
Abstract
Aims/hypothesis
Glycogen accumulation occurs in beta cells of diabetic patients and has been proposed to partly mediate glucotoxicity-induced beta cell dysfunction. However, the role of glycogen metabolism in beta cell function and its contribution to diabetes pathophysiology remain poorly understood. We investigated the function of beta cell glycogen by studying glucose homeostasis in mice with (1) defective glycogen synthesis in the pancreas; and (2) excessive glycogen accumulation in beta cells.
Methods
Conditional deletion of the Gys1 gene and overexpression of protein targeting to glycogen (PTG) was accomplished by Cre-lox recombination using pancreas-specific Cre lines. Glucose homeostasis was assessed by determining fasting glycaemia, insulinaemia and glucose tolerance. Beta cell mass was determined by morphometry. Glycogen was detected histologically by periodic acid–Schiff's reagent staining. Isolated islets were used for the determination of glycogen and insulin content, insulin secretion, immunoblots and gene expression assays.
Results
Gys1 knockout (Gys1 KO) mice did not exhibit differences in glucose tolerance or basal glycaemia and insulinaemia relative to controls. Insulin secretion and gene expression in isolated islets was also indistinguishable between Gys1 KO and controls. Conversely, despite effective glycogen overaccumulation in islets, mice with PTG overexpression (PTGOE) presented similar glucose tolerance to controls. However, under fasting conditions they exhibited lower glycaemia and higher insulinaemia. Importantly, neither young nor aged PTGOE mice showed differences in beta cell mass relative to age-matched controls. Finally, a high-fat diet did not reveal a beta cell-autonomous phenotype in either model.
Conclusions/interpretation
Glycogen metabolism is not required for the maintenance of beta cell function. Glycogen accumulation in beta cells alone is not sufficient to trigger the dysfunction or loss of these cells, or progression to diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
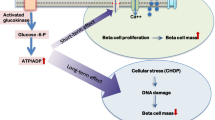
Glycogen is a glucose polymer present mainly in liver and muscle, where it acts as an energy store. Pancreatic beta cells also express the glycogen metabolic machinery and contain detectable amounts of this polysaccharide [1, 2]. However, the precise physiological role of glycogen in beta cells remains elusive. Germ-line deletion of the glycogen synthase 1 (Gys1) gene, which codes for the enzyme responsible for the build up of glycogen in muscle and peripheral tissues, including islets and brain, results in augmented insulin secretion and improved glucose tolerance [3], although the contribution of beta cells to this phenotype has not been established. Additionally, both a reduction in extracellular glucose and pharmacological stimulation of glycogenolysis in beta cells trigger insulin secretion in glycogen-containing islets, thereby suggesting an impact of glycogen on beta cell function [4].
Glucotoxicity is a major contributor to beta cell loss and dysfunction and to the progression of diabetes [5, 6]. Based on the observation that glycogen accumulates in beta cells of diabetic patients and animal models of diabetes [7–11], it has been proposed that glycogen metabolism partly mediates glucotoxic events by altering beta cell glucose fluxes [12]. Thus, in glycogen-rich rodent islets, glycogenolysis stimulates insulin release in the absence of extracellular glucose and, paradoxically, after a rise in glucose concentration, the glycolytic flux is reduced, resulting in decreased insulin secretion. Furthermore, pathological accumulation of glycogen has been implicated in apoptotic neuronal death in the neurodegenerative disorder known as Lafora disease [13, 14]. Likewise, glycogen overaccumulation may negatively affect beta cell survival and potentiate beta cell loss during the course of diabetes.
Here, we studied the role of glycogen in beta cell function. For this purpose, we manipulated beta cell glycogen metabolism using conditional genetic mouse models. We carried out two opposing strategies. On one hand, we generated a pancreas-specific Gys1 knockout (Gys1 KO) model to examine the function of the polysaccharide in beta cells. On the other hand, to establish whether excessive glycogen production is detrimental for beta cell survival, we generated a transgenic model overexpressing the scaffolding protein targeting to glycogen PTG (also known as protein phosphatase 1 regulatory subunit 3C or PPP1R3C) in these cells (PTGOE model). This protein acts as a hub between glycogen synthase (GS), protein phosphatase 1 and glycogen to promote glycogen synthesis [15]. This strategy has been used to successfully enhance glycogen accumulation in several tissues [16–18].
Methods
Animals
For the generation of the Gys1 KO model, conditional Gys1 mice [19] available at our facility were crossed with Pdx1-cre (Tg[Pdx1-cre]6Tuv) mice [20]. Gys1 loxp/loxp cre −/− (WTloxp) mice were used as controls. To drive the expression of PTG to beta cells (PTGOE model), mice conditionally expressing the coding sequence of the Ppp1r3c gene on the action of Cre recombinase [18] were crossed with rat insulin II promoter (RIP)-cre (C57BL/6-Tg[Ins2-cre]25Mgn) mice (Jackson Laboratory, Bar Harbor, ME, USA). RIP-cre mice were used as controls. All procedures were approved by the Animal Ethics/Research Committee of the University of Barcelona and carried out in accordance with the European Community Council Directive and National Institutes of Health guidelines for the care and use of laboratory animals. Mice were maintained on a 12/12 h light/dark cycle in a temperature- and humidity-controlled environment under specific pathogen-free conditions and allowed free access to water and a standard chow diet (Harlan, Indianapolis, IN, USA) or a high-fat diet (HFD) (60% energy from fat, D12492 Research Diets, New Brunswick, NJ, USA) for 16 weeks, starting at 6 weeks of age. After weaning, tail clippings were taken for genotyping (Transnetyx, Cordova, TN, USA). Animals were assigned to experiments randomly on condition of fulfilling the conditions of the experiment (genotype, sex, age). Experimenters were not blind to group assignment or outcome assessment.
Islet isolation and culture
Mouse and rat pancreatic islets were isolated as described previously [21, 22]. Islets were used either fresh or after culture in RPMI medium (Sigma, St Louis, MO, USA) at 5 or 25 mmol/l glucose.
Gene expression analysis
Total islet RNA was purified using RNeasy Micro Kit (Qiagen, Hilden, Germany) and quantified with NanoDrop 1000 (Thermo Scientific, Waltham, MA, USA). SuperScript III Reverse Transcriptase (Life Technologies, Carlsbad, CA, USA) was used to obtain cDNA. Real-time PCR was performed with 7900HT Real-Time PCR (Applied Biosystems, Foster City, CA, USA), with SYBR Green, and data were analysed following the \( {2}^{-\varDelta {\Delta \mathrm{C}}_{\mathrm{t}}} \) method and normalised to Tbp expression. For validation, Actb was used as a second housekeeping gene, though only results for Tbp are shown. Primer sequences are listed in electronic supplementary material (ESM) Table 1.
Western blot
Islets were homogenised in lysis buffer containing protease and phosphatase inhibitors (Roche, Basel, Switzerland) and subjected to three freeze/thaw cycles. Protein was quantified using the Bradford assay (Bio-Rad, Hercules, CA, USA). Proteins were separated in a 10% SDS-PAGE gel, transferred to a PVDF membrane, and immunoblotted with rabbit anti-GS 3893 (1:1,000, Cell Signaling, Danvers, MA, USA), rabbit anti-muscle GS MGS3 [23], rabbit anti-liver GS L1 [24], mouse anti-actin AC-40 (1:25,000, Abcam, Cambridge, UK) and horseradish peroxidase-linked (anti-rabbit from GE, Little Chalfont, UK and anti-mouse from Dako, Glostrup, Denmark) secondary antibodies. Antibodies were validated using tissue/cell protein extracts where the antigen in question is not expressed. Membranes were developed using Pierce ECL (Thermo Scientific). Bands were quantified using ImageJ (http://imagej.nih.gov/ij/).
In vitro insulin secretion
Islets were incubated for 30 min in Hepes-buffer Krebs-Ringer solution [21] with glucose at 2.8 mmol/l. Groups of six islets were incubated in a shaker at 37°C for 90 min in the solution described containing 2.8 or 16.7 mmol/l glucose. Media were collected to quantify secreted insulin. Alcohol-acid solution was added to the islets for insulin content determination. Insulin was measured using the Ultra Sensitive Mouse Insulin ELISA kit (Crystal Chem, Downers Grove, IL, USA). For each animal (n), four replicates (six islets each) per condition were done. Insulin secretion was expressed as percentage of total insulin content.
Glycogen quantification
Glycogen content was determined using a modification of a method described previously [25]. Briefly, 65 μl of 30% KOH was added to 50 islets (frozen in liquid nitrogen and kept at −80°C until the analysis) and heated for 15 min at 100°C. Glycogen was precipitated with cold 66% ethanol overnight, and the pellet was digested with a-amyloglucosidase. Glucose was converted into glucose 6-phosphate and measured using the fluorometric assay [26]. Note that glycogen can be underestimated in fresh-frozen islets as a result of glycogen degradation during isolation.
Histology
Mice were anaesthetised and perfused intracardially with 4% paraformaldehyde. Pancreases were harvested, fixed overnight at 4°C and embedded in paraffin. For immunofluorescence, sections of 5 μm thickness were blocked in 3% donkey serum/0.5% Triton X-100, incubated overnight at 4°C with guinea pig anti-insulin (1:1,000, Dako) and mouse anti-glucagon (1:1,000, Sigma), incubated for 1 h with secondary antibodies Alexa Fluor 555 anti-mouse or anti-guinea pig (1:400, Molecular Probes), Alexa Fluor 488 anti-guinea pig or Cy2 anti-mouse (1:500, Jackson Immunoresearch, West Grove, PA, USA), and then mounted with Hoechst. Specificity of staining was validated using secondary antibodies in the absence of primary antibodies. Images were obtained using a Leica DMI 6000 B inverted microscope with a Leica DFC360 FX camera (Wetzlar, Germany). For morphometric analysis, the insulin- or glucagon-positive area was measured in four sections (at least 90 μm apart) per pancreas using ImageJ, Life-Line version (http://fiji.sc). Beta or alpha cell mass was obtained by multiplying the hormone-positive area relative to total pancreatic area by the weight of the pancreas. The insulin-positive area was divided by the number of insulin-positive cells to obtain beta cell size. Glycogen was detected histologically by periodic acid–Schiff's reagent (PAS) staining.
Metabolic tests
For the i.p. GTT, mice were fasted for 16 h and injected with glucose (2 g/kg body weight for chow-diet-fed mice, 1 g/kg at 8 weeks of HFD and 0.75 g/kg at 16 weeks of HFD). Glycaemia was measured in the tail vein with a glucometer (Glucocard, Arkray, Kyoto, Japan) at the indicated time points. Blood samples were taken to measure insulin using the Ultra Sensitive Mouse Insulin ELISA kit.
Statistical analysis
Two-way analysis of variance was used for GTT data, and the unpaired Student’s t test for other experiments. A value of p < 0.05 was considered significant. Outliers were removed when they were 2 SD from the mean. Data are expressed as mean ± SEM.
Results
Generation of a pancreas-specific Gys1 KO mouse model
As an initial step, we determined the GS isoform expressed in rodent beta cells. In line with previous publications [27], we detected Gys1 but not Gys2 (liver isoform) transcripts in mouse islets (Fig. 1a). Correlating with the mRNA data, muscle GS but not liver GS was detected in total protein extracts from rat islets (ESM Fig. 1). Consequently, to study the role of glycogen metabolism in beta cell function in vivo, we sought to generate beta cell-specific (RIP-cre) Gys1 KO mice. However, no Gys1 KO mice were born, thus precluding the analysis of this model in postnatal life. Previous studies in rodents showed that glycogen infiltration in the pancreas is almost completely restricted to beta cells [10, 28] and that the glycogenic capacity of these cells is much higher than that of any other pancreatic cell type [29, 30]. We therefore undertook an alternative strategy and used a Pdx1-cre transgenic line to delete Gys1 from the whole pancreas from early embryonic stages. Pancreas-specific Gys1 KO mice were born at the expected Mendelian ratio, thus indicating that it was unlikely that failure to generate beta cell-specific Gys1 KO mice was due to beta cell-related embryonic lethality. Therefore, a more reasonable explanation would be that the cre transgene in RIP-cre mice is genetically linked to the Gys1 gene, thus impeding the inheritance of two Gys1 loxp alleles plus one copy of the cre transgene. In isolated islets from Gys1 KO mice, Gys1 mRNA was reduced by 85% and protein was undetectable by western blot (Fig. 1b–d).

Expression of muscle GS in islets and validation of Gys1 KO model. (a) Relative abundance of Gys1 and Gys2 transcripts expressed as \( {2}^{-{\Delta \mathrm{C}}_{\mathrm{t}}} \) using Tbp expression as reference (n = 4). (b) qPCR results for Gys1 in isolated islets normalised to Tbp (n = 8). Control values were set to 1. (c) Western blot showing the absence of MGS protein in Gys1 KO islets using the antibody GS 3893. Actin is shown as a loading control. Muscle protein extract from wild-type animals is shown as control. (d) Densitometric quantification of western blot shown in panel (c). ***p < 0.001. MGS, muscle GS
Gys1 KO mice show no metabolic alterations and have unaltered beta cell function
We characterised whole-body glucose metabolism in Gys1 KO mice at 12 weeks of age. No differences were found in body weight (Fig. 2a) and, after an overnight fast, glycaemia and insulinaemia did not differ from the values of controls (Fig. 2b, c). Glucose tolerance was unaffected in Gys1 KO mice (Fig. 2d). To evaluate beta cell function in the absence of GS, we performed insulin secretion assays in vitro using isolated islets, which revealed no differences in insulin synthesis and in the secretory capacity of Gys1 KO islets compared with controls (Fig. 2f). Likewise, the expression of functionally relevant genes was unchanged in Gys1 KO islets (Fig. 2h). Furthermore, older (45-week-old) Gys1 KO mice continued to exhibit glucose tolerance similar to that of age-matched controls (ESM Fig. 2). In all, these results show that glycogen metabolism is not required for adequate beta cell function and glucose homeostasis under physiological conditions.

Characterisation of Gys1 KO mice. (a) Weight (n = 10); (b) glycaemia (n = 22); (c) insulinaemia (n = 14); and (d) i.p. GTT (n = 9) after overnight fasting (WTloxp, white squares and dashed line; Gys1 KO, black circles and continuous line). (e) Plasma insulin levels at 0 and 30 min of the i.p. GTT (n = 5–6). (f) Glucose-stimulated insulin secretion in freshly isolated islets expressed as percentage of content (n = 5). (g) Insulin content per islet. (h) qPCR results for beta cell genes in isolated islets normalised to Tbp (n = 7). Control values were set to 1. WTloxp, white bars; Gys1 KO, black bars
Generation of a mouse model overexpressing PTG in beta cells
To study the contribution of glycogen overaccumulation to the progression of diabetes, we generated mice overexpressing PTG specifically in beta cells (PTGOE). Isolated pancreatic islets of this model had a 30-fold increase in mRNA levels of Ptg (also known as Ppp1r3c) (Fig. 3a) and 3.5-fold higher glycogen levels than islets from control mice (Fig. 3b). The latter measurement might be an underestimation because of glycogen degradation during islet isolation. In addition, PAS-positive staining confirmed increased glycogen accumulation in PTGOE mice and revealed a considerable degree of heterogeneity in the levels of glycogen among islets and individual beta cells within the same islet (Fig. 3d). In contrast, no differences in the levels of hypothalamic glycogen were found (ESM Fig. 3).

Validation of the PTGOE model. (a) qPCR results for Ppp1r3c in isolated islets normalised to Tbp (n = 3). Control values were set to 1. (b) Glycogen quantification in freshly isolated islets (fed n = 8, fasted n = 2). (c) Glycogen quantification in islets cultured at the indicated glucose concentrations for 24 h (5 mmol/l n = 3, 25 mmol/l n = 8). RIP-cre, white bars; PTGOE, black bars. (d) PAS staining of pancreatic sections obtained from 18-week-old mice. Glycogen is shown in purple/magenta. Scale bars, 100 μm. **p < 0.01 and ***p < 0.001
To determine whether glycogen stores in beta cells of PTGOE mice can be mobilised, we quantified glycogen in islets isolated from mice fasted overnight. Interestingly, glycogen levels were negligible in both groups of mice after this period (Fig. 3b). Next, we compared glycogen accumulation in islets from PTGOE mice fed ad libitum and control islets cultured in either low (5 mmol/l) or high (25 mmol/l) glucose for 24 h. Glycogen levels were almost undetectable in both groups at 5 mmol/l glucose, while at 25 mmol/l the islets from PTGOE mice presented 6.4 times more glycogen than controls (Fig. 3c), reaching values that were nearly three times higher than those in freshly isolated islets from fed mice. Thus, in contrast to other models of PTG overexpression [31], glycogen accumulation in beta cells of PTGOE mice is subject to regulation and can be modulated both in vivo in response to fasting and in vitro by incubation at a range of glucose concentrations.
PTGOE mice show fasting hypoglycaemia and hyperinsulinaemia but unaltered glucose tolerance
We next assessed whether increased glycogenic capacity in beta cells from PTGOE mice resulted in alterations in whole-body glucose homeostasis. At 14 weeks of age, PTGOE mice and controls showed similar body weights (Fig. 4a). In response to fasting, the former exhibited lower glycaemia and increased insulinaemia (Fig. 4b, c). Despite these differences, glucose tolerance curves were comparable between groups (Fig. 4d).

Characterisation of PTGOE mice. (a) Weight (n = 11); (b) glycaemia (n = 18); (c) insulinaemia (n = 8); and (d) i.p. GTT (n = 9) after overnight fasting (RIP-cre, white squares and dashed line; PTGOE, black circles and continuous line). (e) Plasma insulin levels at 0 and 30 min of the i.p. GTT (n = 4). Note that mice carrying the RIP-cre transgene exhibit blunted in vivo glucose-induced insulin secretion [40]. (f) Glucose-stimulated insulin secretion in freshly isolated islets expressed as percentage of content (n = 8). (g) Insulin content per islet. (h) qPCR results for beta cell genes in isolated islets normalised to Tbp (n = 3). RIP-cre, white bars; PTGOE, black bars. **p < 0.01 and ***p < 0.001
The stimulation of beta cell glycogenolysis under low glucose conditions may provide fuel to stimulate insulin secretion when glucose availability is low [32]. Hence, we assessed whether islets isolated from ad libitum-fed PTGOE mice (high glycogen levels in beta cells) showed enhanced insulin secretion under non-stimulatory conditions. Islet insulin content and insulin secretion at 2.8 and 16.7 mmol/l glucose (Fig. 4f, g) or in response to forskolin (ESM Fig. 4) were indistinguishable between PTGOE and controls, thereby indicating that glycogen does not affect insulin production or islet secretory function. At the mRNA level, we observed no changes in the expression of genes that are essential for sustaining beta cell function (Fig. 4h).
Glycogen accumulation is not detrimental for beta cell mass in PTGOE mice
Given that both beta cell death and glycogen accumulation are observed in the course of diabetes, we addressed whether there is a causal relationship between these two events. We examined whether increased glycogen deposition in PTGOE mice affected beta cell survival. We performed morphometric quantification of beta cell area in fixed pancreases from 6-week-old mice. We did not find differences in beta cell mass, beta cell size or islet size distribution between controls and transgenic animals (Fig. 5a–c). Similarly, alpha cell mass was unaltered in PTGOE mice (Fig. 5d). We considered the possibility that a detrimental effect of glycogen overaccumulation in beta cells would manifest in the long term. However, 60-week-old PTGOE mice presented no differences in beta cell mass (Fig. 5e). Islet morphology and architecture did not show obvious alterations (Fig. 5f). Furthermore, whole-body glucose tolerance was similar between aged PTGOE mice and controls (ESM Fig. 5). Therefore, sustained glycogen storage alone does not result in significant beta cell loss during normal ageing, at least at the glycogen levels reached in our PTGOE model.

Histological evaluation of PTGOE mice. (a, b) Beta cell mass and beta cell size at 6 weeks of age (RIP-cre n = 3, PTGOE n = 4). (c) Islet size distribution. RIP-cre, white bars; PTGOE, black bars. (d) Alpha cell mass at 6 weeks of age (n = 3). (e) Beta cell mass at 60 weeks of age (n = 5). (f) Representative images of the pancreas showing unaffected islet structure in PTGOE mice. Nuclei (blue), insulin (green) and glucagon (red). Scale bars, 50 μm
An HFD does not reveal a beta cell phenotype in Gys1 KO or PTGOE mice
Finally, to study whether stress conditions induce a phenotype in our transgenic models, we exposed them to an HFD. After 16 weeks of diet, Gys1 KO mice weighed more, had increased fasting glycaemia and showed a tendency towards higher insulinaemia and worsened glucose tolerance (Fig. 6a–d). However, insulin secretory function and beta cell mass did not differ from that found in controls also on an HFD (Fig. 6e–g). Thus, the worsened glucose tolerance of Gys1 KO mice is probably related to exacerbated insulin resistance (possibly due to hypothalamic deletion of Gys1 by expression of cre under the Pdx1 promoter [33]) rather than to a beta cell-autonomous role. In support of this notion, after 8 weeks on an HFD, when body weights were similar between Gys1 KO and controls (Fig. 6h), fasting glycaemia, fasting insulinaemia and glucose tolerance were indistinguishable between groups (Fig. 6i–k). PTGOE mice fed an HFD for 16 weeks exhibited no significant differences in body weight, fasting glycaemia, insulinaemia or glucose tolerance relative to controls (Fig. 7).

Characterisation of Gys1 KO mice on an HFD. (a–g) 16 weeks of HFD. (h–k) 8 weeks of HFD. (a) Weight; (b) glycaemia; (c) i.p. GTT after overnight fasting; and (d) insulin levels during the i.p. GTT (WTloxp n = 9–12, Gys1 KO n = 10–13). (e) Glucose-stimulated insulin secretion in freshly isolated islets expressed as percentage of total insulin content (WTloxp n = 5, Gys1 KO n = 3). WTloxp, white bars; Gys1 KO, black bars. (f) Insulin content per islet. (g) Beta cell mass (n = 3). (h) Weight; (i) glycaemia; (j) insulinaemia; and (k) i.p. GTT (WTloxp n = 11, Gys1 KO n = 12) after overnight fasting. (c, k) WTloxp, white squares and dashed line; Gys1 KO, black circles and continuous line. *p < 0.05, **p < 0.01 and ***p < 0.001; † p = 0.06

Characterisation of PTGOE mice after 16 weeks on an HFD. (a) Weight; (b) glycaemia; (c) i.p. GTT after overnight fasting; and (d) insulin levels during the i.p. GTT (n = 4). RIP-cre, white squares and dashed line; PTGOE, black circles and continuous line
Discussion
Here, we sought to shed new light on the long-standing debate regarding the relevance of glycogen metabolism in beta cells and the potential role of this process as a mediator of glucotoxicity, which leads to beta cell dysfunction and death. Previous studies [2, 4, 12, 32] had relied mainly on ex vivo and in vitro cellular models. Here, we used conditional genetic mouse models that exhibit impaired glycogen synthesis (Gys1 KO) or excessive glycogen accumulation (PTGOE) in beta cells. Our results show that the incapacity of beta cells to synthesise glycogen does not compromise whole-body glucose homeostasis or insulin secretion. Conversely, excessive glycogen accumulation in beta cells is associated with higher fasting insulin levels but not with augmented beta cell dysfunction or death under normal physiological conditions or in response to an HFD.
Pederson et al [3] showed that whole-body deletion of Gys1 in mice has a beneficial effect on glucose tolerance. This finding was attributed to increased circulating levels of insulin after a glucose challenge, which was proposed to result from enhanced glucose 6-phosphate levels and/or improved beta cell oxidative capacity. However, our data demonstrate that loss of GS does not affect insulin secretion in a beta cell-autonomous manner, thus implying that the high insulin response in constitutive Gys1 KO mice was probably secondary to alterations in other organs. In this regard, IL-6, which is more potently secreted by the muscle in conditions of low glycogen availability [34], has been shown to stimulate insulin release through glucagon-like peptide 1 (GLP-1)-dependent mechanisms [35]. Our data strongly support the notion that glycogen synthesis in beta cells is not relevant for the maintenance of proper glucose homeostasis. In contrast, our observation that exacerbated glucose intolerance of Gys1 KO mice after an HFD is associated with increased weight gain in the absence of obvious pancreatic changes points to a role of glycogen metabolism in the brain on the control of whole-body energy homeostasis.
However, it is still possible that glycogen serves to fine-tune biochemical pathways and energy metabolism in beta cells. Thus, only under specific challenging situations (other than diet-induced obesity) might the inability of beta cells to synthesise glycogen have a measurable impact on beta cell function and whole-body glucose homeostasis. For instance, tissue-specific functions of glycogen have been observed in adipocytes, where it provides glycerol for triacylglycerol synthesis in the fasting to feeding transition [36] and modulates lipid metabolism after a switch from a high-fat to a standard chow diet [37]. Likewise, in neurons, glycogen has been shown to contribute to tolerance to hypoxia [38].
In the opposite scenario, when glycogen overaccumulates in beta cells, glucose tolerance is also not affected, further supporting the minor impact of beta cell glycogen on whole-body glucose homeostasis. It should be noted that in beta cells of PTGOE mice, in contrast to other mouse models of PTG overexpression [31], glycogen stores can be mobilised when extracellular glucose is low. In line with prior work [4], glycogenolysis in beta cells of PTGOE mice after an overnight fast may stimulate insulin release and explain increased insulin levels when these animals are subjected to fasting. However, these differences in basal glycaemia and insulinaemia were not present when PTGOE mice received an HFD. These subtle differences may be masked by the characteristic hyperglycaemia and hyperinsulinaemia of this condition. Remarkably, basal insulin release was similar in isolated islets of PTGOE and control mice. Thus, other signals such as circulating factors or neural innervation may influence insulin release under low glucose conditions in PTGOE mice. Alternatively, temporal differences between the in vivo (16 h) and in vitro (1.5 h) assays may explain the divergence in insulin output. PTG is known to modify the phosphorylation and activity of glycogen phosphorylase [39], resulting in reduced glycogenolytic rates [18]. It is thus plausible that the short incubation times of the in vitro insulin secretion assays precludes detection of the effects of glycogenolysis in this setting.
The PTGOE mice did not show any signs of compromised beta cell survival. We acknowledge the possibility that the amount of glycogen deposited in the islets of these animals may be below levels found in islets from diabetic counterparts and thus not be pathological. Unfortunately, because of the high post-mortem turnover of glycogen, there is no quantitative information on how much of this polysaccharide is stored in beta cells of diabetic patients. Of note, the incubation of rat islets at extremely high glucose concentration (83.3 mmol/l) [4] results in glycogen levels that are within the range stored in the islets of the PTGOE model used in the present study. It is thus reasonable to consider that these levels may represent the maximum glycogen storage capacity of beta cells (which are not ‘designed’ as an energy storage tissue). Alternative genetic manoeuvres, including the overexpression of GS itself, may be needed to enhance further glycogen synthesis.
In conclusion, our present data support a marginal role of beta cell glycogen in whole-body glucose homeostasis, insulin secretion and beta cell survival under physiological conditions and in response to an HFD. The availability of the genetic mouse models used in this study warrants further work to address the impact of altered glycogen metabolism in glucose homeostasis under other pathological conditions.
Abbreviations
- GS:
-
Glycogen synthase
- Gys1 KO :
-
Gys1 knockout
- HFD:
-
High-fat diet
- PAS:
-
Periodic acid–Schiff's reagent
- PTG:
-
Protein targeting to glycogen
- PTGOE :
-
PTG overexpression
- RIP:
-
Rat insulin II promoter
References
Carpenter AM, Lazarow A (1967) Effects of hyper- and hypoglycemia on beta cell degranulation and glycogen infiltration in normal, subdiabetic and diabetic rats. Diabetes 16:493–501
Hellman B, Idahl LA (1969) Presence and mobilization of glycogen in mammalian pancreatic beta cells. Endocrinology 84:1–8
Pederson BA, Schroeder JM, Parker GE et al (2005) Glucose metabolism in mice lacking muscle glycogen synthase. Diabetes 54:3466–3473
Malaisse WJ, Sener A, Koser M et al (1977) The stimulus-secretion coupling of glucose-induced insulin release. Insulin release due to glycogenolysis in glucose-deprived islets. Biochem J 164:447–454
Weir GC, Laybutt DR, Kaneto H et al (2001) Beta cell adaptation and decompensation during the progression of diabetes. Diabetes 50:S154–S159
Robertson RP, Harmon J, Tran PO, Poitout V (2014) Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 53:S119–S124
Toreson WE (1951) Glycogen infiltration (so-called hydropic degeneration) in the pancreas in human and experimental diabetes mellitus. Am J Pathol 27:327–347
Malaisse WJ, Like AA, Malaisse-Lagae F et al (1968) Insulin secretion in vitro by the pancreas of the sand rat (Psammomys obesus). Diabetes 17:754–759
Ravelli RBG, Kalicharan RD, Avramut MC et al (2013) Destruction of tissue, cells and organelles in type 1 diabetic rats presented at macromolecular resolution. Sci Rep 3:1804
Graf R, Klessen C (1981) Glycogen in pancreatic islets of steroid diabetic rats. Histochemistry 73:225–232
Sasaki M, Arai T, Usui T, Oki Y (1991) Immunohistochemical, ultrastructural, and hormonal studies on the endocrine pancreas of voles (Microtus arvalis) with monosodium aspartate-induced diabetes. Vet Pathol 28:497–505
Malaisse WJ, Marynissen G, Sener A (1992) Possible role of glycogen accumulation in B cell glucotoxicity. Metabolism 41:814–819
Duran J, Gruart A, García-Rocha M et al (2014) Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet 23:3147–3156
Duran J, Tevy MF, Garcia-Rocha M et al (2012) Deleterious effects of neuronal accumulation of glycogen in flies and mice. EMBO Mol Med 4:719–729
Printen JA, Brady MJ, Saltiel AR (1997) PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science 275:1475–1478
Jurczak MJ, Danos AM, Rehrmann VR et al (2007) Transgenic overexpression of protein targeting to glycogen markedly increases adipocytic glycogen storage in mice. Am J Physiol Endocrinol Metab 292:E952–E963
Villarroel-Espíndola F, Maldonado R, Mancilla H et al (2013) Muscle glycogen synthase isoform is responsible for testicular glycogen synthesis: glycogen overproduction induces apoptosis in male germ cells. J Cell Biochem 114:1653–1664
López-Soldado I, Zafra D, Duran J et al (2015) Liver glycogen reduces food intake and attenuates obesity in a high-fat diet-fed mouse model. Diabetes 64:796–807
Duran J, Saez I, Gruart A et al (2013) Impairment in long-term memory formation and learning-dependent synaptic plasticity in mice lacking glycogen synthase in the brain. J Cereb Blood Flow Metab 33:550–556
Hingorani SR, Petricoin EF, Maitra A et al (2003) Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4:437–450
Altirriba J, Gasa R, Casas S et al (2010) The role of transmembrane protein 27 (TMEM27) in islet physiology and its potential use as a beta cell mass biomarker. Diabetologia 53:1406–1414
Fernandez-Ruiz R, Vieira E, Garcia-Roves PM, Gomis R (2014) Protein tyrosine phosphatase-1B modulates pancreatic β-cell mass. PLoS One 9:1–10
Cid E, Cifuentes D, Baqué S et al (2005) Determinants of the nucleocytoplasmic shuttling of muscle glycogen synthase. FEBS J 272:3197–3213
García-Rocha M, Roca A, de la Iglesia N et al (2001) Intracellular distribution of glycogen synthase and glycogen in primary cultured rat hepatocytes. Biochem J 357:17–24
Chan TM, Exton JH (1976) A rapid method for the determination of glycogen content and radioactivity in small quantities of tissue or isolated hepatocytes. Anal Biochem 71:96–105
Zhu A, Romero R, Petty HR (2009) An enzymatic fluorimetric assay for glucose-6-phosphate: application in an in vitro Warburg-like effect. Anal Biochem 388:97–101
Nishimura M, Yokoi N, Miki T et al (2004) Construction of a multi-functional cDNA library specific for mouse pancreatic islets and its application to microarray. Mol Cell 323:315–323
Graf R, Tölken M (1984) Ultrastructural distribution of glycogen in pancreatic islets of steroid diabetic rats. Basic Appl Histochem 28:391–397
Malaisse WJ, Ladrière L, Cancelas J et al (2001) Pancreatic and hepatic glycogen content in normoglycemic and hyperglycemic rats. Mol Cell Biochem 219:45–49
Doherty M, Malaisse WJ (2001) Glycogen accumulation in rat pancreatic islets: in vitro experiments. Endocrine 14:303–309
Berman HK, O’Doherty RM, Anderson P, Newgard CB (1998) Overexpression of protein targeting to glycogen (PTG) in rat hepatocytes causes profound activation of glycogen synthesis independent of normal hormone- and substrate-mediated regulatory mechanisms. J Biol Chem 273:26421–26425
Malaisse WJ, Maggetto C, Leclercq-Meyer V, Sener A (1993) Interference of glycogenolysis with glycolysis in pancreatic islets from glucose-infused rats. J Clin Invest 91:432–436
Wicksteed B, Brissova M, Yan W et al (2010) Conditional gene targeting in mouse pancreatic beta cells. Diabetes 59:3090–3098
Keller C, Steensberg A, Pilegaard H et al (2001) Transcriptional activation of the IL-6 gene in human contracting skeletal muscle: influence of muscle glycogen content. FASEB J 15:2748–2750
Ellingsgaard H, Hauselmann I, Schuler B et al (2011) Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 17:1481–1489
Tuerkischer E, Wertheimer E (1942) Glycogen and adipose tissue. J Physiol 100:385–409
Markan KR, Jurczak MJ, Allison MB et al (2010) Enhanced glycogen metabolism in adipose tissue decreases triglyceride mobilization. Am J Physiol Endocrinol Metab 299:E117–E125
Saez I, Duran J, Sinadinos C et al (2014) Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab 34:945–955
Green AR, Aiston S, Greenberg CC et al (2004) The glycogenic action of protein targeting to glycogen in hepatocytes involves multiple mechanisms including phosphorylase inactivation and glycogen synthase translocation. J Biol Chem 279:46474–46482
Lee JY, Ristow M, Lin X et al (2006) RIP-Cre revisited, evidence for impairments of pancreatic b-cell function. J Biol Chem 281:2649–2653
Acknowledgements
The authors thank N. Plana, E. Veza and A. Adrover from IRB Barcelona (Spain) and A. García and Y. Esteban from CIBERDEM (Spain) for their technical assistance. Thanks also go to T. Yates (IRB Barcelona) for correcting the English version of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This work has been supported by grants from the Spanish Ministerio de Economía y Competitividad (BFU2011-30554 to JJG and SAF2014-5254-P to JJG), the Ministerio de Economía y Competitividad/Instituto de Salud Carlos III (PI13/01500 to RGa), CIBER de Diabetes y Enfermedades Metabólicas Asociadas and Instituto de Salud Carlos III (to JJG and RGo), the Generalitat de Catalunya (2014 SGR659 to RGo) and the European Foundation for the Study of Diabetes (EFSD/Novo Nordisk Programme 2012 to JJG).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
JMC, JD, FS and MGR designed and performed the experiments. JJG, RGo and RGa designed the study. JMC, JD, FS and RGa analysed and interpreted the data. JMC, JD and RGa wrote the manuscript. All authors critically reviewed the paper and approved the final version to be published. JJG is responsible for the integrity of this work as a whole.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 50 kb)
ESM Fig. 2
(PDF 25.4 kb)
ESM Fig. 3
(PDF 19.2 kb)
ESM Fig. 4
(PDF 63 kb)
ESM Fig. 5
(PDF 141 kb)
ESM Table 1
(PDF 285 kb)
Rights and permissions
About this article

Cite this article
Mir-Coll, J., Duran, J., Slebe, F. et al. Genetic models rule out a major role of beta cell glycogen in the control of glucose homeostasis. Diabetologia 59, 1012–1020 (2016). https://doi.org/10.1007/s00125-016-3871-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3871-1