
Abstract
Aims/hypothesis
Glucagon-like peptide (GLP-1), an intestinal incretin produced in L cells through proglucagon processing, is released in response to meal intake. The intracellular mechanism by which L cells sense the organism energy level to coordinate the production of GLP-1 remains unclear. Mechanistic target of rapamycin (mTOR) is an intracellular fuel sensor critical for energy homeostasis. In this study, we investigated whether intestinal mTOR regulates GLP-1 production in L cells.
Methods
The effects of mTOR on GLP-1 production were examined in lean- or high-fat diet (HFD) induced diabetic C57/BL6, db/db, Neurog3-Tsc1 −/− mice, and STC-1 cells. GLP-1 expression was investigated by real-time PCR and western blotting. Plasma GLP-1 and insulin were detected by enzyme immunoassay and radioimmunoassay, respectively.
Results
Fasting downregulated mTOR activity, which was associated with a decrement of intestinal proglucagon and circulating GLP-1. Upon re-feeding, these alterations returned to the levels of fed animals. In HFD induced diabetic mice, ileal mTOR signalling, proglucagon and circulating GLP-1 were significantly decreased. Inhibition of mTOR signalling by rapamycin decreased levels of intestinal and plasma GLP-1 in both normal and diabetic mice. Activation of the intestinal mTOR signalling by l-leucine or Tsc1 gene deletion increased levels of intestinal proglucagon and plasma GLP-1. Overexpression of mTOR stimulated proglucagon promoter activity and GLP-1 production, whereas inhibition of mTOR activity by overexpression of tuberous sclerosis 1 (TSC1) or TSC2 decreased proglucagon promoter activity and GLP-1 production in STC-1 cells.
Conclusions/interpretation
mTOR may link energy supply with the production of GLP-1 in L cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glucagon-like peptides (GLPs) and glucagon are hormones encoded by the same proglucagon gene (Gcg) and liberated by tissue-specific post-translational processing [1]. Glucagon is predominantly generated in pancreatic alpha cells by prohormone convertase (PC)-2, whereas GLP-1 and GLP-2 are mainly produced in the intestine and central nervous system after cleavage by PC-1/3 [2–4]. GLP-1, a potent glucose-dependent insulinotropic hormone derived from intestinal L cells, is primarily produced during the postprandial state to promote insulin secretion and to decrease glucagon secretion [1]. The insulinotropic activity of GLP-1 in type 2 diabetes mellitus therefore offers great potential for treatment of hyperglycaemia without causing hypoglycaemia [5, 6]. Targeting an increase in endogenous GLP-1 production through modulation of proglucagon gene transcription in enteroendocrine L cells provides an exciting new therapeutic opportunity. In mammals, the major fragment of proglucagon mRNA contains two glucagon-like sequences named GLP 1 and 2. The proglucagon promoter contains several transcriptional control elements localised in the 2.5 kb 5′-upstream sequence of the transcriptional initiation site [7]. A number of transcription factors control proglucagon gene expression in a tissue-specific manner, allowing physiologically precise regulation of production of different active peptides [8]. Insulin and glycogen synthase kinase (GSK)-3β inhibitors, such as lithium, stimulate G2 response element activity specifically in enteroendocrine L cells through the β-catenin/T cell factor (TCF)-4 signalling pathway, resulting in increased GLP-1 production [9]. Nutrients, vagal nerve innervation and peptides constitutively regulate GLP-1 secretion [10]. The intracellular signalling mechanisms by which enteroendocrine L cells coordinate luminal nutrients with GLP-1 production remain poorly understood.
Mechanistic target of rapamycin (mTOR), a highly conserved serine–threonine kinase, has been reported to serve as a fuel sensor [11]. Aberrant mTOR activity is linked to the development of cancer, diabetes and obesity [12]. Downstream targets of mTOR include ribosomal protein S6 kinases (S6Ks), S6 and eukaryotic translation initiation factor 4E binding protein 1 [12]. In contrast, deletion of S6K1 protects against diet induced obesity and improves insulin sensitivity in mice [13]. There are two mTOR complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 is responsible for nutrient-sensing functions and is composed of mTOR, G protein-subunit-like protein and raptor. mTORC2 phosphorylates Akt and contains mTOR and rictor [14]. mTORC1 also coordinates the overall energy supply with energy intake. Our previous studies demonstrate that gastric mTOR activity is altered by changes in energy status such as fasting and obesity. Change in gastric mTOR activity affects the expression and secretion of ghrelin and nesfatin-1/nucleobindin 2 (NUCB2), with subsequent alteration in food intake and glucose homeostasis [15–18]. Here, we present evidence that mTOR signalling regulates ileal production of GLP-1. Our data identify intestine specific regulation of proglucagon gene expression as a new mechanism of action for mTOR in glucose metabolism, thus expanding its interest as a target for the treatment of type 2 diabetes mellitus.
Methods
Materials
Details of materials can be found in the electronic supplementary material [ESM] Methods.
Animal experiments
Sixteen-week-old male C57BL/6J mice were divided into control, fasted and refed groups. Male C57BL/6J, db/m and db/db mice fed with normal chow diet (NCD) or 45% high-fat diet (HFD) were separated into DMSO, rapamycin, saline and leucine groups. Sixteen-week-old male Neurog3-Tsc1 −/− mice and Tsc1 loxP/loxP littermates fed with NCD were used. Animals used in this study were handled in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996). All animal protocols were approved by the Animal Care and Use Committee of Jinan University. Details of animals used can be found in the ESM Methods.
Molecular biology methods
Western blot analysis, quantitative real-time PCR, glucose tolerance and insulin tolerance tests were performed as described previously [15, 16]. See ESM Methods for further details. Measurements of GLP-1 and insulin secretion were performed according to the manufacturer’s protocol. See ESM Methods for further details. Rat GLP-1 promoter-luciferase expression vectors were constructed by PCR amplification using rat genomic DNA (BD Biosciences, Franklin Lakes, NJ, USA) and subcloned into the promoterless pGL3-basic luciferase reporter vector at the XhoI/Hind-III cloning sites (Promega, Madison, WI, USA). Luciferase activity with the dual-luciferase reporter assay system was measured using an aluminometer (Monolight 2010, San Diego, CA, USA), according to the manufacturer’s instructions. See ESM Methods for further details.
Tissue sample preparations and immunofluorescence analysis
C57BL/6J mice were deeply anaesthetised using pentobarbital. The ileums and pancreas were quickly removed and rinsed thoroughly with PBS. The tissues were fixed in 4% paraformaldehyde (wt/vol.), dehydrated, embedded in wax and sectioned at 6 μm. Paraffin embedded sections were de-waxed, re-hydrated and rinsed in PBS. After boiling for 10 min in 10 mmol/l sodium citrate buffer (pH 6.0), sections were blocked in 1% BSA (wt/vol.) in PBS for 1 h at room temperature, then incubated overnight with rabbit anti-TSC1 (1:100) antibody or goat anti-insulin (1:100) antibody. Tissue sections were then incubated at 22°C for 2 h with the following secondary antibodies: chicken anti-rabbit Texas Red conjugated IgG (1:100) or donkey anti-goat Texas Red conjugated IgG (1:100). Controls included substituting primary antibody with rabbit IgG and goat IgG. The nuclei were visualised by staining with Hoechst 33258 for 10 min. Photomicrographs were taken under a confocal laser-scanning microscope (Leica, Germany). Image analysis (Model Leica Q550CW, Leica) was performed to quantify the immunostaining signals of insulin.
Statistical analysis
All values are expressed as means ± SEM. Statistical differences were evaluated by two-way ANOVA and Newman–Student–Keuls test. Comparisons between two groups involved use of the Student’s t test. A p value < 0.05 denotes statistical significance.
Results
Effects of organism energy status on ileal mTOR signalling and GLP-1 production in mice
To examine the effect of fasting on mTOR signalling and GLP-1 production, male C57BL/6J mice were divided into control, fasted and refed group (see ESM Methods for details). Phosphorylation of S6, a downstream target of mTOR [14], in ileum was markedly decreased in fasted mice relative to fed animals but was reversed by 2 h re-feeding (ESM Fig. 1a). Both mRNA and protein levels of ileal proglucagon were significantly decreased after fasting, and returned to basal levels with re-feeding (ESM Fig. 1b, a). The alteration in ileal proglucagon was accompanied by a similar change in levels of circulating GLP-1 (6.2 ± 1.0, 3.4 ± 0.6, 13.2 ± 3.5 pmol/l for normal feeding, fasting and re-feeding, respectively; ESM Fig. 1c). A significant positive correlation was found between ileal levels of phospho-S6 and proglucagon (Pearson’s = 0.8; p < 0.05).
The effects of long-term changes in nutritional status on ileal mTOR signalling were examined using the male mice fed 45% HFD. As shown in ESM Fig. 1d, there was a significant decrease in ileal phospho-S6 in mice fed HFD relative to the animals fed NCD. The alteration in mTOR signalling was associated with a decrement in ileal proglucagon mRNA and protein, and plasma GLP-1 (ESM Fig. 1d–f) in mice fed HFD relative to those fed NCD.
Effects of ileal mTOR signalling on GLP-1 production in mice
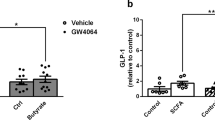
If mTOR signalling is linked to the production of GLP-1, then changes in ileal mTOR activity would be predicted to alter the production and secretion of GLP-1. The effects of rapamycin, a well-characterised mTOR inhibitor, were examined in normal C57BL/6J mice, HFD induced or db/db diabetic mice. In both normal (NCD) and diabetic (HFD) mice, i.p. injection of rapamycin significantly decreased ileal phospho-S6 (Ser235/236; Fig. 1a), ileal proglucagon mRNA (Fig. 1b) and protein (Fig. 1a), and circulating GLP-1 (Fig. 1c). The change of plasma GLP-1 was associated with impaired glucose metabolism and glucose disposal curves typifying insulin resistance induced by rapamycin (Fig. 1d, e). Similar alteration was observed for db/db diabetic mice (Fig. 1f–i).

Modulation of GLP-1 production by rapamycin in mice. C57BL/6J mice, HFD induced diabetic mice (a–e), db/m and db/db mice (f–i) were administrated (i.p.) with DMSO or rapamycin (Rapa, 1 mg/kg) for 6 days to determine the effects of mTOR inhibition. Representative western blots are shown for pS6 and proglucagon in the ileum and the quantified results expressed as means ± SEM (a, f). S6 and β-actin were used as loading controls. Proglucagon mRNA (b, g), plasma GLP-1(active) (c, h), results of OGTTs (d, i) and insulin tolerance tests (e) in mice receiving DMSO (dashed lines, white circles for NCD + DMSO or db/m + DMSO; dashed lines, white triangles for HFD + DMSO or db/db + DMSO) or rapamycin (Rapa, 1 mg/kg: solid lines, black squares for NCD + Rapa or db/m + Rapa; solid lines, black triangles for HFD + Rapa or db/db + Rapa) are expressed as means ± SEM. n = 6. *p < 0.05 vs NCD or db/m control; **p < 0.01 vs NCD or db/m control; † p < 0.05 vs HFD or db/db control
Because inhibition of mTOR signalling in the ileal mucosa leads to downregulation of GLP-1 expression and GLP-1 secretion, it is expected that activation of mTOR signalling in the ileal mucosa would stimulate production of GLP-1. Consistent with this prediction, intragastric administration of l-leucine (3.5 mmol/kg for 6 days), a branched-chain amino acid documented to activate mTOR signalling [15], significantly increased phospho-S6 (Fig. 2a). The increment in phosphorylation of S6 was associated with an upregulation in ileal proglucagon mRNA (Fig. 2b) and protein (Fig. 2a), and plasma GLP-1 (Fig. 2c) in both normal C57BL/6J and HFD induced diabetic mice compared with saline administration. The change of plasma GLP-1 was associated with improved glucose metabolism (Fig. 2d) and glucose disposal curves typifying improved insulin sensitivity (Fig. 2e). Similar findings were observed when l-leucine was injected i.p. (ESM Fig. 2).

Stimulation of GLP-1 by activation of ileal mTOR signalling. Effects of l-leucine or deletion of Tsc1 in neurogenin 3 positive cells are shown in (a–e) and (f–h), respectively. l-leucine (3.5 mmol/kg) was administrated daily by intragastric gavage into C57BL/6J mice or HFD induced diabetic mice for 6 days. Male 16-week-old Tsc1 loxP/loxP (WT) mice were used as the control for Neurog3-Tsc −/− mice (TN). Results are expressed as means ± SEM. n = 6. *p < 0.05 vs NCD + saline or Tsc1 loxP/loxP (WT) mice; † p < 0.05 vs HFD + saline. (a, f) Representative western blots for TSC1, phospho-S6 (pS6) and proglucagon. (b, g) Proglucagon mRNA was analysed by real-time PCR. (c, h) Plasma GLP-1(active) was measured by enzyme immunoassay. (d, e) OGTTs and insulin tolerance tests (dashed lines, white circles for NCD + saline; solid lines, black squares for NCD + leucine; dashed lines, white triangles for HFD + saline; solid lines, black triangles for HFD + leucine)
To further determine the effect of mTOR activation on the production of GLP-1, we used a mouse strain Neurog3-Tsc1 −/− in which Tsc1 gene is deleted under the control of the neurogenin 3 promoter [19]. TSC1 immunoreactivity was detected mainly in the basal 1/3 of ileal epithelium in Tsc1 loxp-loxp wild-type (WT) mice, whereas it demonstrated a limited expression in Neurog3-Tsc1 −/− (TN) mice (ESM Fig. 3). Phosphorylation of S6 was detected at a relative high level in the ileums of Neurog3-Tsc1 −/− mice compared with WT mice (Fig. 2f), suggesting an activation of mTOR in the ileums. Activation of mTOR signalling was associated with an upregulation of ileal proglucagon mRNA and protein (Fig. 2g, f), and circulating GLP-1 (Fig. 2h) compared with WT mice.
Effects of rapamycin on GLP-1 and insulin secretion in response to glucose administration
In rodents and humans, GLP-1 is released rapidly into the circulation after oral nutrient ingestion [1]. To determine whether rapamycin regulates GLP-1 secretion in response to glucose administration, we analysed the dynamic change of plasma GLP-1 induced by glucose administration in mice treated with rapamycin (see ESM Methods for details). Both basal and glucose-induced increments in levels of plasma GLP-1 were significantly attenuated in mice treated with rapamycin relative to control animals (3.7 ± 0.2 vs 2.4 ± 0.2 pmol/l for basal GLP-1 and 6.2 ± 0.3 vs 4.5 ± 0.5 pmol/l for maximal increase of GLP-1 after glucose administration, p < 0.001; ESM Fig. 4a).
Plasma insulin markedly increased after glucose administration in control mice treated with DMSO. In contrast, glucose failed to stimulate an increment in plasma insulin levels in mice treated with rapamycin, suggesting the impairment in response to glucose in these animals (ESM Fig. 4b). The changes of plasma GLP-1 and insulin were accompanied with glucose intolerance and insulin resistance induced by rapamycin (ESM Fig. 4c, d). No significant difference in plasma insulin was observed in mice treated with rapamycin relative to control mice at 0 time point (68.9 ± 14.9 vs 51.1 ± 13.9 pmol/l, p = 0.36; ESM Fig. 4b). Furthermore, no significant difference in islet morphology and insulin immunoreactivity was observed (ESM Fig. 5).
In vitro effects of mTOR signalling on GLP-1 production
We next examined the direct effect of mTOR signalling on GLP-1 in cultured STC-1 cells, an L-like cell line. Exposure of STC-1 cells to rapamycin at the doses of 0.625–10 nmol/l for 24 h caused a concentration-dependent decrease in S6 phosphorylation (Fig. 3a), proglucagon mRNA (Fig. 3b) and protein (Fig. 3a), and medium content of GLP-1 (Fig. 3c). Rapamycin at 5 nmol/l significantly decreased S6 phosphorylation, proglucagon mRNA and protein levels, and GLP-1 secretion in a time-dependent manner in STC-1 cells. Significant inhibition occurred as early as 12 h and lasted for up to 36 h (Fig. 3d–f).

Regulation of mTOR signalling and GLP-1 expression by rapamycin in STC-1 cells. Representative western blots of phospho-S6 (pS6), proglucagon, S6 and β-actin in STC-1 cells exposed to varying concentrations of rapamycin for 24 h (a), or exposed to rapamycin (5 nmol/l) for the indicated times (d). Normalised intensity of phospho-S6 (pS6) and proglucagon (a, d), proglucagon mRNA (b, e) and medium GLP-1(active) concentration (c, f) are expressed as means ± SEM. Experiments were repeated three times, each time in triplicate. *p < 0.05 vs control
l-leucine induced a concentration-dependent increase in GLP-1 production. l-leucine, at doses of 9.375–150 mmol/l for 24 h, caused a concentration-dependent increase in S6 phosphorylation, proglucagon mRNA and protein levels (Fig. 4a–c). Treatment of cells with 75 mmol/l leucine for 24 h significantly enhanced the GLP-1 mRNA expression and GLP-1 release by 300 ± 15% and 200 ± 27%, respectively (p < 0.05; Fig. 4b, a). l-leucine also time-dependently stimulated phosphorylation of S6 (Fig. 4d–f), expression of proglucagon mRNA (Fig. 4e) and protein (Fig. 4d), and GLP-1 secretion (Fig. 4f) in STC-1 cells. Stimulation of GLP-1 production and secretion occurred as early as 12 h and persisted for up to 36 h.

Effects of l-leucine on mTOR signalling and GLP-1 expression in STC-1 cells. Representative western blots of phospho-S6 (pS6), proglucagon, S6 and β-actin in STC-1 cells incubated for 24 h with increasing concentrations of leucine (a), or exposed to leucine (75 mmol/l) for the indicated times (d). Quantification of ileal pS6 and proglucagon (a, d), proglucagon mRNA (b, e) and medium GLP-1(active) concentration (c, f) in STC-1 cells are expressed as means ± SEM. *p < 0.05 vs control. Experiments were repeated three times, each time in triplicate
Consistent with the pharmacological blockage of mTOR signalling, overexpressing TSC1 or Tsc2 significantly reduced proglucagon expression and GLP-1 secretion (Fig. 5a–c). Both TSC1 and TSC2 function to inhibit mTOR signalling (Fig. 5a) in cultured STC1 cells. Further, activation of mTOR signalling by overexpression of mTOR or S6K1 significantly increased the expression and secretion of GLP-1 in cultured STC1 cells (Fig. 5a–f).

Modulation of GLP-1 production by mTOR signalling in STC-1 cells. (a–c) Effects of overexpression of mTOR, TSC1 or Tsc2. STC1 cells were transfected for 48 h with one of the following plasmids: mTOR, TSC1 or Tsc2, or GFP using lipofectamine. Phospho-S6, proglucagon, S6 and β-actin (a), proglucagon mRNA (b) and medium GLP-1(active) concentration (c) were measured and expressed as means ± SEM. Experiments were repeated three times, each time in triplicate. *p < 0.05 vs control, **p < 0.01 vs control. (d–f) Effects of overexpression of S6K1. STC-1 cells were infected with Ad-S6K1 or Ad-GFP adenovirus (106 pfu) for 48 h. Alterations in pS6, proglucagon, S6 and β-actin protein levels (d), proglucagon mRNA (e) and GLP-1 secretion (f) were detected and results expressed as means ± SEM. n = 3. *p < 0.05 vs control, ***p < 0.001 vs control
In vitro effects of mTOR signalling on the activity of proglucagon promoter
The short form of proglucagon gene promoter (about 1,250 nucleotides) directs its gene expression to pancreatic islets and brain, whereas additional upstream sequences extending to ∼2,250 nucleotides are required for its expression in intestinal endocrine cells [20]. To determine whether mTOR signalling modulates the transcription of the proglucagon gene in L-like cells, we assayed the activity of the 2.3 kb rat proglucagon promoter. As shown in Fig. 6a, rapamycin concentration-dependently inhibited the 2,288 bp but not the 1,250 bp proximal promoter activity in STC-1 cells. This observation suggests that downregulation of mTOR signalling specifically decreases the intestinal proglucagon promoter activity. Consistent with this observation, inhibition of mTOR signalling by overexpression of TSC1 or Tsc2 significantly decreased the 2,288 bp promoter activity (Fig. 6b), whereas activation of mTOR signalling by overexpression of mTOR stimulated the 2,288 bp GLP-1 promoter activity (Fig. 6b) in STC-1 cells. Further, alteration of mTOR activity by rapamycin or overexpression of mTOR, TSC1 or Tsc2 did not affect the promoter activity of 2,288 bp constructs in MIN6 cells (Fig. 6c, d).

Modulation of proglucagon promoter activity by mTOR signalling. (a, c) Inhibition of proglucagon (Gcg) promoter activity by rapamycin. STC-1 (a) or MIN6 (c) cells were transfected with pGL3 basic (white bars), Gcg (−1,250 bp, black bars) or Gcg (−2,288 bp, grey bars) and treated with rapamycin. (b, d) Effects of mTOR, TSC1 or Tsc2 overexpression on Gcg promoter activity. STC-1 (b) or MIN6 (d) cells were cotransfected with the proglucagon promoter and one of the following plasmids: GFP, mTOR, TSC1 or Tsc2. Relative luciferase activity was measured and normalised to the internal control pSV-β-galactosidase. Results are expressed as means ± SEM. Experiments were repeated by three separate transfections, each transfection in triplicate. *p < 0.05 vs control
Discussion
The major finding of the present study is that mTOR signalling in L cells functions to alter the expression and secretion of GLP-1. This conclusion is supported by the following distinct observations: (1) a positive relation exists between intestinal mTOR signalling and the expression and secretion of GLP-1 during changes in energy status; (2) inhibition of mTOR signalling suppresses ileal proglucagon mRNA and protein content in both normal and diabetic mice; (3) conversely, activation of intestinal mTOR signalling by leucine or deletion of Tsc1 gene increases the expression and secretion of GLP-1; (4) inhibition of intestinal mTOR signalling by rapamycin inhibits plasma active GLP-1 and insulin release in response to glucose administration; (5) activation of mTOR increases the −2,288 bp proglucagon promoter activity, while blocking mTOR signalling decreases its activity in STC1 cells.
Enteroendocrine L cells, mainly located in the distal ileum and colon [21], are open-type endocrine cells with a slender triangular form with the base resting on the basal lamina. L cells can sense the luminal nutrients and transform this information into a stimulation of GLP-1 secretion [22]. We thus speculate that GLP-1 biosynthesis can be modulated by nutrient-sensing molecules. Our studies indicate that intestinal mTOR may coordinate luminal nutrients with production of GLP-1. First, mTOR signalling molecules exist within the intestinal mucosa. Second, alterations in intestinal mTOR signalling during short-term energy deficiency and long-term energy surplus suggest that intestinal L cells may recognise energy status at the organism level and trigger the coordinate expression and secretion of GLP-1. Furthermore, the reciprocal suppression in intestinal mTOR signalling and GLP-1 production is detected in HFD induced diabetic and db/db mice. This observation is consistent with a previous report showing an inverse correlation between obesity and GLP-1 secretion [23]. Third, and most convincingly, alteration of mTOR activities in both intestinal mucosa and cultured L-like cells leads to a reciprocal change in GLP-1 production. Rapamycin suppresses, while l-leucine enhances, GLP-1 biosynthesis both in mice and in cultured cells. Neurog3-Tsc1 −/− transgenic mice in which mTOR signalling is activated by deletion of Tsc1 gene in L cells demonstrate a significant increase in GLP production. Transcription factor neurogenin 3 specifically drives stem cells into enteroendocrine cell types such as L cells [24], thus deletion of Tsc1 driven by the neurogenin 3 promoter is supposed to activate mTOR signalling mainly in enteroendocrine cells including the intestinal L cells. Indeed, we have observed the islet hypertrophy and hyperinsulinaemia in Neurog3-Tsc1 −/− transgenic mice (data not shown). The increase in GLP-1 levels induced by activation of mTOR in the intestinal L-like cells may partially contribute to the hyperinsulinaemia observed in Neurog3-Tsc1 −/− transgenic mice. Our data thus provide evidence that both pharmacological and genetic interventions of mTOR signalling in intestinal L cells alter GLP-1 biosynthesis in mice. The effect of mTOR signalling on GLP-1 production appears to occur through its regulation on the long form proglucagon promoter. These observations collectively show that direct control of GLP-1 expression and secretion can occur at the level of intestinal mucosa, and that mTOR signalling is a crucial control point in GLP-1 expression and secretion.
Limitations exist for our approaches that seek to assess the physiological role of mTOR signalling in the control of GLP-1 production. Pharmacological intervention of mTOR activity may have a profound effect in cells other than L-like cells. Previous studies have demonstrated that alteration of mTOR signalling by rapamycin or l-leucine may affect insulin release through either a direct effect on islet beta cells or an indirect effect secondary to its modulation of glucose metabolism in liver, skeletal muscle and adipose tissue [25–28]. While our data on Neurog3-Tsc1 −/− mice and cultured cells provide evidence of the direct influence of GLP-1 by mTOR signalling, complete exclusion of the secondary effects from mTOR signalling in other cells requires a conditional manipulation of mTOR signalling in L-like cells. In addition, the slow response to a high dose of l-leucine in our study indicates that mTOR is unlikely to underlie the acute triggering of postprandial GLP-1 secretion after nutrient ingestion. Instead, it may function as a mechanism for chronic adaption to energy supplies. Absence of insulin response after rapamycin treatment also indicates the GLP-1-independent effects on islet function. Functional data from islets isolated from mice treated for 6 days with rapamycin will address this potential.
As an insulinotropic and glucagonostatic hormone that inhibits appetite and reduces body weight, GLP-1 is currently under active investigation as a therapy for type 2 diabetes [29]. Current efforts focus on GLP-1 supplement and attenuation of GLP-1 breakdown. Liraglutide and exenatide are two GLP-1-mimetic drugs currently approved for clinical use to treat type 2 diabetes [30, 31]. While GLP-1 analogues can efficiently reduce blood glucose level in patients with type 2 diabetes, they have adverse effects on thyroid glands because GLP-1 receptors are expressed in thyroid glands of humans [32] as well as rodents [33]. In preclinical animal studies, liraglutide was associated with an increased risk of thyroid C-cell focal hyperplasia and C-cell tumours [33]. Another safety concern of liraglutide is a risk of pancreatitis [34]. Active forms of GLP-1 such as GLP-1(7-36) and GLP-1(7-37) are susceptible to cleavage the enzyme dipeptidyl peptidase IV (DPP-IV) [35]. The metabolite thus generated, GLP 9-36 amide or GLP 9-36, is inactive and may act as a competitive antagonist at the GLP-1 receptor [36]. Inhibition of DPP-IV increases blood concentration of GLP-1, which portrays a very low risk of hypoglycaemia [37]. DPP-IV inhibitors can significantly reduce levels of glycosylated haemoglobin (HbA1c), either as monotherapy or as a combination with other glucose-lowering agents [38, 39]. Adverse side effects such as pancreatitis and precancerous pancreatic lesion have been reported to be associated with DPP-IV inhibitors [40–42]. Our study suggests that increasing production of GLP-1 by targeting mTOR signalling may provide a novel strategy for the treatment of type 2 diabetes. Pharmacological and genetic inhibition of mTOR signalling decreases the production of GLP-1, which is associated with the impairment in glucose tolerance, glucose-stimulated insulin secretion and insulin sensitivity induced by rapamycin in mice. If the relationship of mTOR signalling with GLP-1 production exists in normal and diabetic patients, it might be feasible to target mTOR fuel-sensing mechanisms in the intestinal mucosa to treat type 2 diabetes.
In conclusion, our study indicates the physiological relevance of ileal mTOR to glucose homeostasis. mTOR signalling in intestinal mucosa may coordinate overall fuel levels with production and secretion of GLP-1. The existence of a fuel-sensing pathway within the intestinal mucosa thus provides an alternative opportunity of a therapy for type 2 diabetes.
Abbreviations
- DPP-IV:
-
Dipeptidyl peptidase IV
- GLP-1:
-
Glucagon-like peptide 1
- HFD:
-
High-fat diet
- mTOR:
-
Mechanistic target of rapamycin
- mTORC:
-
mTOR complexes
- NCD:
-
Normal chow diet
- PC:
-
Prohormone convertase
- S6:
-
Ribosomal protein S6
- S6K:
-
S6 kinase
- TSC:
-
Tuberous sclerosis
- WT:
-
Wild-type
References
Baggio LL, Drucker DJ (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132:2131–2157
Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF (1986) Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J Biol Chem 261:11880–11889
Drucker DJ, Mojsov S, Habener JF (1986) Cell-specific post-translational processing of preproglucagon expressed from a metallothionein-glucagon fusion gene. J Biol Chem 261:9637–9643
Philippe J, Mojsov S, Drucker DJ, Habener JF (1986) Proglucagon processing in a rat islet cell line resembles phenotype of intestine rather than pancreas. Endocrinology 119:2833–2839
Elahi D, McAloon-Dyke M, Fukagawa NK et al (1994) The insulinotropic actions of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (7-37) in normal and diabetic subjects. Regul Pept 51:63–74
Meier JJ, Gallwitz B, Salmen S et al (2003) Normalization of glucose concentrations and deceleration of gastric emptying after solid meals during intravenous glucagon-like peptide 1 in patients with type 2 diabetes. J Clin Endocrinol Metab 88:2719–2725
Jin T, Drucker DJ (1995) The proglucagon gene upstream enhancer contains positive and negative domains important for tissue specific proglucagon gene transcription. Mol Endocrinol 9:1306–1320
Jin T (2008) Mechanisms underlying proglucagon gene expression. J Endocrinol 198:17–28
Yi F, Brubaker PL, Jin T (2005) TCF-4 mediates cell type-specific regulation of proglucagon gene expression by beta-catenin and glycogen synthase kinase-3beta. J Biol Chem 280:1457–1464
Mansour A, Hosseini S, Larijani B, Pajouhi M, Mohajeri-Tehrani MR (2013) Nutrients related to GLP-1 secretory responses. Nutrition 29:813–820
Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G (2001) Mammalian TOR: a homeostatic ATP sensor. Science 294:1102–1105
Inoki K, Corradetti MN, Guan KL (2005) Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 37:19–24
Um SH, Frigerio F, Watanabe M et al (2007) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431:200–205
Yang Q, Guan KL (2007) Expanding mTOR signaling. Cell Res 17:666–681
Xu G, Li Y, An W et al (2009) Gastric mammalian target of rapamycin signaling regulates ghrelin production and food intake. Endocrinology 150:3637–3644
Xu G, Wang Z, Li Y et al (2012) Ghrelin contributes to derangements of glucose metabolism induced by rapamycin in mice. Diabetologia 55:1813–1823
Li Z, Xu G, Li Y, Zhao J, Mulholland MW, Zhang W (2012) mTOR-dependent modulation of gastric nesfatin-1/NUCB2. Cell Physiol Biochem 29:493–500
Li Z, Gao L, Tang H et al (2013) Peripheral effects of nesfatin-1 on glucose homeostasis. PLoS One 8, e71513
Li D, Han L, Li Y, Zhao J, He P, Zhang W (2014) Neurogenin 3–directed Cre deletion of Tsc1 gene causes pancreatic acinar carcinoma. Neoplasia 16:909–917
Lee YC, Asa SL, Drucker DJ (1992) Glucagon gene 5′-flanking sequences direct expression of simian virus 40 large T antigen to the intestine, producing carcinoma of the large bowel in transgenic mice. J Biol Chem 267:10705–10708
Mortensen K, Christensen LL, Holst JJ, Orskov C (2003) GLP-1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regul Pept 114:189–196
Eissele R, Göke R, Willemer S et al (1992) Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur J Clin Investig 22:283–291
Verdich C, Toubro S, Buemann B, Lysgård Madsen J, Juul Holst J, Astrup A (2001) The role of postprandial releases of insulin and incretin hormones in meal-induced satiety–effect of obesity and weight reduction. Int J Obes Relat Metab Disord 25:1206–1214
Schonhoff SE, Giel-Moloney M, Leiter AB (2004) Minireview: development and differentiation of gut endocrine cells. Endocrinology 145:2639–2644
Fraenkel M, Ketzinel-Gilad M, Ariav Y et al (2008) mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 57:945–957
Barlow AD, Nicholson ML, Herbert TP (2013) Evidence for rapamycin toxicity in pancreatic β-cells and a review of the underlying molecular mechanisms. Diabetes 62:2674–2682
Yang J, Dolinger M, Ritaccio G et al (2012) Leucine stimulates insulin secretion via down-regulation of surface expression of adrenergic α2A receptor through the mTOR (mammalian target of rapamycin) pathway: implication in new-onset diabetes in renal transplantation. J Biol Chem 287:24795–24806
Fuhrmann A, Lopes P, Sereno J et al (2014) Molecular mechanisms underlying the effects of cyclosporin A and sirolimus on glucose and lipid metabolism in liver, skeletal muscle and adipose tissue in an in vivo rat model. Biochem Pharmacol 88:216–228
Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W (1993) Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7-36 amide) in type 2 (noninsulin-dependent) diabetic patients. Diabetologia 36:741–744
Buse JB, Rosenstock J, Sesti G et al (2009) Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet 374:39–47
Kolterman OG, Buse JB, Fineman MS et al (2003) Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 88:3082–3089
Gier B, Butler PC, Lai CK, Kirakossian D, DeNicola MM, Yeh MW (2012) Glucagon like peptide-1 receptor expression in the human thyroid gland. J Clin Endocrinol Metab 97:121–131
Bjerre Knudsen L, Madsen LW, Andersen S et al (2010) Glucagon-like peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation. Endocrinology 151:1473–1486
Parks M, Rosebraugh C (2010) Weighing risks and benefits of liraglutide–the FDA’s review of a new antidiabetic therapy. N Engl J Med 9:774–777
Deacon CF, Johnsen AH, Host JJ (2002) Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab 80:952–957
Knudsen LB, Pridal L (1996) Glucagon-like peptide-1-(9-36) amide is a major metabolite of glucagon-like peptide-1-(7-36) amide after in vivo administration to dogs, and it acts as an antagonist on the pancreatic receptor. Eur J Pharmacol 318:429–435
Mulvihill EE, Drucker DJ (2014) Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev 35:992–1019
Charbonnel B, Karasik A, Liu J, Wu M, Meininger G (2006) Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 29:2638–2643
Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, de Williams-Herman (2006) Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 29:2632–2637
Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC (2011) Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 141:150–156
Matveyenko AV, Dry S, Cox HI et al (2009) Beneficial endocrine but adverse exocrine effects of sitagliptin in the human islet amyloid polypeptide transgenic rat model of type 2 diabetes: interactions with metformin. Diabetes 58:1604–1615
Butler AE, Campbell-Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC (2013) Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes 62:2595–2604
Acknowledgements
We thank K. Guan (University of California San Diego, San Diego, CA, USA) for providing mTOR, TSC1 and Tsc2 plasmids.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31401001 and 81330010), the Guangdong Medical Science Research Foundation (A2014375), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (20141685), the Fundamental Research Funds for the Central Universities (11614318) and American Diabetes Association Grant 1-13-BS-225.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author contributions
WZ, GX and ZL designed the research; GX, LD, HT, SG, HL and HW contributed to the acquisition of data; GX, ZL, LD and HW analysed the data; and GX and WZ wrote the paper. All authors contributed to the discussion and revised the article, and all approved the final version of the manuscript. WZ is responsible for the integrity of the work as a whole.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 147 kb)
ESM Fig. 1
(PDF 568 kb)
ESM Fig. 2
(PDF 314 kb)
ESM Fig. 3
(PDF 538 kb)
ESM Fig. 4
(PDF 216 kb)
ESM Fig. 5
(PDF 868 kb)
Rights and permissions
About this article

Cite this article
Xu, G., Li, Z., Ding, L. et al. Intestinal mTOR regulates GLP-1 production in mouse L cells. Diabetologia 58, 1887–1897 (2015). https://doi.org/10.1007/s00125-015-3632-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3632-6