an environment with localized contamination and S saturation and little to no transport of sulfide xenomelt. Additionally, such low R factor values would require rapid segregation of the sulfide liquid from the komatiite magma, suggesting that flow of the komatiite melt was not vigorous and at most only weakly turbulent (Lesher and Campbell 1993). These processes could have contributed to a relatively low Ni-tenor in the Hart deposit with only an average of 3.77 % Ni in 100 % sulfide (Barnes and Naldrett 1987).
Conclusions
Through the use of sulfur and iron isotopes, we can distinguish between the two potential crustal sources of contamination for the komatiitic flow, which could have provided sulfur required for the genesis of the mineralization at the Hart deposit. Both the exhalite and graphitic argillite lithologies in the footwall to the deposit are characterized by negative Δ33S and δ56Fe values, suggesting low-temperature, potentially biological processing of both the S and Fe forming the sulfides in these rocks.
The Δ33S and δ34S data suggests different sulfur sources for the main and eastern extension mineralized zones of the Hart deposit. The main zone has sulfur likely derived from the exhalite unit, whereas the eastern extension derived its sulfur from the graphitic argillite unit. A minor local crustal sulfur source with positive Δ33S and δ34S values, possibly a disseminated sulfide in shale, also contributed sulfur to the komatiite, but not in large enough amounts to produce economic sulfide mineralization. Samples from the footwall of the deposit analyzed in this study do not match this local source.
Δ33S, δ34S, and δ56Fe values trend within the flow from the signature of crustal contamination toward mantle values away from the deposits, both laterally and vertically. This trend allows for vectoring toward mineralization within mineralized flows, even from a distance of a few hundred meters from the main mass of mineralization.
R factor calculations, involving Δ33S and δ56Fe values, and Pt and Ni concentrations, suggest a low R factor for the Hart deposit, between 5 and 50. This is lower than the estimates of R factor for several other komatiite-associated deposits and suggests that formation of the deposit resulted from a local contamination and rapid segregation of sulfide xenomelt from the komatiite magma.
References
Archer C, Vance D (2006) Coupled Fe and S isotope evidence for Archean microbial Fe(III) and sulfate reduction. Geology 42:153–156
Arndt NT, Lesher CM, Barnes SJ (2008) Komatiite. Cambridge University Press, New York, p 467
Barnes SJ, Fiorentini ML (2012) Komatiite magmas and sulfide nickel deposits: a comparison of variably endowed Archean terranes. Econ Geol 107:755–780
Barnes SJ, Naldrett AJ (1987) Fractionation of the platinum-group elements and gold in some komatiites of the Abitibi Greenstone Belt, Northern Ontario. Econ Geol 82:165–183
Barnes SJ, Lesher CM, Sproule RA (2007) Geochemistry of komatiites in the Eastern Goldfields superterrane, Western Australia and the Abitibi greenstone belt, Canada, and the implications for the distribution of associated Ni-Cu-PGE deposits. Appl Earth Sci 116:167–187
Barnes SJ, Heggie GJ, Fiorentini ML (2013) Spatial variation in platinum group element concentrations in ore-bearing komatiite at the Long-Victor deposit, Kambalda Dome, Western Australia: enlarging the footprint of nickel sulfide orebodies. Econ Geol 108:913–933
Beard BL, Johnson CM, Skulan JL, Nealson KH, Cox L, Sun H (2003) Application of Fe isotopes to tracing the geochemical and biological cycling of Fe. Chem Geol 195:87–117
Bekker A, Barley ME, Fiorentini ML, Rouxel OJ, Rumble D, Beresford SW (2009) Atmospheric sulfur in Archean komatiite-hosted nickel deposits. Science 326:1086–1089
Bekker A, Slack J, Planavsky N, Krapez B, Hofmann A, Konhauser KO, Rouxel OJ (2010) Iron formation: the sedimentary product of a complex interplay among mantle, tectonic, oceanic, and biospheric processes. Econ Geol 105:467–508
Bekker A, Grokhovskaya TL, Hiebert R, Sharkov EV, Bui TH, Stadnek KR, Chashchin VV, Wing BA (2015) Multiple sulfur isotope and mineralogical constraints on the genesis of Ni-Cu-PGE magmatic sulfide mineralization of the Monchegorsk Igneous Complex, Kola Peninsula, Russia, Mineralium Deposita, published online
Bennett SA, Rouxel O, Schmidt K, Garbe-Schonberg D, Statham PJ, German CR (2009) Iron isotope fractionation in a buoyant hydrothermal plume, 5 degrees S Mid-Atlantic Ridge. Geochim Cosmochim Acta 73:5619–5634
Butler IB, Archer C, Vance D, Oldroyd A, Rickard D (2005) Fe isotope fractionation on FeS formation in ambient aqueous solution. Earth Planet Sci Lett 236:430–442
Campbell IH, Naldrett AJ (1979) The influence of silicate: sulfide ratios on the geochemistry of magmatic sulfides. Econ Geol 75:1503–1506
Chaussidon M, Albarede F, Sheppard SMF (1989) Sulfur isotope heterogeneity in the mantle from ion microprobe measurements of sulfide inclusions in diamonds. Nature 330:242–244
Dauphas N, Rouxel O (2006) Mass spectrometry and natural variations of iron isotopes. Mass Spectrom Rev 25:515–550
Dauphas N, vanZuilen M, Wadhwa M, Davis AM, Marty B, Janney PE (2004) Clues from Fe isotope variations on the origin of early Archean BIFs from Greenland. Science 306:2077–2080
Dauphas N, Teng F-Z, Arndt NT (2010) Magnesium and iron isotopes in 2.7 Ga Alexo komatiites: mantle signatures, no evidence for Soret diffusion, and identification of diffusive transport in zoned olivine. Geochim Cosmochim Acta 74:3274–3291
Farquhar J, Wing BA (2003) Multiple sulfur isotopes and the evolution of the atmosphere. Earth Planet Sci Lett 213:1–13
Farquhar J, Bao H, Thiemens M (2000) Atmospheric influence of Earth’s earliest sulfur cycle. Science 289:756–758
Farquhar J, Wing BA, McKeegan KD, Harris JW, Cartigny P, Thiemens MH (2002) Mass-independent sulfur of inclusions in diamond and sulfur recycling on early earth. Science 298:2369–2372
Fiorentini M, Beresford S, Barley M, Duuring P, Bekker A, Rosengren N, Cas R, Hronsky J (2012a) District to camp controls on the genesis of komatiite-hosted nickel sulfide deposits, Agnew-Wiluna greenstone belt, Western Australia: insights from the multiple sulfur isotopes. Econ Geol 107:781–796
Fiorentini ML, Bekker A, Rouxel O, Wing BA, Maier W, Rumble D (2012b) Multiple sulfur and iron isotope composition of magmatic Ni-Cu-(PGE) sulfide mineralization from Eastern Botswana. Econ Geol 105:107–116
Guilbaud R, Butler IB, Ellam RM (2011) Abiotic pyrite formation produces a large Fe isotope fractionation. Science 332:1548–1551
Habicht KS, Gade M, Thamdrup B, Berg P, Canfield DE (2002) Calibration of the sulfate levels in the Archean ocean. Science 298:2372–2374
Hanski E, Huhma H, Rastas P, Kamenetsky VS (2001) The Palaeoproterozoic komatiite-picrite association of Finnish Lapland. J Petrol 42:855–876
Helt KM, Williams-Jones AE, Clark JR, Wing BA, Wares RP (2014) Constraints on the genesis of the Archean oxidized, intrusion-related Canadian Malartic gold deposit, Quebec, Canada. Econ Geol 109:713–735
Hiebert RS, Bekker A, Wing BA, Rouxel OJ (2013) The role of paragneiss assimilation in the origin of the Voisey’s Bay Ni-Cu sulfide deposit, Labrador: multiple S and Fe isotope evidence. Econ Geol 108:1459–1469
Hofmann A, Bekker A, Dirks P, Gueguen B, Rumble D, Rouxel OJ (2014) Comparing orthomagmatic and hydrothermal mineralization models for komatiite-hosted nickel deposits in Zimbabwe using multiple-sulfur, iron, and nickel isotope data. Mineral Deposita 49:75–100
Houlé MG, Lesher CM (2011) Komatiite-associated Ni-Cu-(PGE) deposits, Abitibi greenstone belt, Superior Province, Canada; in Magmatic Ni-Cu and PGE deposits: geology, geochemistry, and genesis. Society of Economic Geologists. Rev Econ Geol 17:89–121
Houlé MG, Gibson HL, Lesher CM, Davis PC, Cas RAF, Beresford SW, Arndt NT (2008) Komatiitic sills and multigenerational peperite at Dundonald Beach, Abitibi greenstone belt, Ontario: volcanic architecture and nickel sulfide distribution. Econ Geol 103:1269–1284
Houlé MG, Préfontaine S, Fowler AD, Gibson HL (2009) Endogenous growth in channelized komatiite lava flows: evidence from spinifex-textured sills at Pyke Hill and Serpentine Mountain, western Abitibi greenstone belt, northeastern Ontario, Canada. Bull Volcanol 71:881–901
Houlé MG, Lesher CM, Gibson HL, Ayer JA, Hall LAF (2010a) Localization of komatiite-associated Ni-Cu-(PGE) deposits in the Shaw Dome, Abitibi greenstone belt, Superior Province. In: Abstracts, 11th International Platinum Symposium, 21–24 June 2010, Sudbury, Ontario, Canada, Ontario Geological Survey, Miscellaneous Release—Data 269
Houlé MG, Lesher CM, Préfontaine S, Ayer JA, Berger BR, Taranovic V, Davis PC, Atkinson B (2010b) Stratigraphy and physical volcanology of komatiites and associated Ni-Cu-(PGE) mineralization in the western Abitibi greenstone belt, Timmins area, Ontario: a field trip for the 11th International Platinum Symposium; Ontario Geological Survey, Open File Report 6255, p 99
Houlé MG, Lesher CM, Davis PC (2012) Thermomechanical erosion at the Alexo Mine, Abitibi greenstone belt, Ontario: implications for the genesis of komatiite-associated Ni-Cu-(PGE) mineralization. Mineral Deposita 47:105–128
Jamieson JW, Wing BA, Farquhar J, Hannington MD (2013) Neoarchean seawater sulphate concentrations from sulphur isotopes in massive sulphide ore. Nat Geosci 6:61–64
Johnson CM, Beard BL, Beukes NJ, Klein C, O’Leary JM (2003) Ancient geochemical cycling in the Earth as inferred from Fe isotope studies of banded iron formations from the Transvaal Craton. Contrib Mineral Petrol 144:523–547
Kerr A (2003) The calculation and use of sulfide metal contents in the study of magmatic ore deposits: a methodological analysis. Explor Min Geol 10:289–301
Konnunaho JP, Hanski EJ, Bekker A, Halkoaho TAA, Hiebert RS, Wing BA (2013) The Archean komatiite-hosted, PGE-bearing Ni-Cu sulfide deposit at Vaara, eastern Finland: evidence for assimilation of external sulfur and post-depositional desulfurization. Mineral Deposita 48:967–989
Labidi J, Cartigny P, Birck JL, Assayag N, Bourrand JJ (2012) Determination of multiple sulfur isotopes in glasses: a reappraisal of the MORB delta S-34. Chem Geol 334:189–198
Labidi J, Cartigny P, Moreira M (2013) Non-chondritic sulphur isotope composition of the terrestrial mantle. Nature 501:208–211
Labidi J, Cartigny P, Jackson MG (2015) Multiple sulfur isotope composition of oxidized Samoan melts and the implications of a sulfur isotope ‘mantle array’ in chemical geodynamics. Earth Planet Sci Lett 417:28–39
Lesher CM (1989) Komatiite-associated nickel sulfide deposits. In: Whitney JA, Naldrett AJ (eds) Ore deposition associated with magmas. Society of Economic Geologists, Dordrecht, pp 45–102
Lesher CM, Arndt NT (1995) REE and Nd isotope geochemistry, petrogenesis and volcanic evolution of contaminated komatiites at Kambalda, Western Australia. Lithos 34:127–157
Lesher CM, Burnham OM (2001) Multicomponent elemental and isotopic mixing in Ni-Cu-(PGE) ores at Kambalda, Western Australia. Can Mineral 39:421–446
Lesher CM, Campbell IH (1993) Geochemical and fluid dynamic modeling of compositional variations in Archean komatiite-hosted nickel sulfide ores in Western Australia. Econ Geol 88:804–816
Lesher CM, Groves DI (1986) Controls on the formation of komatiite-associated nickel-copper sulfide deposits: geology and metallogenesis of copper deposits; Proceedings of the Twenty-Seventh International Geological Congress, Berlin, Springer Verlag, pp 43–62
Lesher CM, Keays RR (2002) Komatiite-associated Ni–Cu–(PGE) deposits: mineralogy, geochemistry, and genesis. In: Cabri LJ (ed) The geology, geochemistry, mineralogy, and mineral beneficiation of the platinum-group elements. Canadian Institute of Mining, Metallurgy and Petroleum, special volume 54. Canadian Institute of Mining, Metallurgy and Petroleum, Westmount, pp 579–617
Lesher CM, Burnham OM, Keays RR (2001) Trace-element geochemistry and petrogenesis of barren and ore-associated komatiites. Can Mineral 39:673–696
Marin-Carbonne J, Rollion-Bard C, Bekker A, Rouxel O, Agangi A, Cavalazzi B, Wohlgemuth-Ueberwasser CC (2014) Coupled Fe and S isotope variations in pyrite nodules from Archean shale. Earth Planet Sci Lett 392:67–79
Mavrogenes JA, O’Neill HSC (1999) The relative effects of pressure, temperature and oxygen fugacity on the solubility of sulfide in mafic magmas. Geochim Cosmochim Acta 63:1173–1180
Maynard JB, Sutton SJ, Rumble D III, Bekker A (2013) Mass-independently fractionated sulfur in Archean paleosols: a large reservoir of negative Δ33S anomaly on the early Earth. Chem Geol 362:74–81
McDonough WF, Sun SS (1995) The composition of the Earth. Chem Geol 120:223–253
Moeller K, Schoenberg R, Grenne T, Thorseth IH, Drost K, Pedersen RB (2014) Comparison of iron isotope variations in modern and Ordovician siliceous Fe oxyhydroxide deposits. Geochim Cosmochim Acta 126:422–440
Nebel O, Campbell IH, Sossi PA, Van Kranendonk MJ (2014) Hafnium and iron isotopes in early Archean komatiites record a plume-driven convection cycle in the Hadean Earth. Earth Planet Sci Lett 397:111–120
Ono S, Eigenbrode JL, Pavlov AA, Kharecha P, Rumble D III, Kasting JF, Freeman KH (2003) New insights into Archean sulfur cycle from mass-independent sulfur isotope records from the Hamersley Basin, Australia. Earth Planet Sci Lett 213:15–30
Ono S, Beukes NJ, Rumble D (2009) Origin of two distinct multiple-sulfur isotope compositions of pyrite in the 2.5 Ga Klein Naute Formation, Griqualand West Basin, South Africa. Precambrian Res 169:48–57
Planavsky N, Rouxel OJ, Bekker A, Hofmann A, Little CTS, Lyons TW (2012) Iron isotope composition of some Archean and Proterozoic iron formations. Geochim Cosmochim Acta 80:158–169
Pyke DR, Naldrett AJ, Eckstrand OR (1973) Archean ultramafic flows in Munro Township, Ontario. Geol Soc Am Bull 84:955–978
Ripley EM (1999) Systematics of sulphur and oxygen isotopes in mafic igneous rocks and Cu-Ni-PGE mineralization; in Dynamic processes in magmatic ore deposits and their application in mineral exploration. Geological Association of Canada, Short Course Notes, 13:111–158
Ripley EM, Li C (2003) Sulfur isotope exchange and metal enrichment in the formation of magmatic Cu-Ni-(PGE) deposits. Econ Geol 98:635–641
Rouxel O, Dobbek N, Ludden J, Fouquet Y (2003) Iron isotope fractionation during oceanic crust alteration. Chem Geol 202:155–182
Rouxel OJ, Bekker A, Edwards KJ (2005) Iron isotope constraints of the Archean and Paleoproterozoic ocean redox state. Science 307:1088–1091
Rouxel O, Shanks Iii WC, Bach W, Edwards KJ (2008) Integrated Fe- and S-isotope study of seafloor hydrothermal vents at East Pacific Rise 9-10°N. Chem Geol 252:214–227
Scheussler JA, Schoenberg R, Behrens H, von Blanckenburg F (2007) The experimental calibration of the iron isotope fractionation factor between pyrrhotite and peralkaline rhyolitic melt. Geochim Cosmochim Acta 71:417–433
Sharman ER, Taylor BE, Minarik WG, Dubé B, Wing BA (2014) Sulfur isotope and trace element data from ore sulfides in the Noranda district (Abitibi, Canada): implications for volcanogenic massive sulfide deposit genesis. Mineral Deposita 50:1–18
Sproule RA, Lesher CM, Houlé MG, Keays RR, Ayer JA, Thurston PC (2005) Chalcophile element geochemistry and metallogenesis of komatiitic rocks in the Abitibi greenstone belt, Canada. Econ Geol 100:1169–1190
Teng F-Z, Dauphas N, Helz RT (2008) Iron isotope fractionation during magmatic differentiation in Kilauea Iki lava lake. Science 320:1620–1623
Teng F-Z, Dauphas N, Huang S, Marty B (2013) Iron isotope systematics of oceanic basalts. Geochim Cosmochim Acta 107:12–26
Thurston PC (2008) Depositional gaps in Abitibi greenstone belt stratigraphy: a key to exploration for syngenetic mineralization. Econ Geol 103:1097–1134
Wendlandt RF (1982) Sulfide saturation of basalt and andesite melts at high pressures and temperatures. Am Mineral 67:877–885
Weyer S (2008) What drives iron isotope fractionation in magma? Science 320:1600–1601
Williams HM, Peslier AH, McCammon C, Halliday AN, Levasseur S, Teutsch N, Burg J-P (2005) Systematic iron isotope variations in mantle rocks and minerals: the effects of partial melting and oxygen fugacity. Earth Planet Sci Lett 235:435–452
Wing BA, Halevy I (2014) Intracellular metabolite levels shape sulfur isotope fractionation during microbial sulfate respiration. Proc Natl Acad Sci 111:18116–18125
Yamaguchi KE, Johnson CM, Beard BL, Ohmoto H (2005) Biogeochemical cycling of iron in the Archean-Paleoproterozoic Earth: Constraints from iron isotope variations in sedimentary rocks from the Kaapvaal and Pilbara Cratons. Chem Geol 218:135–169
Acknowledgments
We would like to express our appreciation to Northern Sun Mining Corp. (formerly Liberty Mines Ltd.) for their logistical support, access to properties, information, and discussions with staff throughout this project. Discussions with, and advice from, Dr. C. Michael Lesher (Laurentian University) during the course of this study are greatly appreciated. We also greatly appreciate the helpful reviews by S.J. Barnes and M. Fiorentini, whose comments significantly added to this contribution, and editorial suggestions by Georges Beaudoin. Financial support for this project has been provided by the Targeted Geoscience Initiative 4 of the Geological Survey of Canada and Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery and Accelerator Grants to AB. OR acknowledges the technical support of Y. Germain and E. Ponzevera (Ifremer) and funding from LabexMer (ANR-10-LABX-19-01). The McGill Stable Isotope Laboratory is supported by NSERC through Research Tools and Infrastructure and Discovery Grants to BAW as well as operating funds from FQRNT through the GEOTOP research center. We thank Thi Hao Bui for technical assistance in the McGill Stable Isotope Laboratory.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: M. Fiorentini and G. Beaudoin
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(XLSX 32 kb)
Rights and permissions
About this article

Cite this article
Hiebert, R.S., Bekker, A., Houlé, M.G. et al. Tracing sources of crustal contamination using multiple S and Fe isotopes in the Hart komatiite-associated Ni–Cu–PGE sulfide deposit, Abitibi greenstone belt, Ontario, Canada. Miner Deposita 51, 919–935 (2016). https://doi.org/10.1007/s00126-016-0644-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00126-016-0644-1