
Abstract
The petrology of base metal sulfides and associated accessory minerals in rocks away from economically significant ore zones such as the Merensky Reef of the Bushveld Complex has previously received only scant attention, yet this information is critical in the evaluation of models for the formation of Bushveld-type platinum-group element (PGE) deposits. Trace sulfide minerals, primarily pyrite, pyrrhotite, pentlandite, and chalcopyrite are generally less than 100 microns in size, and occur as disseminated interstitial individual grains, as polyphase assemblages, and less commonly as inclusions in pyroxene, plagioclase, and olivine. Pyrite after pyrrhotite is commonly associated with low temperature greenschist alteration haloes around sulfide grains. Pyrrhotite hosted by Cr- and Ti-poor magnetite (Fe3O4) occurs in several samples from the Marginal to Lower Critical Zones below the platiniferous Merensky Reef. These grains occur with calcite that is in textural equilibrium with the igneous silicate minerals, occur with Cl-rich apatite, and are interpreted as resulting from high temperature sulfur loss during degassing of interstitial liquid. A quantitative model demonstrates how many of the first-order features of the Bushveld ore metal distribution could have developed by vapor refining of the crystal pile by chloride–carbonate-rich fluids during which sulfur and sulfide are continuously recycled, with sulfur moving from the interior of the crystal pile to the top during vapor degassing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The current debate on the origin of the major platinum-group element (PGE) deposits of the Bushveld, Stillwater, and other layered intrusions is unresolved; one group argues that the ores form by contamination, magma mixing, and sulfide saturation models (e.g., Campbell et al. 1982; Cawthorn 1999; Naldrett et al. 2009); another group suggests that the PGE and S may have been scavenged by upward-percolating fluids moving through the underlying solidifying crystal pile (e.g., Stumpfl and Tarkian 1976; Ballhaus and Stumpfl 1985, 1986; Boudreau and McCallum 1992; Willmore et al. 2000). Much of this debate has focused on a detailed understanding of the petrographic, textural, and geochemical features of the ore zones themselves or their immediately surrounding environment. Away from the ore zones, most studies have attempted to use bulk rock analyses of S, base and noble metals, and other elements to infer whether or not the magma was sulfide-saturated at the time these rocks were crystallizing (e.g., Maier and Barnes 1999; Barnes and Maier 2002; Barnes et al. 2009). With few exceptions (most notably, Liebenberg 1970) the petrography of the sulfide assemblages and the accessory minerals with which they may be associated is rarely described, and even in these cases the authors focus on the more sulfur-rich parts of the Bushveld Complex (e.g., Von Gruenewaldt 1976; Li et al. 2004).
Petrographic studies of the accessory sulfides and associated minerals in the rocks stratigraphically above and below the ore zones have the potential to elucidate when the magma became sulfide saturated or if sulfur and the other ore elements had been remobilized from these rocks (e.g., Larocque et al. 2000; Andersen 2006). This study reports a survey of the base-metal sulfide and associated mineral assemblages in the rocks that make up the bulk of the Bushveld Complex, stratigraphically away from the ore zones. This work also presents evidence for the original presence of a reactive high temperature chloride–carbonate fluid in the rocks beneath the Merensky Reef.
Finally, a vapor-refining model is presented which explains many of the first-order features of the PGE-sulfide distribution of the Lower to Main Zones of the Bushveld Complex. We have previously suggested that metal transport during degassing of a solidifying crystal pile can lead to the formation of PGE-rich rocks by a chromatographic/zone refining process (e.g., Meurer et al. 1998; Boudreau and Meurer 1999; Boudreau 2004). This earlier work mainly looked at the effect of chromatographic effects as S and the ore elements were moved entirely within the crystal pile as the pile underwent degassing. Interaction of fluid with the overlying magma column was suggested but not explicitly accounted for. In addition, the fluid was ~100 % effective at removing the ore elements if all sulfide was lost as the crystal pile degassed. This new modeling allows some ore components to remain behind. It also allows for fluid to drive sulfide saturation in the lower part of the magma column above a vapor-saturated crystal pile. This new model more accurately reproduces the ore element stratigraphic distribution, particularly S and Pd.
Geology and previous work
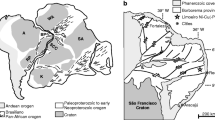
The Bushveld Complex in the Republic of South Africa is the most significant source of platinum in the world. As summarized by Eales and Cawthorn (1996, and references therein), the layered mafic–ultramafic intrusion formed 2,054 Ma ago and comprises approximately 66,000 km2 of the northern Kaapvaal craton. The complex crops out as three distinct regions: the Western, Northern, and Eastern lobes. The intrusion is 7–9 km thick and is also referred to as the Rustenburg Layered Suite. It is subdivided into five distinct zones from bottom to top based mainly on major changes in mineral modes (Fig. 1): basal Marginal Zone, Lower Zone, Critical Zone, Main Zone, and Upper Zone. The Marginal Zone (MR) is developed as a chilled basal norite, and is overlain by the ~1,200-m-thick Lower Zone (LZ), which consists of mainly of orthopyroxenite and harzburgite. This followed by the ~300-m-thick Lower Critical Zone (LCZ), which consists predominantly of bronzitite and hosts the lower group of chromitites, and the overlying ~750-m-thick Upper Critical Zone (UCZ), which consists mainly of norites anorthosites and the upper group of chromitites.

General geologic setting and stratigraphic subdivisions of the Bushveld Complex, South Africa
The Main Zone is ~3,300 m thick and is made up largely of gabbronorite. Two marker horizons, the Upper Mottled Anorthosite and the Pyroxenite Marker, subdivide the Main Zone into subzones A, B, and C (Von Gruenewaldt 1973). The reappearance of primary orthopyroxene at the Pyroxenite Marker coincides with a significant break in most geochemical parameters, including the initial 87Sr/86Sr ratio (Sharpe 1985), which Kruger et al. (1987) proposed is the true magmatic boundary between the Main and Upper Zones.
The ~1,500-m-thick Upper Zone consists mainly of ferrogabbro, anorthosite, and interlayered magnetitites. Iron-rich olivine and apatite appear in the upper part of this sequence (Von Gruenewaldt 1973). The rocks of the Upper Zone also contain a considerable quantity of base metal sulfides (Liebenberg 1970; Von Gruenewaldt 1976).
Although sulfides occur in minor quantities throughout all of the layered mafic rocks of the complex, above-average concentrations are restricted to a few layers or areas. Of these, three very extensive deposits are of economic importance: the Merensky Reef and the UG2 chromitite layer, both of which occur towards the top of the Upper Critical Zone, and the Platreef in the Potgietersrus area. Liebenberg (1970) considered that variations in the S content of the mafic magma were responsible for varying concentration of sulfides. He presented evidence for a gradual decrease in the S content until it became so low that a mainly Cu sulfide immiscible liquid formed during the crystallization of the norite and anorthosite below the Merensky Reef. The Cu content was insufficient to remove all the sulfur with the result that the sulfur content of the magma gradually increased.
As noted by Barnes et al. (2009), bulk rock sulfur in barren rocks of the Bushveld Complex reaches its highest abundance in the Upper Zone, in some cases exceeding 1.0 wt.%. In a section from the base of the Subzone A to Subzone D of the Upper Zone, Von Gruenewaldt (1976) found that pyrrhotite increased from ~55 % to 95 % as a percentage of the of the sulfide minerals whereas chalcopyrite decreased from 40 % to 3 % and pentlandite decreased from 6 to <1 % toward the top of the Upper Zone. He attributed the abundance of sulfide in the Upper Zone as compared with the Main Zones as being due a number of processes, including iron enrichment in the magma.
Apart from those rocks with economically viable concentrations, the data of Maier and Barnes (1999) and show that all of the rocks in the lower part of the Bushveld Complex are unusually enriched in PGE. As summarized by Naldrett et al. (2009), the median values for Pt and Pd, respectively, are 78 and 21 ppb in the Upper Critical Zone, 27 and 14 ppb in the Lower Critical Zone, and 10 and 12 ppb in the Lower Zone. These values contrast with median values of 5 ppb Pt and 4 ppb Pd in the Main Zone. They are also higher than the parent (marginal and sill rocks) magma concentrations of ~30 ppb combined Pt + Pd reported by Davies and Tredoux (1985). Furthermore, Barnes et al. (2009) interpret the rocks below the Merensky Reef of the Bushveld Complex to have a “cumulus” sulfide component based on moderate enrichments in the PGE, although S contents are too low for the magma to have been S saturated. They also note that magmatic Se/S ratios preclude low temperature loss of S, and they suggested three possible models to explain the contradiction: (1) migration of sulfide out of the crystal pile, (2) removal of sulfide suspended in a sulfide-saturated magma prior to emplacement, and (3) loss of S and Se to a high temperature degassing event.
Methods
Because the focus of the study was on complex-wide processes, sampling concentrated on typical Bushveld rocks that make up the bulk of the complex. Approximately two hundred surface hand samples and drill cores were collected throughout the Bushveld Complex, particularly concentrated on the Marginal to Main Zones. A sample of a marginal sill type B2 (Sharpe and Hulbert 1985) was also investigated. A total of 50 samples from this collection were selected to characterize the phase assemblages present in this barren stratigraphy. These were chosen to be free of observable low temperature alteration in hand sample.
Because of the small size of many of the sulfide grains, sulfide identification was undertaken by compound petrographic microscope and electron microprobe. Compositional data on base metal sulfides, both by microanalyses of minerals and X-ray composition mapping, was completed using a Cameca CAMEBAX electron microprobe using a 4Pi energy dispersive X-ray spectrometer (later upgraded to a silicon drift detector) at the Department of Earth and Ocean Sciences at Duke University. The analytical conditions were 15–20 keV accelerating voltage and 15 nA beam current. Counting times were approximately 120 s using a focused to 10 μm beam diameter. Pure metal standards were used for iron, nickel, copper, cobalt, and zinc. In-house natural mineral standards include PY-1 (FeS2) for sulfur, ASPY (FeAsS) for arsenic, STBW (Sb2S3) for antimony, and OLV74 (Mg,Fe)2SiO4 for silicon and oxygen.
Petrology and geochemistry of the sulfide assemblages
Early-crystallized igneous minerals such as olivine, orthopyroxene, clinopyroxene, and plagioclase are the dominant silicate minerals, and chromite and magnetite are locally common accessory oxide phases. Late-crystallized anhydrous minerals such as quartz and orthoclase are present as minor interstitial minerals. High temperature volatile-bearing minerals, which typically amount to <1 % (but in some samples can be about 1 %), including biotite, apatite, and calcite, the latter of which is discussed below. The major disseminated sulfides occurring throughout the complex are pyrite (Py–FeS2), pyrrhotite (Po–FeS), pentlandite (Pn–(Fe,Ni)9S8), and chalcopyrite (Ccp–CuFeS2), whereas millerite (NiS), siegenite (Ni,Co)3S4, and possibly troilite (FeS) are more rarely observed. Examples of sulfide associations are illustrated in Fig. 2.

Three examples of base metal sulfide assemblages of the Bushveld Complex. From left to right, the samples are shown in plane polarized light, crossed polarizers, plane polarized reflected light, and a composite X-ray composition map (512 × 512 pixel resolution). Abbreviations: Bt biotite, Cc calcite, Ccp chalcopyrite, Mt magnetite, Opx orthopyroxene, Pl plagioclase, Po pyrrhotite, Pn pentlandite, q quartz. In the X-ray maps, yellow is S (defining sulfides), blue is Ca (defining calcite and/or clinozoisite), white is Si (defining quartz); other mixed colored minerals are as labeled. Sample LZ 216 (Lower Zone orthopyroxenite): intergrown Ccp + Pn interstitial to pristine grains of orthopyroxene. Sample 45-74 (Main Zone gabbro): Po + Py + Pn with interstitial quartz and biotite. Note minor alteration in the orthopyroxene and the calcite intergrown with the sulfide assemblage evident in the reflected light and composite X-ray map. Sample 06-168 (Main Zone gabbro): Py with alteration halo of mainly very fine-grained clinozoisite, quartz, and disseminated Ccp grains in surrounding plagioclase
Where fresh, single-, and poly-phase sulfide grains occur as isolated interstitial grains at the contacts between unaltered grains of early-crystallizing plagioclase, orthopyroxene, clinopyroxene, and olivine, and more rarely as inclusions near the margins of the same minerals (e.g., sample LZ 216 in Fig. 2). Grains comprising a single mineral species can also occur as inclusions along cleavage planes and fractures of fresh igneous minerals. The shape of individual grains are typically subhedral and the sizes vary from 0.1 to 100 μm. Single grains of sulfide and polyphase assemblages also commonly occur with late-crystallizing quartz, biotite, apatite, and locally calcite (e.g., sample 45-74 in Fig. 2).
Although all of the samples were chosen to be as fresh as possible, under the microscope almost all samples show some degree of localized, low temperature alteration that is typically developed as a halo of alteration in the silicate phases surrounding the sulfide minerals (e.g., sample 06-168 in Fig. 2). The alteration assemblage, where present, consists of typical greenschist assemblages including talc, tremolite, epidote/clinozoisite, and chlorite, and is similar to that described in the Merensky Reef by Li et al. (2004). While larger sulfide grains that form the core of these halos are similar in size to those in unaltered areas of the same sample, finely disseminated sulfides can occur throughout the alteration halo. Sulfide shapes are otherwise variable and range from anhedral to euhedral. This association of sulfide with variably developed halos of greenschist alteration is generally more common than are pristine assemblages.
Pyrite is commonly associated with more extensive alteration, indicating that it is a replacement of high temperature sulfides. However, regions where alteration is absent or minor may also have pyrite as part of the sulfide assemblage. This leads to some uncertainty as to whether or not pyrite is part of the original assemblage developed during crystallization and cooling of a high temperature sulfide solution or if it formed subsequently (e.g., compare samples 45-74 and 06-168 in Fig. 2).
An association of very fine-grained pyrrhotite (<1 μm) hosted by magnetite (Mt) occurs in some samples from the Marginal, Lower, and Lower Critical Zones (Fig. 3). Both pyrrhotite and magnetite commonly occur with calcite; the calcite has an apparent equilibrium texture with the surrounding igneous silicate minerals (pyroxene and plagioclase). The rocks also contain up to 1–2 % biotite, much higher than the zero to trace amounts more commonly observed. The presence of magnetite as part of the sulfide mineral assemblage crystallizing or exsolving from a high temperature immiscible sulfide liquid is not uncommon in igneous rocks (Naldrett 2004). For example, magnetite is observed in sulfide globules in MORB (Mathez 1976). However, in the case of the MORB globules, the magnetite is relatively minor (~8 %) and is hosted by modally dominant S-rich phases, usually pyrrhotite. In the Bushveld samples, the magnetite may comprise 50 % or more of the Po–Mt assemblage. In this regard, the mineralogy and textures of the Fe–S–O assemblage are more similar to that observed in a variety of igneous rocks as summarized by Larocque et al. (2000), who interpreted it to be the result of preferential S loss during open system magma degassing or circulating high temperature hydrothermal fluids.

Example of pyrrhotite-magnetite-calcite assemblages in a leuco-orthopyroxenite from the Lower Critical Zone of the Bushveld Complex (sample LCZ-105). Abbreviations: Opx orthopyroxene, Cpx clinopyroxene, Cc calcite, Po pyrrhotite, Mt magnetite, Pl plagioclase, p plucked region. a Plane polarize light. b Crossed polarizers. c Reflected polarized light showing detail of pyrrhotite (bright phase) hosted by magnetite (slightly darker gray phase) associated with the larger calcite grain shown in a and b. d Composite X-ray phase map (1,024 × 1,024 pixel resolution) for which red is Mg (defining opx), gray is Al (defining plagioclase), blue is Ca (defining calcite), green is Fe (defining magnetite), and yellow is S (defining pyrrhotite). Note the unaltered nature of silicates surrounding the two calcite grains
Qualitatively, the amount of sulfide observed in the stratigraphic section is consistent with the whole rock S abundance reported by others (e.g., Barnes et al. 2009). For example, in the generally S-poor Critical Zone, only a few grains of base metal sulfides, typically no larger than 10–100 microns in size, are present in each thin section, and many sections from the Critical Zone do not contain any sulfide. Attempts to quantify modal proportions of the sulfide minerals were not made as they would likely be strongly biased by the paucity of sulfide grains and the nugget effect. However, it is qualitatively observed that chalcopyrite is the most commonly observed sulfide; it is found in 31 of the 50 samples, although other sulfide minerals may be locally dominant (ESM Table 1). Pyrrhotite is the second most commonly observed sulfide, found in 18 of the 50 samples. Pyrite is found mainly in the Marginal and Main Zones, and less commonly in the Upper and Lower Critical Zones. The most common sulfide association is pyrrhotite intergrown with chalcopyrite and/or pentlandite along the margins. Intergrowths of two or more of the four sulfide minerals Py + Po + Ccp + Pn are found in every zone.
Stratigraphic trends
Stratigraphic trends in the major base metal sulfide assemblages are shown in Fig. 4. The results of this study broadly agree with the major sulfide minerals and modes described by Liebenberg (1970). The samples of the Marginal Zone at the base of the intrusion are primarily composed of orthopyroxenite and melanorite. Bent and broken pyroxenes and plagioclase grains are common. Biotite, olivine, and chromite are locally present as accessory interstitial phases. Sulfide grains are typically anhedral to subhedral and very fine- to fine-grained in size. Alteration is common in sample MR 98, where the sulfide assemblages are clustered as rims wrapping around subhedral–euhedral altered plagioclase crystals. Most of the sulfide crystals in the Marginal Zone (sample MR 77, MR 82, MR 86, MR 96, MR 97) are observed as interstitial minerals at the contact zone of fresh primary igneous minerals. A few crystals of chalcopyrite in MR 77 and MR 97 are present as inclusions. Chalcopyrite is the most abundant sulfide, whereas pentlandite is the least. Several pyrrhotite crystals in sample MR 86 and MR 77 are intergrown with grey magnetite grains as interstitial phases between orthopyroxene and plagioclase. Very fine grained (<1 μm) pyrrhotite crystals hosted by magnetite are locally present.

Distribution of base metal sulfides and composition of iron sulfides (pyrrhotite and pyrite) and associated magnetite in the different zones of the Bushveld Complex, comparing to Cl-end member concentration of apatite (Willmore et al. 2000, and references therein) and bulk rock S and Pt concentrations (Barnes et al. 2009). Also shown are the relative sulfide mineral percentages of the sulfide assemblage (after Liebenberg 1970). Stratigraphic section is that of Von Gruenewaldt et al. (1985)
In the rocks transitional from melanorites of the Marginal Zone to the ultramafic rocks of the Lower Zone, equigranular orthopyroxenite and harzburgite become the main rock types sampled. An- to subhedral grains of orthopyroxene and olivine are the predominant minerals, whereas the plagioclase content (5 % or less) is negligible. Biotite and quartz are locally abundant as interstitial minerals. Anhedral to euhedral chromite grains are present in samples LZ 214, LZ 216, 13P, and C6. Sulfide minerals in this section of the stratigraphy are largely present as interstitial phases at the contacts between fresh primary igneous minerals. Pentlandite is the most abundant sulfide and pyrrhotite is the second. A few of the pyrrhotite crystals in sample LZ 216 and LZ 222 are also intergrown with magnetite grains as described above. The shape of sulfides is mostly subhedral and the size is smaller (typically <100 μm) than those in the Marginal Zone.
Equigranular orthopyroxenite and mafic norite samples of the Lower Critical Zone contain slightly more plagioclase than the Lower Zone. Biotite is present in abundances as high as 1 %, whereas the olivine content is present in only trace amounts. On the other hand, chromite becomes abundant in this zone (samples 7, 6R, 5, 5P2, and 8P). Chalcopyrite is the most commonly observed sulfide and no pentlandite was observed. Micron-scale pyrrhotite grains are intergrown with fine magnetite ± calcite in samples LCZ 101, LCZ 105, and LCZ 133. A few very fine-grained pyrite and chalcopyrite crystals occur as inclusions in orthopyroxene and plagioclase, typically near the margins. However, no sulfide minerals are observed in samples 7, 6R, 5, 5P2, 4R2, 4P, and 8P. The paucity of sulfur in chromitite has been noted by Maier and Barnes (1999); the absence of sulfides in chromitite-bearing rocks of the current study indicates that this feature extend to rocks that have higher than normal amounts of chromitite.
Plagioclase-rich rocks such as anorthosite and norite dominate in the sampling of the Upper Critical Zone. Melanorite and orthopyroxenite are also present. Orthopyroxene and clinopyroxene contents drastically drop relative to those in the previously described zones. Biotite, olivine, and quartz are rarely observed accessory/interstitial minerals. The rocks range from equigranular, spotted to mottled anorthosite and norite. The primary igneous minerals are generally medium grained, although some are coarse-grained. Chalcopyrite is the most commonly observed sulfide and is largely interstitial to (now altered) plagioclase and pyroxenes; a few are observed as inclusions in altered primary igneous silicate minerals. The paucity of sulfide minerals other than chalcopyrite in this section was also noted by Liebenberg (1970). Chromite crystals are rarely observed and no intergrowths between magnetite and pyrrhotite were found. Pentlandite was observed in only one sample, continuing the Ni-poor nature of the assemblage in the Lower Critical Zone.
Samples from the Main Zone are mostly composed of gabbronorite, melano- and leuconorite, and anorthosite. Plagioclase, orthopyroxene, and clinopyroxene are present in all samples with minor olivine and biotite scattered throughout the zone. The sulfide assemblages of samples 45-74 and 06-168 are associated with variable amounts of alteration in the surrounding primary igneous minerals. Chalcopyrite and pentlandite are common sulfide minerals and pyrrhotite and pyrite may occur together. The sulfide crystals are generally very fine-grained to fine-grained with subhedral to euhedral shape. The sulfide assemblages occur as both interstitial minerals and inclusions in fresh igneous silicate minerals.
The Upper Zone is dominated by plagioclase-rich rock such as gabbro and norite. Except near the very top of the intrusion, base-metal sulfide minerals are generally scarce in the small number of thin sections studied. Pyrrhotite is the major sulfide, occurring as in interstitial grains and as inclusions in altered primary igneous minerals and magnetite. The grains are typically fine-grained and anhedral to subhedral in shape. The sulfides of this section have been well described by Von Gruenewaldt (1976).
Sulfide mineral compositions
Selected microprobe analyses of sulfide minerals are listed in ESM Tables 2 and 3 and are plotted in Figs. 4 and 5. Measured variations in molar Fe–S in pyrrhotite ranges from 0.77 to 1.02 (Fig. 3), with less than 1 wt.% Ni. Some compositions approach troilite (Fe–S ≈ 1), but in part this may also reflect other elements substituting for Fe. The mole ratio of Fe–S in pyrite ranges from (0.74–1.04):2, but generally is close to stoichiometric FeS2 (Fig. 3). Within the scatter of the data, there are no stratigraphic trends observed in pyrrhotite or pyrite compositions. Chalcopyrite also shows no systematic stratigraphic trends, with Cu–Fe–S ranging from 1:1:2 to (0.62–1.10):(0.91–1.37):2 (Fig. 5). Pentlandite (ideally (Fe,Ni)9S8) compositions are largely variable with (Fe + Ni):S ratios ranging from 9:8 to (6.58-10.76):8 There is a suggestion that Ni decreases and Fe increases in pentlandite up section (Fig. 5), but the trends are obscured by local scatter. Cobalt may also substitute for iron.

Stratigraphic variation in the molar Fe/S and Cu/S ratios of chalcopyrite (left column) and the molar Fe/S and Ni/S ratios of pentlandite (right column) from the Bushveld Complex
Analyses of the pyrrhotite + magnetite mixed phase in sample LCZ-105 from the Lower Critical Zone are listed in ESM Table 3. The intergrowth is too fine grained to resolve the different minerals using the electron microprobe, thus the analysis are mixtures of Po + Mt. There is no significant Ni, Ti, or Cr, but a few analyses contain more than 1 wt.% Cu. With the exception of minor Cu, the bulk composition can be approximated as a simple mixture of Po + Mt, as shown in ESM Table 3.
Discussion
Bushveld base metal sulfides are dominated by pyrrhotite, pyrite, chalcopyrite, and pentlandite (Liebenberg 1970). There is also a general trend of decreasing pentlandite/chalcopyrite modal ratio up to the level of the Merensky Reef. Pentlandite again becomes common in the Lower Main Zone, but is then rare or absent in the upper half of the Main Zone and in the Upper Zone. Locally, the sulfides show two main types of alteration (1) an apparently relatively high temperature alteration of pyrrhotite to magnetite and (2) a lower temperature alteration of pyrrhotite to pyrite during localized greenschist-grade hydration events. The former appears to be more common in rocks generally below the Merensky Reef, whereas the latter may occur in any rock. These are further discussed below.
Overall the types of minerals observed and the range in sulfide mineral compositions are similar to those reported in mid-ocean ridge gabbro (e.g., Miller and Cervantes 2002), except that pyrite is more common and troilite less common in the Bushveld suite. In contrast, the sulfide assemblages are markedly different from those of the Skaergaard Intrusion. Although both the Bushveld and the Skaergaard are shallow crustal intrusions, the Skaergaard is much smaller intrusion and was emplaced as a single injection of magma, whereas the Bushveld is much larger and was most likely emplaced as a series of multiple injections (e.g., Cawthorn 1998). Wager et al. (1957, 1958) note that sulfide minerals in the Skaergaard Layered Series range from a Cu-rich assemblage dominated by bornite, digenite, and chalcocite with minor chalcopyrite; at the very top of the Layered Series the assemblage is Fe-rich and dominated by pyrrhotite. They explained the Cu-rich and Ni-poor composition of the sulfide assemblage by late-stage sulfide saturation and segregation.
Miller and Andersen (2002) suggest that the differences between intrusions (such as the Bushveld and Skaergaard intrusions) are largely a result of different liquid lines of descent of the parent magmas. Crystallization of olivine and magnetite removes Ni prior to sulfide saturation, whereas Cu behaves incompatibly until sulfide saturation is achieved. Because sulfide saturation in the Skaergaard was achieved relatively late, this formed a Cu-rich sulfide assemblage. However, Andersen (2006) noted that Skaergaard bulk rock S concentrations are lower than the expected saturation values and suggested that the formation of Cu-rich assemblages represent post-magmatic oxidation and S loss. He suggested the conversion of pyrrhotite to Fe-oxides and chalcopyrite to Fe-oxides and bornite, digenite, chalcocite, and (locally) native Cu by reaction with late hydrothermal fluids.
The Bushveld suite is complicated by possible multiple injections of one or more magma types as the intrusion expanded and crystallized. Thus, although olivine is abundant in the Lower Zone and could readily remove Ni, new injections of primitive magma at different stratigraphic levels could explain why pentlandite is found into the Main Zone. In contrast, the paucity of pentlandite compared to chalcopyrite in the Critical Zone implies the Critical Zone did not see significant influx of more primitive magma and that the sulfides grew from magma or interstitial liquid that was relatively depleted in Ni. However, a simple loss of Ni to silicate minerals and increase in S and Cu during sulfide-undersaturated fractional crystallization of the Lower and Critical Zones should produce an approximately constant Cu/S ratio (or ~constant Ccp/Po ratio) in the sulfides once the trapped liquid reaches sulfide saturation, which is not observed. Both this study and that of Liebenberg (1970) have found that Ccp is the only sulfide mineral in much of the Upper Critical Zone.
Evidence for high temperature ore element remobilization in the Lower and Critical Zones
Several studies have shown that the sulfide-rich rocks of the Merensky Reef and UG-2 have been locally modified by both high and low temperature events (e.g., McLaren and De Villiers 1982; Li et al. 2004). For example, Kinloch (1982) notes that the Pt-Pd sulfides in both the Merensky Reef and the underlying UG-2 chromitite are replaced by Pt-Fe alloys in and around potholes. Kinloch attributed these features to late loss of S to high temperature volatiles preferentially streaming through the pothole structures. In another example, Peyerl (1982) noted that the discordant Driekop pipe has evidently modified the type and abundance of PGE minerals in the UG-2 chromitite, changing from Pt–Pd sulfide minerals in normal assemblages to Pt–Fe alloy and Pt–Pd–As–Sb minerals towards the pipe over a distance of at least 1.3 km. He attributed this change to volatiles migrating outward from the pipe. It seems reasonable to conclude that, if the late fluids emanating from the crystal pile and discordant pipes can affect the S-rich reefs, they should also have some effect on the sulfide assemblages in the rocks away from the ore zones that they presumably migrated through.
Two lines of evidence indicate that the Bushveld sulfides below the major reefs are likely to have been modified during cooling and degassing of the intrusion. First, as noted in the introduction, preferential S loss from the Lower and Critical Zones can explain the otherwise puzzling the high PGE and low S bulk rock concentrations of the these zones. The observed replacement of sulfide by magnetite is consistent with this S loss. Second, preferential loss of S and enrichment in Cu, as suggested by Andersen (2006) in the case of the Skaergaard mineral assemblages, can explain the low bulk rock S and the relatively more common occurrences of chalcopyrite in the Lower and Critical Zones.
The lack of Ti and Cr in the magnetite of the pyrrhotite-magnetite-carbonate assemblage in the rocks stratigraphically below the Merensky Reef suggests that the reactive agent was a fluid and not a silicate liquid. In general, on reaching sulfide saturation on cooling, the interstitial silicate liquid is unlikely to become S-undersaturated with further crystallization in the absence of an exsolving vapor phase. This is because co-precipitation of silicates and oxides will continue to keep S abundances at saturation levels and falling temperatures will generally lead to lower S saturation concentrations (e.g., Haughton et al. 1974; Li and Ripley 2009). Hence cooling and crystallization of the interstitial silicate liquid should lead to more sulfide precipitation over time. However, if the silicate liquid were to be heated, perhaps by moving into hotter overlying rocks by compaction or buoyant advective flow, the liquid could become sulfide undersaturated and resorb/replace the earlier precipitated sulfides with magnetite. But even if this were the case, one would expect a Ti or Cr signature in the magnetite, perhaps similar to co-precipitating spinel as observed in Uraguayan dikes (Prichard et al. 2004). Nor is this intergrowth observed in the Main Zone where Fe–Ti oxides are closer to becoming saturated in the magma. Instead, it is suggested that the assemblage developed later and the process may be broadly analogous to the formation of the pothole reef assemblage of PGE- and Fe-alloys. The occurrence of pyrrhotite as isolated grains hosted by magnetite suggests replacement of pyrrhotite and loss of S by a redox reaction involving the degassing of the interstitial silicate liquid and loss of a vapor phase, perhaps as follows:
The above reaction is similar that proposed by Larocque et al. (2000) to explain magnetite-sulfide intergrowths observed in a variety of plutonic rocks.
Although one should not discount the possibility that low temperature minerals may polymorph an original high temperature mineral phase, several lines of evidence point to a relatively high-temperature origin for the calcite–magnetite–pyrrhotite assemblage. While it has long been known that fluids exsolved from the Bushveld Complex have a carbon component and precipitate graphite (e.g., Mathez et al. 1989), specific evidence that carbonates may also be precipitated by these fluids include the following:
-
(1)
The calcite grains in Fig. 3 occur with pyroxene and plagioclase that are typical of a higher temperature paragenesis and are texturally in equilibrium with no evidence of replacement of the surrounding igneous silicate minerals. In some cases, the calcite appears to be present as inclusions along the margins of the silicate minerals; evidence that that the calcite was present before the growth of the pyroxene rim. Although calcite- and magnetite-bearing assemblages have been described with sulfide alteration in the Merensky Reef and elsewhere in the Bushveld Complex (e.g., Li et al. 2004 and sample 45-74 in Fig. 2), these typically also include other low temperature (greenschist) minerals such as actinolite, tremolite, chlorite, talc, and epidote. In contrast, the silicates in Fig. 3 do not contain any of these low temperature minerals. The textural equilibrium of calcite with the igneous assemblage and the lack of greenschist minerals that otherwise readily form at low temperatures implies that the fluid infiltrated at a relatively high temperature.
-
(2)
Although the sulfide is being replaced by magnetite, there is no evidence that this fluid is otherwise strongly reactive. As noted previously, when there is some alteration in a rock, it is the sulfide minerals that tend to be the loci for the halo of greenschist assemblages in the surrounding silicates, implying that the presence of sulfide can enhance silicate susceptibility to alteration by greenschist-grade fluids. Acids formed by reaction of infiltrating fluids with sulfides (e.g., H2SO4) at low temperature are more likely to dissociate and result in fluids with high H+ concentrations that attack the immediately surrounding silicates. In contrast, higher temperature fluids have a larger proportion of neutral associated complexes owing to a lower dielectric constant of water (cf. Walther 1997, and references therein), and hence a more neutral pH, than fluids of the same composition at a lower temperature. In addition, the work of Mathez and Webster (2005) and Hanley et al. (2008) suggest that early Stillwater fluids where hypersaline and may have contained very little free H2O, further diminishing the ability of acids to dissociate. The textural equilibrium of calcite with the anhydrous igneous assemblage and the lack of greenschist minerals that otherwise readily form at low temperatures is consistent with fluid infiltration and S-loss occurred at a relatively high temperature in the presence of a H2O-poor fluid.
-
(3)
There are examples in other an intrusion in which calcite has been observed as part of a high temperature assemblage. For example, calcite, pyroxene and an Fe-oxide (in this case hematite) have been described as part of the mineral assemblage found in Cl- and CO2-bearing fluid inclusions in quartz in gabbronorite pegmatoids from the Stillwater Complex (Hanley et al. 2008). These pegmatoids occur in roughly similar stratigraphic positions as the Bushveld calcite-bearing samples, and is consistent with formation from an igneous, CO2/carbonatitic fluid. Hanley et al. describe the Stillwater calcite inclusions as “accidental,” with the implication that the fluid was precipitating calcite prior to or simultaneously with the formation of the fluid inclusion. This “late magmatic” precipitation of calcite is also consistent with the presence of calcite grains interstitial to the coarse silicate minerals observed in the Stillwater pegmatoids (J.S. Scoates, personal communication, 2011). More recently has been the discovery of high-temperature carbonates in association with sulfides below the platiniferous J-M Reef of the Stillwater Complex. The carbonate assemblage consists of dolomite with exsolved calcite and is found in contact with sulfide minerals: chalcopyrite and pyrrhotite in the Peridotite Zone; and pyrrhotite with pentlandite, pyrite and chalcopyrite in Gabbronorite Zone I of the Lower Banded Series (Aird and Boudreau 2012).
-
(4)
Newton and Manning (2002) note that calcite solubility in NaCl–H2O solutions at magmatic and mantle conditions increases greatly with increasing salinity and temperature and less so with pressure. The Bushveld calcite occurs in that part of the complex where the apatite is unusually Cl-rich (Fig. 4), approaching chlorapatite endmember compositions in many samples. These Cl-rich compositions imply unusually high Cl/F and Cl/OH activity/fugacity ratios from any silicate liquid or fluid from which they may have precipitated (e.g., Boudreau and Simon 2007). Inferences from experimental studies (e.g., Mathez and Webster 2005; Safonov et al. 2007—the latter of which is done at higher pressure than applicable to crustal intrusions but is consistent) indicate that the presence of chlorapatite would require the presence of late stage (igneous) carbonate-saline fluids.
-
(5)
Barnes et al. (2009) noted that the average Se and S contents in rocks from the Lower Zone and Critical Zones below the UG-1 chromitite are 0.03 and 99 ppm, respectively, and that the average S/Se ratio (3,300) is similar to the mantle ratio (3,400). They also note that any low temperature S loss event would increase this ratio, given the more soluble nature of S relative to Se in low temperature fluids (e.g., Dreibus et al. 1995). In contrast, Barnes et al. noted the study of Wulf et al. (1995), who heated meteorites above ~1,050 °C and found that S and Se were equally depleted. Regardless, the magmatic S/Se ratio is consistent with the S loss observed in the Lower Critical Zone being a relatively high temperature event.
Figure 6 shows two semi-quantitative pathways by which an “initial sulfide” (labeled) may be altered by both low and high temperature fluids. The upper path shows the case illustrated by sample 06-168 of Fig. 2, in which pyrite is associated with greenschist-grade assemblages replacing the original igneous minerals in surrounding silicates. Prior to the introduction of the low temperature fluid, cooling of the original sulfide to lower temperature allows the sulfide to stay within the pyrrhotite field. However, interaction with a low temperature fluid to produce the greenschist assemblage in the surrounding silicate minerals allows an increase in sulfur fugacity to stabilize pyrite over pyrrhotite. It is suggested that hydration of the silicates to form the greenschist assemblages decreases H2O and consequently increases S and fS2 such as to drive the conversion of pyrrhotite to pyrite. The observation that the alteration occurs as haloes about the sulfide suggests that the presence of sulfide enhances the formation of the greenschist assemblage by the local production of sulfuric acid as the infiltrating fluid reacts with sulfide:

Semi-quantitative interpretation of Bushveld assemblages plotted on Fe–S–O system as a function of Temperature (°C). The Po–Mt boundary is calculated for f(O2) defined by the QFM buffer. Initial sulfide (labeled), with two possible cooling paths shown: a low temperature pyrrhotite to pyrite alteration event (upper path) and a high temperature S-loss event leading to replacement of pyrrhotite by magnetite (lower path). Temperatures shown are approximate and illustrative only. See text for additional discussion
The incorporation of the H+ ions produced by the above reaction into hydrous alteration minerals would drive the reaction to the right, producing more pyrite with increasing alteration in surrounding silicates.
In contrast, the lower path shown in Fig. 6 shows the high temperature S loss implied by reaction (1). Loss of S during open system degassing of the interstitial silicate liquid lowers local fS2 and drives the pyrrhotite to the pyrrhotite–magnetite boundary. On cooling (and in the absence of late, low temperature alteration) the pyrrhotite–magnetite assemblage avoids intersection with the pyrite field.
These two paths represent the high and low temperature extremes, and it is likely that these reactions occur on a continuum as the Bushveld Complex cooled. Indeed, the low grade assemblages could have developed at any time after the complex cooled to ambient temperature.
A phase diagram interpretation that is broadly compatible with the presence of high temperature calcite and Cl-rich apatite in the Lower and Critical Zones is shown in Fig. 7. A calcite-saturated fluid starting at point A would begin with a relatively low NaCl content. On cooling and precipitating calcite the fluid would eventually intersect the two-fluid field at the point F1. The fluid would then split into two fluids, one a carbonatitic liquid (F1) and the other a NaCl-rich aqueous solution (F2). The former would become slightly more carbonate-rich on further cooling while the aqueous solution would become less saline and less carbonate. These two fluids would continue to precipitate calcite until the carbonate fluid is used up. Once the carbonatitic fluid is exhausted, the remaining aqueous solution continues to precipitate calcite until the fluid reaches halite saturation, at which point the fluid precipitates both calcite and halite and continues to become richer in H2O. The presence of increasingly H2O-rich fluids that remain in the rocks to lower temperature would also lead to the formation of the hydrous, greenschist-grade assemblage. However, the low temperature alteration could have formed at any time after the intrusion cooled as well.

The calcite–H2O–NaCl system, showing a possible crystallization path for a Bushveld carbonatitic liquid. See text for details. Modified after Newton and Manning (2002)
Although not shown in Fig. 7, an alternative is that the fluid starts in the halite saturation field. The fluid would then precipitate halite until the fluid became saturated in calcite, at which time the fluid would precipitate calcite and halite and also become richer in H2O. This implies that that halite could be a high temperature interstitial phase, but is lost during thin section manufacture. A study of samples prepared without water might resolve this possibility.
Implications for the PGE distribution in the Bushveld Complex
Recent experimentally determined (volatile fluid)/(silicate liquid) partition coefficients for Pt can range across more than four orders of magnitude (<101 to ~104; Simon and Pettke 2009; Blaine 2010; Blaine et al. 2011), with values of 103 to 104 favored by more Cl-rich fluids and by more mafic over more felsic silicate liquid compositions. Blaine (2010) reports a partition coefficient of 1,100 for Ir partitioning into Cl- and CO2-rich fluids. These studies suggest that chloride–carbonate fluids can be more effective for moving both the PPGE (Rh, Pt, Pd) and the IPGE (Ir, Ir, Ru) than can more dilute H2O-dominant fluids.
The unusually high PGE concentrations in the rocks below the Merensky Reef and UG-2 indicate that they are a potential source rock for the metals in these zones. The PGE content of the Merensky Reef has a near-constant grade (5–8 g/t total PGE) over a thickness of 40–120 cm in both the Western and Eastern Limbs, and those of the UG-2 are broadly similar (Wagner 1929; Vermaak 1976; Lee 1996; Barnes and Maier 2002; Cawthorn et al. 2002). Using the compiled data summary of Barnes and Maier (2002) for the Western Limb, one can calculate how much higher the observed footwall noble metal concentration need to have been to account for the metals now in the Merensky Reef and the UG-2 chromitite (ESM Table 4 and Fig. 8). The calculation assumes a conservative value for the noble metal concentration in ore rocks of twice observed values. Adding the ore metals back into 1,000 m of average footwall would increase the footwall concentrations from 8 % to 28 %, with the higher values for the PPGE. Put another way, any fluid migrating through the footwall would only need to be from 8 % to 28 % efficient at removing the noble metals from the footwall to form the UG-2 and the Merensky Reef, assuming all transported metals were deposited in the ore zones. If the metals came from a thinner or thicker footwall section these numbers would change proportionally. The greater loss of the PPGE relative to the IPGE required to form the ore zones is consistent with the higher vapor/(silicate liquid) partition coefficients for Pt relative to Ir noted above. The preferential loss of the PPGE from the footwall also can explain the flatter mantle-normalized patterns of the footwall rocks compared with either the reefs or high-Mg basaltic andesite parent liquid.

Mantle-normalized noble metal concentrations illustrating the change in the average footwall assuming the footwall was the source of the reef metals. “Footwall” = thickness-normalized average concentrations of the Lower and Critical Zones. “Footwall + reefs” = average footwall + 2× (UG-2 + Merensky Reef) added to 1,000 m of footwall rock. “High–Mg basaltic andesite” (dash line) = average composition of the high-Mg basaltic andesite from the Bushveld marginal sill-dike suite, shown for reference. All data are for the Western Limb of the Bushveld Complex from data compiled by Barnes and Maier (2002); mantle values are from McDonough and Sun (1995)
The evidence that the sulfide assemblage of the Bushveld Complex has been affected by both low temperature and especially high temperature alteration events has implications for the bulk rock S and PGE abundances of the Bushveld Complex. As noted above, the high PGE concentrations in the Lower and Critical Zones indicate that the rocks had a “cumulus” sulfide component; however, the S contents of the rocks are too low for the magma to have been S saturated (e.g., Barnes et al. 2009). It is possible that the magmas that formed the Lower and especially the Critical Zones were abnormally enriched in the PGE (and became more so with height in the intrusion). If so, these PGE-rich compositions are not seen in the sills and chilled marginal rocks that are thought to be samples of the magma(s) that were involved in the crystallization of the Bushveld Complex. As noted above, Davies and Tredoux (1985) found that the magnesian basaltic andesites and a tholeiitic suite rocks most likely to represent Bushveld liquid compositions generally fall into the range of basalts and chilled margins of other layered intrusions, although the magnesian basaltic andesites are on the high end for a magma (containing an average 17 ppb Pt and 12 ppb Pd; Fig. 8.). These concentrations are too low to account for the Pt and Pd concentrations beneath the major reefs, as the trapped liquid component of the bulk rock would be about an order of magnitude lower than these values.
It has also been suggested that new magma injections carried in a partly-resorbed, PGE-enriched sulfide (e.g., Barnes et al 2009). If so, these transported sulfides would necessarily not have carry in significant Ni, as the pentlandite-poor sulfide assemblage in the rocks of the Lower and Critical Zones are generally consistent with an early loss of Ni to silicate minerals (e.g., olivine) and enrichment in Cu during the crystallization of these zones and/or preferential S loss to degassing. The results of this study point to the second possibility, viz: the rocks lost sulfur to a high temperature carbonate- and chloride-rich fluid. Sulfide saturation during crystallization of the Lower and Critical Zones with a later S loss (and some of the PGE) could explain the observed relatively high PGE and low S concentrations of these rocks.
The Bushveld Complex represented the crystallization of a system open to magma gain or loss. Thus, one cannot categorically exclude the possibility that a PGE-rich magma evolved elsewhere prior to its introduction into the chamber. In the following, however, a model is proposed by which the ore element distribution profile could have arisen solely by processes happening within the Bushveld magma chamber.
A model
The quantitative vapor refining model of Boudreau and Meurer (1999) and Boudreau (2004) has been modified to account for the broad features of the Bushveld PGE-S concentrations and is shown schematically in Fig. 9. At some point during the solidification of the crystal pile, a vapor is exsolved from the crystallizing interstitial liquid. The separation and loss of this vapor removes sulfur and, if present, can result in partial resorption of any preexisting sulfide and removal of some of the metals they contain. This vapor will migrate upward where it will encounter hotter, vapor undersaturated silicate liquid. If the fluid is composed predominately of a low solubility component like CO2, then the redissolution of vapor will quickly drive the small amount of interstitial liquid to become vapor-saturated, leading to a rapid upward migration of the vapor-saturation front in the crystal pile. Eventually the vapor saturation front may reach the top of the crystal pile.

Cartoon of the ore element transport and sulfur recycling in a growing crystal pile at the two time steps t 1 and t 2. See text for discussion
Owing to its larger mass, the magma above the pile can readily absorb the relatively small amount of vapor escaping from the crystal pile without becoming vapor saturated. However, the increase in S concentration as the fluid redissolves in the magma can drive a thin layer of magma just above the top of the pile to sulfide saturation. The precipitation of sulfide can then lead to modest enrichments of the PGE being incorporated into the growing pile. As the pile grows sulfur is continuously recycled, moving from the interior of the crystal pile to the top to produce a thin, ephemeral zone of sulfide-saturated magma. On solidification what is produced are rocks that are relatively enriched in the PGE but with lower than cotectic sulfide abundance. An advantage of this model is that it does not require the entire Bushveld magma to be sulfide saturated during the crystallization of the Lower and Critical Zones.
A modified version of the PALLADIUM program (Boudreau 2004) was used to quantify the model. The program models the growth and compaction of a crystal pile and also allows for vapor separation and migration through the pile. It uses simple portioning behavior between silicate liquid, vapor, immiscible sulfide liquid, Pd metal, and silicate minerals, coupled with heat and mass transport equations. The major modifications include (a) the inclusion of sulfide mineral modes (pyrrhotite, pentlandite, chalcopyrite, and chalcocite), (b) the option of allowing the vapor-induced sulfide saturation at the top of the pile to equilibrate metals from some thickness of magma at the bottom of the magma column, and (c) the program allows a “vapor trap” to develop within the pile that can trap upward-migrating vapor.
In regards to the first modification, the program calculates sulfide mode based on element partitioning. For example, if sulfide liquid/silicate liquid partition coefficient for Cu is 1,000 and the silicate liquid has 100 ppm Cu, the sulfide liquid will contain 100,000 ppm (or 10 %) Cu. This would be the equivalent of a sulfide assemblage containing ~29 % chalcopyrite. In regards to the stratigraphic vapor “trap,” this is accomplished in the model by keeping the water concentration of the interstitial liquid at zero at and above the level of the trap to prevent vapor separation. This mimics a real permeability trap in the crystal pile in which vapor comes into a zone faster than it can leave. However there are other possible traps as well. As note previously (e.g., Boudreau and Meurer 1999), the “trap” could also be the top of the crystal pile if the escaping vapor, on mixing with the resident magma, causes a significant lowering of the solidus such that the magma temporarily ceases to crystallize. In this case, the crystal pile may degas more or less completely without significant growth of the pile.
Values for the many variables one can set in the program are shown in ESM Table 5. In brief, the program uses the “slow compaction” values from Shirley (1986). Partition coefficients are similar to those in the literature (e.g., Boudreau 2004; Naldrett 2009). For example, as noted above PGE vapor/(silicate liquid) partition coefficients can vary widely as a function of fluid and melt composition. A value of 1,000 was taken for the Pd vapor/(silicate liquid) partition coefficient under the assumption that the higher values of 103–104 noted by Blaine et al. (2011) for Pt partitioning between a Cl- and CO2-rich fluid, and a basaltic silicate liquid are appropriate for the Bushveld. Finally it is noted that the program does not consider that the exsolved vapor might be a mixed CO2–H2O vapor whose solubility may vary as degassing continues. Instead, the solubility of a mixed fluid (denoted “H2O” for simplicity) is assumed constant and the amount of H2O has been adjusted so that not all sulfur is lost during degassing of the interstitial liquid in the crystal pile.
The model results are shown in Fig. 10 for the crystallization of a 1-km-thick magma column at the four model times, from bottom to top, of 615, 1,839, 2,358, and 6,344 years. The two profiles of most importance to the following discussion are the weight fraction mineral modes (leftmost column at each time step) and the bulk ore element concentrations (second column from the right).

PALLADIUM model results for the crystallization and degassing of a 1-km-thick magma column at the four model times, from bottom to top, of 615, 1,839, 2,358, and 6,344 years. The six columns at each time step shows, from left to right: (1) the weight fraction (f) of silicate liquid (solid line), sulfide liquid (dashed line), and vapor (heavy dotted line; the values for the latter two are defined on the lower scale). The straight vertical light dotted line is the cotectic sulfide mode for a sulfide-saturated magma, shown for reference. (2) The compaction velocity of the solid matrix (solid line) and the liquid (dashed line) in the compacting part of the mush. (3) The temperature profile. (4) The “H2O” concentration in the bulk rock and of the liquid. (5) The concentrations of Ni (dash–dot line), Cu (dotted line), Pd (solid line), and S (dashed line). Metal concentrations are shown relative to starting bulk composition, whereas S is plotted relative to sulfur concentration as sulfide liquid saturation. (6) The calculated wt.% of pentlandite, chalcopyrite, and pyrrhotite in the sulfide assemblage. The region labeled “mush zone” at each time step is the region of silicate liquid + crystals; below this zone, the rocks have solidified completely and above this zone the system is all liquid. A volatile “trap” (labeled) initially develops at the top of the mush zone when the crystal pile is 400 m thick after ~2,100 years (not shown); at 2,358 and 6,344 years compaction has caused this “trap” to move to a stratigraphically lower position
615 years
At this time, crystallization of the magma has produced a crystal pile about 150 m in thickness, of which the lowermost ~70 m has already solidified, leaving a mush zone about 80 m thick. In terms of the vapor and sulfide history of the pile, it can be divided into two parts as well. Below about 80 m, including the completely solidified part of the crystal pile and the lower part of the still-partial liquid mush, the sulfide abundance reflects mainly that which crystallized from the “trapped liquid,” after some loss of S to degassing.
Prior to this time, degassing of the pile resulted in the vapor-saturation front first reaching the top of the mush pile when the pile was about 80–100 m thick. Vapor redissolving into the vapor-undersaturated silicate liquid at the top of the pile enriches the silicate liquid in S to the point it becomes sulfide saturated. This leads to an increase in the strongly chalcophilic Pd such that rocks in the upper part of the mush have concentrations about twice that of the original magma.
Sulfide abundance in the upper part of the crystal pile is a function of three competing processes: (1) compaction, which leads to an increase in the wt.% of immiscible sulfide but a decrease in S-saturated silicate liquid, (2) degassing of interstitial liquid, which leads to S-loss and a decrease in sulfide liquid as it re-dissolves to maintain the liquid at saturation concentrations, and (3) heat loss, which leads to crystallization of silicate minerals, and increase in S concentration in the liquid and the precipitation of more sulfide liquid. Loss of silicate liquid to compaction and solidification leads to less and less vapor generated as a function of depth within the mush zone (and is zero in the fully solidified portion). As shown in the plot of sulfide mode and the plot of S/Ssat concentration, sulfide mode increases downward in the upper part of the crystal mush but never reaches the expected bulk saturation values because some sulfur is always lost from the lower part of the pile and transported to the top of the pile.
1,839 years
At this time, the top of the crystal pile has reached about 375 m and the mush zone is about 160 m thick. The vapor refining of the mush zone continues, with sulfur leached from the mush leading to sulfide saturation at the top of the pile. Note the slight decrease in bulk Pd in the pile as Pd-depleted liquid from the bottom of the magma column mixes with the rest of the magma above.
2,358 years
Just prior to this time, the upward-migrating vapor has encountered the vapor “trap” which prevents the vapor from rising higher in the column. This trap, originally at 400 m, has moved down to ~375 m as a result of compaction of the solid matrix. Because all sulfur and vapor-transported metals are eventually precipitated at this level, a pronounced peak in the concentrations of all of these elements occurs at the trap.
6,344 years
The solidification front has now moved past the trap and the bottom of the liquid + crystal mush is now at ~525 m. Below this one can see the final metal profile in the fully solidified rock. S and sulfide modes both above and below the peak at the trap are always less than the expected values for a S-saturated magma. The same is generally true for a chalcophile element like Cu with an intermediate D sulfide liquid/silicate liquid partition coefficient. In contrast, Pd has a very high D sulfide liquid/silicate liquid partition coefficient, is two to three times its initial concentration below the trap but then falls to relative concentrations similar to the other elements. Ni, which in the model is assumed to be the only element that significantly partitions into the silicate minerals host (e.g., olivine), is largely defined by fractional crystallization of the silicate solids.
The Pd concentrations at the peak reach about 2 orders of magnitude higher than the original magmatic concentrations. This is actually reduced somewhat by an uncorrected numerical diffusion artifact in the model that causes peaks—which should be one node or 1 m thick—to spread across several nodes during compaction. A visual integration of the peak area to one node suggests an actual enrichment of about 3 orders of magnitude. In other words, a magma initially containing a few to several tens of ppb Pd can form a deposit with a bulk concentration of a few to several tens of ppm over 1 m.
A comparison of the general trends of the model profile with that of the Bushveld Complex of Fig. 4 shows a number of first-order similarities and differences:
-
(1)
Modeled bulk rock sulfur distribution shows that background S concentrations are generally below expected saturation levels, except at the trap. This is similar to that of the Bushveld, where S below and above the reefs of the Upper Critical Zone are generally low. Indeed the low S concentrations of the Main Zone generally continue the low (~100 ppm) values in much of the Lower and Critical Zones. There is no expected jump if the magma had become sulfide saturated at the level of the Merensky Reef.
-
(2)
Modeled Cu concentrations parallel S concentration trends. Although Bushveld Cu trends are not shown in Fig. 4, this is also observed in the Bushveld Complex (e.g., Maier and Barnes 1999). Also, except for a modest increase in modeled chalcopyrite mode as a proportion of the sulfide assemblages associated with the run-up of the degassing front near the base, chalcopyrite mode varies only modestly with stratigraphic position. In contrast, Ni can partition into the silicate minerals and is depleted from the liquid as the pile grows. This broad trend of decreasing pentlandite/chalcopyrite mimics the trend in Bushveld sulfides up to the Main Zone, although in the Bushveld Complex new injections of magma can cause a more erratic appearance of pentlandite (e.g., at the base of the Main Zone).
-
(3)
In contrast to S and Cu, modeled Pt values below the trap are about a half-order of magnitude higher than the parent magma concentrations. This mimics the high background Pt concentrations in the Bushveld Complex. Again, the exception is near the base when the model degassing front has not yet reached the top of the pile. Above the trap, Pt concentrations drop off to lower values reflecting the amount of trapped liquid in the final rock. Because the model does not incorporate new influxes of magma, the Pt concentration does drop slightly towards the trap (owing to Pt depletion from the magma). In contrast background Pt concentrations in the Bushveld Complex increases from the Lower Zone into the Critical Zone. If the high Pt concentration are due to the mechanism proposed here, this may reflect either more extensive scavenging with height in the Bushveld (e.g., sulfide equilibrates with a thicker magma layer above the top of the pile) or new magma inputs that replenish the PGE, or both.
-
(4)
In the model, vapor is present at and below the vapor trap, but not above. Using the Cl concentration in apatite as a proxy for the presence of fluids (e.g., Boudreau et al. 1986; Mathez and Webster 2005), the Bushveld apatite shows a similar break to more F-rich compositions above the Merensky Reef.
Conclusions
This work largely confirms the Bushveld trace sulfide distribution as described by Liebenberg (1970). It expands this earlier study by documenting evidence for high temperature S-loss in rocks below the Merensky Reef and replacement by magnetite. The finding of calcite as a high temperature (igneous) phase was an unexpected result and has not been reported in the Bushveld Complex before, as far as we are aware. In regards to possible ore element mobility during solidification and degassing, the presence of carbonates in the part of the Bushveld stratigraphic section also characterized by Cl-rich apatite is consistent with experimental studies (e.g., Blaine 2010) that Cl- and CO2-rich fluids can be effective at transporting the PGE. Quantitative modeling of a growing and degassing crystal mush illustrates how the stratigraphic distribution of the ore elements in the Bushveld Complex could have developed as S is continuously recycled from the interior of the mush to the top of the crystal pile.
If one grants the possibility that PGE can be effectively moved by such fluids, then variations in fluid composition potentially can explain variations in the PPGE/IPGE ratio of different deposits. For example, the PGE zones of the Lac des Iles (e.g., Hanley and Gladney 2011 and references therein) and the Wengeqi (e.g., Su and Lesher 2012) show a much stronger IPGE/PPGE fractionation than do the Bushveld or Stillwater Complexes. In the Lac Des Iles, Hanley and Gladney (2011) have described primary carbonic-rich fluid inclusions in pegmatoids, but also note that they do not appear to contain appreciable Cl. This is in contrast to fluid inclusion data from the Stillwater Complex where carbonate-brine inclusions occur in pegmatoids beneath the platiniferous J-M Reef and halide inclusions occur within olivine from the Ultramafic Series (Hanley et al. 2008). It is suggested that Cl- and CO2-rich nature of the Stillwater and Bushveld fluids produced much less fractionation of the PPGE from the IPGE as compared with the Lac des Iles. Future work focusing on detailed descriptions of the trace mineral assemblages in these and other intrusions could further elucidate their degassing history, fluid compositional evolution, and the possibility of significant remobilization of the ore elements.
References
Aird H, Boudreau AE (2012) High-temperature carbonates in the Stillwater Complex, Montana, USA. 35th Mineral Deposits Studies Group, Cardiff University, UK, 3-6 January. Abstract
Andersen JCØ (2006) Postmagmatic sulphur loss in the Skaergaard Intrusion: implications for the formation of the Platinova Reef. Lithos 92:198–221
Ballhaus CG, Stumpfl EF (1985) Occurrence and petrological significance of graphite in the Upper Critical zone, western Bushveld complex, South Africa. Earth Planet Sci Lett 74:58–68
Ballhaus CG, Stumpfl EF (1986) Sulfide and platinum mineralization in the Merensky reef: evidence from hydrous silicates and fluid inclusions. Contrib Mineral Petrol 94:193–204
Barnes S-J, Maier WD (2002) PGE distributions in the Rustenburg Layered Suite of the Bushveld Complex, South Africa. In: Cabri L (ed) The geology, geochemistry, mineralogy and mineral beneficiation of PGE. Can Inst Metallurgy Spec 54:553–580
Barnes S-J, Savard D, Bédard LP, Mairer WD (2009) Selenium and sulfur concentrations in the Bushveld Complex of South Africa and implications for formation of the platinum-group element deposits. Miner Depos 44:647–664
Blaine, FA (2010) The Effect of Volatiles (H2O, Cl and CO2) on the Solubility and Partitioning of Platinum and Iridium in Fluid-Melt Systems. PhD Dissertation University of Waterloo 170 pp
Blaine FA, Rl L, Holtz F, Bruegmann GE (2011) The effect of Cl on Pt solubility in haplobaslatic melt: implications for micronugget formation and evidence for fluid transport of the PGEs. Geochim Cosmochim Acta 75:7792–7805
Boudreau AE (2004) PALLADIUM—a program to model the chromatographic separation of the platinum-group elements, base metals and sulfur in a solidifying igneous crystal pile. Can Mineral 42:393–403
Boudreau AE, McCallum IS (1992) Concentration of Platinum-group elements by magmatic fluids in layered intrusions. Econ Geol 87:1830–1848
Boudreau AE, Meurer WP (1999) Chromatographic separation of the platinum-group elements, gold, base metals and sulfur during degassing of a compacting and solidifying crystal pile. Contrib Mineral Petrol 134:174–185
Boudreau AE, Simon A (2007) Halogen variations and degassing in the Basement Ferrar sill, Antartica. J Petrol 48:1369–1386
Boudreau AE, Mathez EA, McCallum IS (1986) Halogen geochemistry of the Stillwater and Bushveld Complexes: evidence for transport of the platinum-group elements by Cl-rich fluids. J Petrol 27:967–986
Campbell IH, Naldrett AJ, Barnes SJ (1982) A model for the origin of the platinum-rich sulfide horizons in the Bushveld and Stillwater Complexes. J Petrol 24:133–165
Cawthorn RG (1998) Emplacement and crystallization time for the Bushveld Complex. J Pet 39:1669–1687
Cawthorn RG (1999) Platinum-group element mineralization in the Bushveld Complex; a critical reassessment of geochemical models. S Afr J Geol 102:268–281
Cawthorn RG, Merkle RKW, Viljoen MJ (2002) Platinum-group element deposits in the Bushveld Complex, South Africa. In: Cabri LJ (ed) The geology, geochemistry, mineralogy and beneficiation of platinum-group elements. Canadian Institute of Mining, Metallurgy and Petroleum, Special Volume 54:389–430
Davies G, Tredoux M (1985) The platinum-group element and gold contents of the marginal rocks and sills of the Bushveld Complex. Econ Geol 80:838–848
Dreibus G, Palme H, Spettel B, Zipfel J, Wänke H (1995) Sulfur and selenium in chondritic meteorites. Meteoritics 30:439–445
Eales HV, Cawthorn RG (1996) The Bushveld Complex. In: Cawthorn RG (ed) Layered intrusions. Elsevier, Amsterdam, pp 181–229
Hanley JJ, Gladney ER (2011) The presence of carbonic-dominant volatiles during the crystallization of sulfide-bearing mafic pegmatites in the North Roby Zone, Lac des Iles Complex, Ontario. Econ Geol 106:33–54
Hanley JJ, Mungall JE, Pettke T, Spooner ETC, Bray CJ (2008) Fluid and halide melt inclusions of magmatic origin in the Ultramafic and Lower Banded Series, Stillwater Complex, Montana, USA. J Petrol 49:1133–1160
Haughton DR, Roeder PL, Skinner BJ (1974) Solubility of sulfur in mafic magmas. Econ Geol 69:451–467
Kinloch ED (1982) Regional trends in platinum-group mineralogy of the Critical Zone of the Bushveld Complex, South Africa. Econ Geol 77:1328–1347
Kruger FJ, Cawthorn RG, Walsh KL (1987) Strontium isotopic evidence against magma addition in the Upper Zone of the Bushveld Complex. Earth Planet Sci Lett 84:51–58
Larocque ACL, Stimac JA, Keith JD, Huminicki MAE (2000) Evidence for open-system behavior in immiscible Fe-S-O liquids in silicate magmas: implications for contributions of metals as sulfur to ore-forming fluids. Can Mineral 38:1233–1249
Lee CA (1996) A review of mineralization in the Bushveld Complex and some other layered mafic intrusions. In: Cawthorn RG (ed) Layered intrusions. Elsevier, Amsterdam, pp 103–146
Li C, Ripley EM (2009) Sulfur contents at sulfide-liquid or anhydrite saturation in silicate melts: empirical equations and example applications. Econ Geol 104:405–412
Li C, Ripley EM, Merino E, Maier W (2004) Replacement of base metal sulfides by actinolite, epidote, calcite and magnetite in the UG2 and Merensky Reef of the Bushveld Complex, South Africa. Econ Geol 99:173–184
Liebenberg L (1970) The sulfides in the layered sequence of the Bushveld Igneous Complex. Geol Soc S Afr Spec Publ 1:108–208
Maier WD, Barnes SJ (1999) Platinum-group Elements in Silicate Rocks of the Lower, Critical, and Main Zones at Union Section, Western Bushveld Complex. J Petrol 40:1647–1671
Mathez EA (1976) Sulfur solubility and magmatic sulfides in submarine basalt glass. J Geophys Res 81:4269–4275
Mathez EA, Webster JD (2005) Partitioning behavior of chlorine and fluorine in the system apatite-silicate melt-fluid. Geochim Cosmochim Acta 69:1275–1286
Mathez EA, Dietrich VJ, Holloway JR, Boudreau AE (1989) Carbon distribution in the Stillwater Complex and evolution of vapor during crystallization of Stillwater and Bushveld Magmas. J Petrol 30:153–173
McDonough WF, S-s S (1995) The composition of the Earth. Cehm Geol 120:223–253
McLaren CH, De Villiers JPR (1982) The platinum-group chemistry and mineralogy of the UG-2 chromitite layer of the Bushveld Complex. Econ Geol 77:1348–1366
Meurer WP, Willmore CC, Boudreau AE (1998) Metal redistribution during fluid exsolution and migration in the Middle Banded series of the Stillwater Complex, Montana. Lithos 47:143–156
Miller JD Jr, Andersen JCØ (2002) Attributes of Skaergaard-type PGE reefs. In: Boudreau AE (ed) 9th Int Platinum Symp Abstr. Duke University, Durham, pp 305–308
Miller DJ, Cervantes P (2002) Sulfide mineral chemistry and petrography and platinum group element composition in gabbroic rocks from the Southwest Indian Ridge. In: Natland JH, Dick HJB, Miller DJ, Von Herzen RP (eds.) Proceedings of the Ocean Drilling Program, Scientific Results v. 176 doi:10.2973/odp.proc.sr.176.009.2002
Naldrett AJ (2004) Magmatic sulfide deposits: geology, geochemistry and exploration. Springer-Verlag, Berlin 727 pp
Naldrett AJ (2009) Secular variation of magmatic sulfide deposits and their source magmas. Econ Geol 105:669–688
Naldrett AJ, Wilson A, Kinnaird J, Chunnett G (2009) PGE tenor and metal ratios within and below the Merensky Reef, Bushveld Complex: implications for its genesis. J Petrol 50:625–659
Newton RC, Manning CE (2002) Experimental determination of calcite solubility in H2O-NaCl solutions at deep crust/upper mantle pressures and temperatures: implications for metasomatic processes in shear zones. Am Mineral 87:1401–1409
Peyerl W (1982) The influence of the Driekop dunite pipe on the platinum-group mineralogy of the UG-2 chromitite in its vicinity. Econ Geol 77:1432–1438
Prichard HM, Hutchinson D, Fisher PC (2004) Petrology and crystallization history of multiphase sulfide droplets in a Mafic Dike from Uruguay: implications for the Origin of Cu-Ni-PGE Sulfide Deposits. Econ Geol 99:365–376
Safonov OG, Perchuk LL, Litvin YA (2007) Melting relations in the chloride–carbonate–silicate systems at high pressure and the model for formation of alkali diamond-forming liquids in the upper mantle. Earth Planet Sci Lett 253:112–128
Sharpe MR (1985) Strontium isotope evidence for preserved density stratification in the Main Zone of the Bushveld Complex, South Africa. Nature 316:119–126
Sharpe MR, Hulbert LJ (1985) Ultramafic sills beneath the Eastern Bushveld Complex: mobilized suspensions of early Lower Zone Cumulates in a parental magma with boninitic affinities. Econ Geol 80:849–871
Shirley DN (1986) Compaction of igneous cumulates. J Geol 94:795–809
Simon AC, Pettke T (2009) Platinum solubility and partitioning in a felsic melt-vapor-brine assemblage. Geochim Cosmochim Acta 73:438–454
Stumpfl EF, Tarkian M (1976) Platinum genesis: new mineralogical evidence. Econ Geol 71:1451–1460
Su S, Lesher M (2012) Genesis of PGE mineralization in the Wengeqi mafic-ultramafic complex, Guyang Country, Inner Mongolia, China. Miner Deposita 47:197–207
Vermaak CF (1976) The Merensky Reef—thoughts on its environment and genesis. Econ Geol 71:1270–1298
Von Gruenewaldt G (1973) The main and upper zones of the Bushveld Complex in the Roossenekal area, eastern Transvaal. Geol Soc S Afr Trans 76:53–61
Von Gruenewaldt G (1976) Sulfides in the upper zone of the eastern Bushveld Complex. Econ Geol 71:1324–1336
Von Gruenewaldt G, Sharpe MR, Hatton CJ (1985) The Bushveld Complex: introduction and review. Econ Geol 80:803–812
Wager LR, Vincent EA, Smales AA (1957) Sulphides in the Skaergaard Intrusion, East Greenland. Econ Geol 52:855–903
Wager LR, Vincent EA, Smales AA (1958) The behaviour of sulphur during fractionation of basic magma. Geochim Cosmochim Acta 14:165–166
Wagner PA (1929) The platinum deposits and mines of South Africa. Oliver & Boyd, Edinburgh, 326 pp
Walther JV (1997) Determining the activity coefficients of neutral species in supercritical H2O solutions. Geochim Cosmochim Acta 61:3311–3318
Willmore CC, Boudreau AE, Kruger FJ (2000) The halogen geochemistry of the Bushveld Complex, Republic of South Africa: implications for chalcophile element distribution in the lower and critical zones. J Petrol 41:1517–1539
Wulf AV, Palme H, Jochum KP (1995) Fractionation of volatile elements in the early solar system: evidence from heating experiments on primitive meteorites. Planet Space Sci 43:451–468
Acknowledgments
This work was completed as part of a Senior Thesis by the senior author at Duke University. We wish to thank Grant Cawthorn of the University of the Witwatersrand, Gordon Chunnett of Anglo Platinum Ltd., and the geologists at the Rustenburg and Marula platinum mines for some of the samples used in this study. This work was improved by the comments of Ed Ripley, Chusi Li, Sarah Penniston-Dorland, Peter Lightfoot, Jacob Hanley, Mike Lesher, and an anonymous reviewer. This work was supported by NSF grant EAR 04-7928.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: C. M. Lesher
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table 1
A summary of rock samples and major sulfide mineral assemblages (DOC 102 kb)
Table 2
Electron microprobe analysis of sulfide minerals (DOCX 66 kb)
Table 3
Microprobe analysis of mixed magnetite-pyrrhotite phase from sample LCZ-105 (DOCX 12.1 kb)
Table 4
Average Bushveld noble metal values of various units from the Western Limb of the Bushveld Complex (from Barnes and Maier 2002) and calculated theoretical original footwall source concentrations (DOCX 14 kb)
Table 5
Parameters for PALLADIUM model run. All concentrations are in weight fraction, except as noted (DOCX 11.6 kb)
Rights and permissions
About this article
Cite this article
Kanitpanyacharoen, W., Boudreau, A.E. Sulfide-associated mineral assemblages in the Bushveld Complex, South Africa: platinum-group element enrichment by vapor refining by chloride–carbonate fluids. Miner Deposita 48, 193–210 (2013). https://doi.org/10.1007/s00126-012-0427-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00126-012-0427-2