
Abstract
Aims/hypothesis
More than 90% of Chinese familial early-onset type 2 diabetes mellitus is genetically unexplained. To investigate the molecular aetiology, we identified and characterised whether mutations in the KCNJ11 gene are responsible for these families.
Methods
KCNJ11 mutations were screened for 96 familial early-onset type 2 diabetic probands and their families. Functional significance of the identified mutations was confirmed by physiological analysis, molecular modelling and population survey.
Results
Three novel KCNJ11 mutations, R27H, R192H and S116F117del, were identified in three families with early-onset type 2 diabetes mellitus. Mutated KCNJ11 with R27H or R192H markedly reduced ATP sensitivity (E23K>R27H>C42R>R192H>R201H), but no ATP-sensitive potassium channel currents were detected in the loss-of-function S116F117del channel in vitro. Molecular modelling indicated that R192H had a larger effect on the channel ATP-binding pocket than R27H, which may qualitatively explain why the ATP sensitivity of the R192H mutation is seven times less than R27H. The shape of the S116F117del channel may be compressed, which may explain why the mutated channel had no currents. Discontinuation of insulin and implementation of sulfonylureas for R27H or R192H carriers and continuation/switch to insulin therapy for S116F117del carriers resulted in good glycaemic control.
Conclusions/interpretation
Our results suggest that genetic diagnosis for the KCNJ11 mutations in familial early-onset type 2 diabetes mellitus may help in understanding the molecular aetiology and in providing more personalised treatment for these specific forms of diabetes in Chinese and other Asian patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maturity-onset diabetes of the young (MODY) is a monogenic, autosomal dominant, early-onset form of non-autoimmune diabetes caused by primary pancreatic beta cell dysfunction [1], and more than ten MODY genes have been identified [2, 3]. The clinical phenotype of familial early-onset type 2 diabetes mellitus is often characterised by insulin resistance, in contrast with MODY, which is similar to later-onset type 2 diabetes [4]. Heterozygous activating mutations in KCNJ11 have been reported as a cause of not only permanent neonatal diabetes (PNDM) [2, 5] but also MODY and adult-onset diabetes in a number of studies [6]. This was reaffirmed in a French MODY gene-negative pedigree last year [7], wherein the defined KCNJ11 gene was proposed to be MODY13.
KCNJ11 on 11p15.1 is a single open reading frame encoding a 390-amino acid protein, potassium inwardly-rectifying channel Kir6.2, which contains two putative transmembrane (TM) segments and a pore loop domain, H5 (Fig. 1a) [8, 9]. ATP-sensitive potassium channels (KATP) control electrical signalling by coupling cellular metabolism to potassium ion movement across cell membranes. Pancreatic beta cell KATP channels comprise two components: four subunits of Kir6.2 forming the channel pore, and the sulfonylurea receptor, SUR1, regulating channel gating [10]. The KATP channel is sensitive to ATP and inhibited by sulfonylureas [11], drugs that are widely used to treat type 2 diabetes and regulate insulin secretion by coupling the metabolic state of the cell to membrane potential.

Identification of KCNJ11 mutations. (a) Schematic illustration of KCNJ11 and the corresponding domains in Kir6.2. Numbers refer to the amino acids bordering the domains. Filled arrows indicate the mutations identified in KCNJ11. Dashed arrows indicate single-nucleotide polymorphisms (SNPs) identified in KCNJ11. The gene borders were determined based on mammalian homology using published data [8, 9]. N, N-terminus; C, C-terminus; TM1, first TM domain; H5, pore loop domain; TM2, second TM domain. (b) Alignment of specific regions of KCNJ11/Kir6.2 from different mammals using ClustalX. (1) Entire 68-amino acid N-terminal domain of KCNJ11/Kir6.2; the R27 residue is indicated. (2) Portion of the C-terminal domain of KCNJ11/Kir6.2; the R192 residue is indicated. (c) Fragment of the KCNJ11/Kir6.2 sequence; the S116F117del is indicated. The corresponding amino acid sequences of wild-type Kir6.2 and the S116F117del mutation (mt) are shown; the S116F117del results in truncated Kir6.2 lacking two amino acids (Ser116 and Phe117)
Mutations (e.g. E227K) [7] and variations (e.g. E23K) [12] in KCNJ11 have already been linked to diabetes. In Kir6.2 knockout mice, both glucose- and tolbutamide-induced insulin secretion and membrane depolarisation and calcium influx into beta cells are defective, indicating that the regulation of insulin secretion depends on KATP channel activity [13]. A high frequency of beta cell apoptosis is observed in Kir6.2G132S transgenic mice before the appearance of hyperglycaemia, suggesting that KATP channels also play a significant role in beta cell survival [14].
We screened a cohort of 96 MODY gene-negative probands with early-onset autosomal dominant type 2 diabetes, and their families, for KCNJ11 mutations, and report here three novel heterozygous KCNJ11 mutations associated with MODY gene-negative autosomal dominantly inherited type 2 diabetes.
Methods
Recruitment of diabetic index cases and families
We recruited the families for studies on the genetics of type 2 diabetes at the Shanghai Diabetes Institute, Shanghai Diabetic Clinical Medical Center. Briefly, families were selected if their pattern of type 2 diabetes was consistent with autosomal dominant inheritance. An additional selection criterion was the availability of a large number of family members (with and without diabetes) who agreed to participate in the study. The screening criteria for eligible families were: (1) at least one index case (early-onset type 2 diabetes diagnosed <40 years old; range 12–39 years old); (2) index case treatment by dietary control or oral agents for the first 2 years; (3) diabetes in at least three generations; (4) index cases without the mitochondrial DNA 3243 A-to-G point mutation, as confirmed by PCR–RFLP analysis, using ApaI (Promega, Madison, WI, USA), as described by Fukui et al [15] with slight modifications; (5) index cases without mutations in the following six MODY genes [16]: HNF4α/MODY1, GCK/MODY2, HNF1α/MODY3, PDX1/MODY4, HNF1β/MODY5 and NEUROD1/BETA2/MODY6, confirmed by PCR-direct sequencing, as previously reported with some modifications [17–22]. No deletions of these six MODY genes were excluded by performing multiplex ligation-dependent probe amplification (MLPA) in the set of the 96 early-onset type 2 diabetic probands. To date, we have recruited and examined 96 families of Han Chinese and measured their height, weight and blood pressure, and blood was obtained for DNA extraction and biochemical measurements, including blood glucose, triacylglycerol, total cholesterol, HbA1c, C-peptide and insulin. From participants without diabetes and diabetic patients treated with oral agents or diet, we took blood again 2 h after an OGTT (75 g glucose) to determine blood glucose, C-peptide and insulin. In addition, fasting serum C-peptide in insulin-treated individuals was measured.
We selected 109 non-diabetic participants according to the following criteria: over 65 years of age (age 73.2 ± 5.7 years; BMI 21.1 ± 2.4 kg/m2 [means ± SD]), normal glucose tolerance, HbA1c < 5.6% (38 mmol/mol) and no family history of diabetes. All participants completed medical and family history questionnaires; this information was supplemented with information from medical records. The ADA criteria were used to diagnose diabetes and impaired glucose tolerance (IGT) [23].
All patients underwent a standardised clinical and laboratory evaluation. Serum antibodies against GAD and protein tyrosine phosphatase-like protein (IA-2) were tested using a radioimmunoprecipitation kit with 125I-labelled GAD65 and 125I-labelled IA-2, according to the manufacturer’s recommendations (RSR, Cardiff, UK). The criterion for a positive assay for each antibody was an index >1.0 (2 SDs above the mean value in normal controls). Serum insulin and C-peptide were measured by an RIA (Linco Research, St Charles, MO, USA). The insulin assay showed little cross-reactivity (<2%) with human proinsulin. Triacylglycerol and total cholesterol were determined by enzymatic procedures using an autoanalyser (Hitachi 7600-020; Hitachi, Tokyo, Japan). All the patients in this study had negative results for GAD and IA-2 antibodies and 3243 A-to-G point mutation.
This study was approved by the institutional review board of Shanghai Jiaotong University Affiliated Sixth People’s Hospital. Written informed consent was obtained from all participants.
Sequencing of KCNJ11
We used two pairs of primers to sequence the entire coding sequence and flanking sequences of KCNJ11 (forward, 5′-CGAGAGGACTCTGCAGTGAG-3′, reverse, 5′-GCTTGCTGAAGATGAGGGTC-3′; and forward, 5′-CATCGTGCAGAACATCG-TG-3′, reverse, 5′-TAACACCCTGGATGAGCAG-3′) [6]. PCR was performed using each pair of specific primers at 95°C for 5 min, followed by 30 cycles of denaturation at 95°C for 1 min, annealing at 58°C for 1 min, extension at 72°C for 1 min and a final extension of 10 min on the Gene Amp PCR system 9700 thermocycler (PE Applied Biosystems, Foster City, CA, USA). Amplified DNA was purified using the QIAquick PCR Purification Kit (Qiagen, Shanghai, China) and sequenced from both ends using specific primers and the BigDye Terminator cycle sequencing kit (version 3.1; Applied Biosystems). Electrophoresis was performed on the ABI PRISM 3100 Genetic Analyzer, and ABI PRISM Sequencing Analysis software version 3.7 was used for analysis. We confirmed the sequence of the three mutations by cloning the PCR product of the entire coding sequence of the genes into a TA cloning vector (PCR 2.1; Invitrogen, Shanghai, China) and sequencing. Sequences were compared with the published sequence (NM_000525.3) using Sequence Navigator (Applied Biosystems). Novel mutations were tested for cosegregation with diabetes in other family members and the 109 normal individuals of Han Chinese origin.
Parametric linkage analysis
Genotyping data were calculated using the MLINK subprogram from the LINKAGE package (version 5.1) for two-point linkage analysis. Autosomal dominant inheritance was assumed, with a disease-gene frequency of 0.0001 and 80% penetrance.
Construction of the three KCNJ11 mutants
Mammalian expression plasmids containing the whole coding regions of human KCNJ11 and ABCC8 (also known as SUR1) have been described previously [6, 8]. Human KCNJ11 cDNA was subcloned into pCMV vector (Promega, Madison, WI, USA), and a single-stranded template of pCMV KCNJ11 was prepared using the helper phage R408. Expression plasmids with R27H, R192H or S116F117del mutations were generated by site-directed mutagenesis.
Cell culture and DNA transfection
COS-1 cells were cultured in DMEM supplemented with 10% FBS. For single-channel recordings, 1 × 105 cells were plated in 35 mm dishes, and transfected with 1.5 μmol/l pCMV6c carrying wild-type ABCC8 and 1.5 μmol/l pCMV6b carrying wild-type KCNJ11 or mutated KCNJ11 (R27H, R192H or S116F117del) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions; pEGFP-N1 (Clontech, Palo Alto, CA, USA) encoding green fluorescent protein was also cotransfected as a reporter gene [14]. The cells were cultured for 48–72 h before electrophysiological recordings.
Electrophysiology
Single-channel recordings were performed using the excised inside–out membrane patch configuration, as previously described [14]. Single-channel currents were analysed using a combination of pCLAMP (version 9.0; Axon Instruments, Foster City, CA, USA) and in-house software. The ATP sensitivity of the wild-type and mutant channels was determined using an Axopatch 200B patch-clamp amplifier (Axon Instruments). Sulfonylurea sensitivity was assessed as the ratio between the amplitudes of the KATP channel currents before and after application of 100 μmol/l tolbutamide. Experiments were conducted for both the wild-type (n = 15) and mutant (Kir6.2 R27H/SUR1, n = 10; Kir6.2 R192H/SUR1, n = 9) channels. The results are expressed as means ± SE; Mann–Whitney U tests (detectable rate of the channels) or unpaired Student’s t tests were used to test for statistical significance.
Model building of Kir6.2
To further explore the effect of the mutations on the function of Kir6.2, we constructed a molecular model of the C-terminus of Kir6.2, based on the crystal structure of Kir2.1 [24]. The N-terminal domains of these proteins share 53.1% sequence identity. The sequences were aligned using ClustalX (Clustal X version 2.0, www.clustal.org/clustal2/). Homology models were generated using CPHmodels-2.0 Server and swiss-model. The model of Kir6.2 (residues 1-177) was based on the crystal structures of the bacterial inwardly rectifying K+ channel (KirBac3.1; PDB entry 1U4F) and the intracellular domain of the rat Kir3.1 channel (PDB entry 1X6L). The TM region of Kir channels is highly conserved. The level of conservation between the TM regions of KirBac3.1 and rat Kir3.1 is 36%, indicating that the TM region of KirBac3.1 would be an appropriate model for the TM segment of Kir3.1. The TM region was modelled as a tetramer with fourfold symmetry imposed. The three segments of the model (TMs, N and C domains) were constructed separately and then joined together. The structure constructed is similar to a previous report [25] and is a truncated form of Kir6.2 in the absence of SUR1. However, this predicted Kir6.2 model is hypothetical and requires further verification. Homology modelling was also performed for the pore-forming inwardly rectifying potassium channel Kir6.2 subunits, to provide a rational explanation for the effects of the mutations.
Results
Genetic analysis
Six KCNJ11 variants were identified in the MODY gene-negative families: three known single-nucleotide polymorphisms (SNPs) (E23K [rs5219], A190A [rs5218] and I337V [rs5215]) and three novel mutations (c.80G>A, p.R27H; c.575G>A, p.R192H; and c.348-353delCTTCTC, p.S116F117del) (Fig. 1a). The frequencies of the three SNPs (E23K [rs5219], A190A [rs5218] and I337V [rs5215]) were similar in the 96 index cases and 109 unrelated non-diabetic individuals (39.1% vs 38.5%, 43.7% vs 46.8% and 40.0% vs 38.5%, respectively). The three novel mutations (R27H, R192H and S116F117del) were detected in three individual index cases, but not in the other probands or 109 non-diabetic controls, suggesting that these mutations are not simple polymorphisms.
Bioinformatics and functional analysis of mutant channels
The index case from family a had an amino acid substitution in codon 27 (NM_000525.3: c.80G>A, p.R27H) (Fig. 1b). R27H is located in the cytosolic region of the N-terminal domain close to the ATP-binding sites (R201 and K185) in the C-terminus of a single subunit of Kir6.2 in steric conformation (Fig. 2). As Arg is positively charged, R27H may reduce the ligand-binding affinity of Kir6.2, by reducing positive charge distribution and increasing steric repulsion between two adjacent ATP-binding pocket monomers.

Final modelled ribbon structure of two subunits of Kir6.2, indicating the location of the novel KCNJ11 mutations. (a) The homology model of Kir6.2, which is quite similar to the previously reported structure of the other isoconformer, corresponds most closely to a truncated form of Kir6.2 expressed in the absence of SUR1. The labelled residues shown in yellow stick format are S116, F117, R27 and R192, which are indicated with solid arrows. The ATP-binding site is composed of the residues shown in the dark-blue stick model, R201 and K185, which are indicated with dashed arrows. (b) Schematic of the expected mixture of channels with different subunit compositions when wild-type and mutant Kir6.2 are coexpressed (as in the heterozygous state). The expected relative numbers of channel types for wild-type and mutant subunits are indicated above the figure, and segregate independently (i.e. follow a binomial distribution)
The index case from family b had an amino acid substitution in codon 192 (NM_000525.3: c.575G>A, p.R192H) (Fig. 1b), located in the cytosolic region of the C-terminal domain of Kir6.2. R192 is one of the positively charged donors in the three electrostatic interaction pairs (E229–R314, R301–E292 and R192–E227) between two adjacent subunits of Kir6.2, which are essential for stability of the Kir6.2 tetramer structure and pore-forming channel [25].
PSI-BLAST analysis demonstrated that Arg27 or Arg192 residues are evolutionally well-conserved in mammals, suggesting the functional importance of the two residues. Our proposed structure model (Fig. 2) indicates that R27 faces towards the interface of the Kir6.2 and SUR subunits, suggesting that R27H may have a greater influence on the interaction between Kir6.2 and SUR than R192H. Both R27H and R192H mutations show significant elevations in IC50 compared with the wild-type of Kir6.2, and R192H had a sevenfold lower sensitivity to ATP in vitro than R27H (Fig. 3a), suggesting that both R27H and R192H reduce the sensitivity of the KATP channel to ATP-induced closure.

ATP sensitivity and sulfonylurea sensitivity of the wild-type and mutant KATP channels. (a) ATP sensitivity. Patch-clamp experiments were performed for the wild-type (Kir6.2/SUR1, black circles, n = 10) and mutant (Kir6.2 R27H/SUR1, white squares, n = 10; Kir6.2 R192H/SUR1, white triangles, n = 8) channels. The mutant channels had significantly higher IC50 values than the wild-type channels (92.3 ± 9.6 vs 20.2 ± 2.4 μmol/l, p = 2.66 × 10−5; 652.7 ± 178.0 vs 20.2 ± 2.4 μmol/l, p = 9.14 × 10−7), indicating that the mutant KATP channels had lower ATP sensitivities, especially the Kir6.2 R192H/SUR1 channel. (b) Sulfonylurea sensitivity. Residual KATP channel activities were determined by the ratio between the amplitudes of KATP channel currents before and after 100 μmol/l tolbutamide application. Experiments were conducted for wild-type (white column, n = 15) and mutant (black column, Kir6.2 R27H/SUR1, n = 10; Kir6.2 R192H/SUR1, grey column, n = 9) channels. No significant difference in sulfonylurea sensitivity was observed for the mutant channels compared with the wild-type channels (p > 0.05)
The index case from family c had a heterozygous frame shift mutation, causing deletion of Ser116 and Phe117 (NM_000525.3: c.348-353delCTTCTC, p.S116F117del), resulting in an in-frame deletion of two amino acid residues (Fig. 1c). Because no KATP currents were detected in the loss-of-function S116F117del channel in vitro, we did not further assay ATP sensitivity or sulfonylurea sensitivity for the S116F117del channel. The proposed structure model of Kir6.2 indicates that the evolutionally well-conserved Ser116 and Phe117 are residues located on the pore-forming H5 domain at the exit of the KATP channel on the TM region interface (Fig. 2). Ser116 lies within the end of the extracellular loop (94-116) connecting the TM region with the pore helix, H5; F117 lies at the beginning of the pore-forming helix (117-128) [9]. Deletion of these residues may compress the channel, preventing K+ from flowing freely (Fig. 2), accounting for the absence of detectable currents as well as unregulated insulin secretion in vitro.
Hence the in-frame deletion loss-of-function mutation (p.S116F117del) probably primarily acts as a hyperinsulinism (HI) mutation.
Clinical characteristics of early-onset type 2 diabetic families with mutations
We investigated the presence of these mutations in the probands’ families. Of the five R27H carriers in family a, three had previously been diagnosed with diabetes. In addition, one was diagnosed with diabetes and one was diagnosed with IGT at the time of examination (Fig. 4 and Table 1); the average age of these newly diagnosed carriers at the time of diagnosis was 38.8 (range 32–52) years. These patients were prescribed insulin or gliclazide. None of the members of family a were obese. All R27H carriers had low serum insulin levels, and two (II-1 and II-2) patients who were treated with insulin had undetectable serum C-peptide (Table 1), indicating the absence of endogenous insulin secretion. Patients II-4 and III-2 were treated with a sulfonylurea alone (gliclazide), and achieved perfect control of their diabetes. In addition, a non-carrier, II-3 aged 51, had IGT. This individual most likely displays phenocopy, a phenomenon manifested in MODY pedigrees [17].

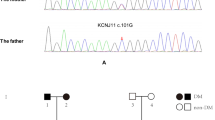
Pedigrees, genotypes and clinical characteristics of families a, b and c. Black circles and squares indicate participants diagnosed with type 2 diabetes; half-filled black circles and squares indicate IGT; white circles and squares indicate normal glucose tolerance (NGT); black arrows indicate the index cases for the three families. Individuals treated with insulin did not undergo the OGTT. The numbers under the symbols are the family members’ identification numbers, followed below by the genotype at codon 27 in family a, codon 192 in family b and codon 116-117 in family c, then age at diabetes diagnosis for affected members and age at examination, followed by treatment for diabetes. nd, not determined. Family a, Pedigree and genotypes of family a. Left pedigree shows the segregation of the R27H mutation. For the genotype, N shows normal allele (Arg); m shows mutant allele (His) at codon 27. The parametric LOD score for this pedigree was 0.83. OHA, oral hypoglycaemic agents, gliclazide. Family b, Pedigree and genotypes of family b. Middle pedigree shows the segregation of the R192H mutation. For the genotype, N shows normal allele (Arg); m shows mutant allele (His) at codon 192. The parametric log10 of odds (LOD) score for this pedigree was 0.60. OHA, glimepiride. Family c, Pedigree and genotypes of family c. Right pedigree shows the segregation of the S116F117del mutation. N shows normal allele S116F117; m shows mutant allele S116F117del (His) at codon116-117. The parametric LOD score for this pedigree was 0.52. OHA, gliclazide
The tolbutamide sensitivity of the R27H, R192H and wild-type channels was not significantly different (p > 0.05, Fig. 3b), suggesting that sulfonylureas could efficiently close the mutant channels and stimulate insulin secretion. Moreover, previous studies indicated that diabetic patients with a KCNJ11 gain-of-function mutation could transfer successfully from insulin to oral sulfonylureas [26, 27]. As the affected R27H carriers in family a (II-4 or III-2) maintained perfect metabolic control with sulfonylureas, we adjusted the treatment of the other two R27H carriers (II-1 and II-2) from insulin to gliclazide, and these patients achieved good glycaemic control (HbA1c, 5.7–6.4% [39–46 mmol/mol]).
In family b, four R192H carriers were previously diagnosed with diabetes; at the time of examination, their diabetes was treated with insulin or sulfonylureas (glimepiride) (Fig 4). Two R192H carriers (II-1 and III-1) were treated with insulin, with or without metformin, and had undetectable serum C-peptide (Table 1), indicating the absence of endogenous insulin secretion. The other two carriers (III-2 and III-3) were treated with sulfonylureas (glimepiride) with or without metformin, and maintained good control of their diabetes, suggesting that sulfonylureas are effective for R192H carriers with diabetes mellitus. We prescribed sulfonylurea therapy (glimepiride) for the other two R192H carriers (II-1 and III-1) who had been on insulin with metformin since diagnosis. Currently, these patients are only taking sulfonylureas and metformin, and their diabetes is well controlled (HbA1c, 5.6–6.2% [38–44 mmol/mol] for II-1, 5.5–6.4% [37–46 mmol/mol] for III-1).
In family c, two S116F117del carriers had previously diagnosed diabetes (Fig. 4 and Table 1). Neither high concentration glucose nor tolbutamide elicits significant insulin secretion in islets isolated from Kir6.2 knockout mice [13]. Tolbutamide-induced insulin secretion is absent in Kir6.2 knockout mice, indicating that sulfonylurea-induced insulin secretion also critically depends on KATP channel activity. At the time of examination, S116F117del carrier II-1 had been effectively treated with insulin. The affected carriers of S116F117del were the proband III-1 (female, gestation 39 weeks, birthweight 4.0 kg, 95–97th percentile) [28], father II-1 (gestation 39 weeks, birthweight 3.8 kg, 75–90th percentile) and son IV-1 (gestation 38 weeks, birthweight 3.95 kg, 95–97th percentile). However, the unaffected sibling, brother III-2, a non-S116F117del carrier was in the normal range of birthweight (male, gestation 40 weeks, birthweight 3.3 kg, 25–50th percentile). In addition, all S116F117del carriers had complained that they had mild symptoms such as tremulousness, hunger and fatigue, which could be remitted by taking in food or glucose, when their blood glucose was determined to be 2.9–3.3 mmol/l (52.2–59.4 mg/dl) for proband III-1, 3.4–3.7 mmol/l (61.2–66.6 mg/dl) for son IV-1, and 3.6–3.9 mmol/l (64.8–70.2 mg/dl) for father II-1 during their childhood; this could be inferred to be largely due to mild hypoglycaemia caused by HI.
The clinical course of the proband III-1 was described as mild hypoglycaemia in childhood, IGT in early adulthood, and diabetes in middle age. Since diagnosis, this patient had been taking sulfonylureas without effective blood glucose control (HbA1c 7.8–9.2% [62–77 mmol/mol]). Treatment of the patients was switched from sulfonylureas to insulin, at which point their blood glucose became normal and was well sustained (HbA1c 5.2–6.3% [33–45 mmol/mol]). The son of this patient, IV-1, was on dietary control for IGT at the time of examination.
Discussion
Previous studies indicated that 90–95% of Chinese familial cases of early-onset type 2 diabetes remain genetically unexplained [29–31]. In this study, a 3.12% prevalence of KCNJ11 mutations was observed in 96 Chinese families with early-onset type 2 diabetes mellitus, suggesting that KCNJ11 may be one of the unidentified monogenic aetiologies in the Chinese population. Although three known KCNJ11 variations were identified in the Chinese probands in this study, none had an association with familial early-onset type 2 diabetes mellitus. However, the E23K variation has been reported to be associated with modest reductions in ATP sensitivity and linked to type 2 diabetes mellitus [12, 32].
We demonstrated that two novel gain-of-function mutations (R27H and R192H) lead to the development of type 2 diabetes by reducing the ATP-binding activity of Kir6.2. On comparison of the IC50 of the most common mutation, R201H [5], the mildest common variation, E23K [33], and the in-between mutation, C42R [6], with that of R192H or R27H, the reduction in ATP sensitivity is R201H>R192H>C42R>R27H>E23K, which is consistent with their clinical phenotypes in diabetes, i.e. the severe phenotype is PNDM, the mildest is type 2 diabetes and the in-between is early-onset type 2 diabetes or MODY. Although these data suggest a strong genotype–phenotype relationship in KCNJ11 mutations, the association is relative, not absolute. It has been reported that, in some families with the C42R or R201H mutation, the clinical presentation and degree of impaired beta cell function are different [5, 6, 34]. Even in families with R27H or R192H mutations, clinical symptoms in the same family were different. This suggests that both genetic and environmental factors may influence the clinical phenotypes, in addition to their responses to sulfonylureas.
One study reported that p.Glu227Lys (i.e. the E227K mutation) located at E227, a single amino acid that governs the electrostatic interaction of the R192–E227 pair, has a functional effect [25]. E227K reduces the ATP sensitivity of the KATP channel and increases the intrinsic open probability of the channel in its mutant heterozygous state in vitro [35]. In addition, several studies have indicated that the E227K mutation causes monogenic neonatal diabetes mellitus [35, 36]. Moreover, E227K has been identified as causal for a French MODY pedigree, and therefore the KCNJ11 gene was proposed to be MODY13 in that study [7]. These E227K studies offer support for R192H, which caused MODY gene-negative autosomal dominantly inherited early-onset type 2 diabetes mellitus in family b, in the same way as E227K in the French MODY pedigree [7]. Further studies are required to confirm whether these effects occur in vivo.
We concluded that the development of type 2 diabetes in R27H and R192H carriers is due to defective glucose-induced insulin secretion via one of three distinct mechanisms: (1) primary impairment of the ATP sensitivity of the mutant KATP channels; (2) the effect of R27H on the interaction between Kir6.2 and SUR1 subunits in the KATP channel; (3) the effect of the mutant R192H on the stability of the Kir6.2 tetramer structure and the pore-forming channel.
Loss-of-function mutations in human SUR1/ABCC8 or Kir6.2/KCNJ11 are the most common causes of congenital HI of infancy [37, 38]. Patients with hyperinsulinaemic hypoglycaemia due to loss-of-function in KATP, such as heterozygous ABCC8 mutations, ‘cross-over’ to diabetes in later life [39]. This is inferred from mouse models where loss-of-function in KATP channel activity led to glucose intolerance and diabetes on high-fat diets [14, 40].
The three affected heterozygous S116F117del carriers in family c were in the upper range of normal birthweight, suggesting that insulin oversecretion might have occurred in their fetal period. In addition, all S116F117del carriers with diabetes or IGT had mild symptoms of childhood hypoglycaemia in this study. Because of poor hospital conditions at the time, no insulin level tests were conducted in local hospitals, and we infer that they may have had HI during their childhood. Therefore the S116F117del mutation may represent a new subtype of autosomal dominantly inherited diabetes with a moderate phenotype, presenting as hyperinsulinaemic hypoglycaemia in childhood, IGT in early adulthood, and diabetes in middle age due to slow, progressive loss of insulin secretory capacity. Interestingly, one previously reported human KCNJ11 mutation affecting S116 is known to be responsible for HI [41], which provides evidence to support the idea that p.S116F117del may have caused HI during childhood in family c (Fig. 4). However, the precise mechanism of the progressive loss of insulin secretory capacity and the reversion of the phenotype is still poorly understood.
Development of type 2 diabetes in S116F117del carriers was associated with defective glucose-induced insulin secretion, which promotes apoptotic beta cell death, and contributes to hyperglycaemia due to unregulated insulin secretion in later life.
As the residues S116, F117 and G132 are all located in the pore-forming domain of Kir6.2, the clinical course of human S116F117del mutation carriers may be similar to Kir6.2G132S transgenic mice [13], which exhibit hypoglycaemia and unregulated insulin secretion as neonates and then develop severe hyperglycaemia with almost no insulin response to glucose. A high frequency of apoptotic beta cells is observed before the appearance of hyperglycaemia in Kir6.2G132S transgenic mice, suggesting that KATP channels may play a significant role in beta cell survival, except for insulin secretion [14, 42]. Whether spontaneous regeneration of pancreatic beta cells can occur in human carriers of the S116F117del mutation, as in Kir6.2G132S transgenic mice, needs further investigation. Moreover, further experiments on S116F117del transgenic mice may help to explain HI or diabetes caused by this mutation, as well as the phenotype and mechanisms by which HI could result in diabetes in later life.
This study has enabled therapeutic improvements. Discontinuation of insulin and implementation of sulfonylureas for R27H or R192H carriers, and continuation/switch to insulin therapy for S116F117del carriers resulted in good control of glycaemic levels. Our results suggest that genetic diagnosis of the KCNJ11 mutations in the autosomal dominantly inherited early-onset type 2 diabetic pedigrees may be helpful for understanding molecular aetiology and providing more personalised treatment for these specific forms of diabetes in Chinese individuals, as well as Asians patients in whom the prevalence of the investigated MODY genes [16] (i.e. HNF4α/MODY1, GCK/MODY2, HNF1α/MODY3, PDX1/MODY4, HNF1β/MODY5 and NEUROD1/MODY6) is as low as 10–20% [30, 31, 43, 44].
Further studies are required to confirm whether these heterozygous mutations disturb pancreatic islet development or decrease beta cell mass. The identification of these novel mutations broadens the spectrum of diabetic phenotypes linked to KCNJ11, suggesting that routine genetic diagnosis for autosomal dominantly inherited early-onset type 2 diabetic families should also include KCNJ11.
Abbreviations
- HI:
-
Hyperinsulinism
- IA-2:
-
Tyrosine phosphatase-like protein
- IGT:
-
Impaired glucose tolerance
- KATP :
-
ATP-sensitive potassium channel
- MODY:
-
Maturity-onset diabetes of the young
- PNDM:
-
Permanent neonatal diabetes
- SNP:
-
Single nucleotide polymorphism
- TM:
-
Transmembrane
References
Bonnefond A, Froguel P, Vaxillaire M (2010) The emerging genetics of type 2 diabetes. Trends Mol Med 16:407–416
American Diabetes Association (2013) Diagnosis and classification of diabetes mellitus. Diabetes Care 36(Suppl 1):S67–S74
Fajans SS, Bell GI (2011) MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care 34:1878–1884
Doria A, Yang Y, Malecki M et al (1999) Phenotypic characteristics of early-onset autosomal-dominant type 2 diabetes unlinked to known maturity-onset diabetes of the young (MODY) genes. Diabetes Care 22:253–261
Gloyn AL, Pearson ER, Antcliff JF et al (2004) Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 350:1838–1849
Yorifuji T, Nagashima K, Kurokawa K et al (2005) The C42R mutation in the Kir6.2 (KCNJ11) gene as a cause of transient neonatal diabetes, childhood diabetes, or later-onset, apparently type 2 diabetes mellitus. J Clin Endocrinol Metab 90:3174–3178
Bonnefond A, Philippe J, Durand E et al (2012) Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS One 7:e37423
Inagaki N, Gonoi T, Clement JP 4th et al (1995) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270:1166–1170
Tucker SJ, Gribble FM, Proks P et al (1998) Molecular determinants of KATP channel inhibition by ATP. EMBO J 17:3290–3296
Girard CA, Wunderlich FT, Shimomura K et al (2009) Expression of an activating mutation in the gene encoding the KATP channel subunit Kir6.2 in mouse pancreatic beta cells recapitulates neonatal diabetes. J Clin Invest 119:80–90
Proks P, Reimann F, Green N, Gribble F, Ashcroft F (2002) Sulfonylurea stimulation of insulin secretion. Diabetes 51(Suppl 3):S368–S376
Gloyn AL, Weedon MN, Owen KR et al (2003) Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 52:568–572
Seino S, Iwanaga T, Nagashima K, Miki T (2000) Diverse roles of KATP channels learned from Kir6. 2 genetically engineered mice. Diabetes 49:311–318
Miki T, Tashiro F, Iwanaga T et al (1997) Abnormalities of pancreatic islets by targeted expression of a dominant-negative KATP channel. Proc Natl Acad Sci U S A 94:11969–11973
Fukui M, Nakano K, Obayashi H et al (1997) High prevalence of mitochondrial diabetes mellitus in Japanese patients with major risk factors. Metabolism 46:793–795
American Diabetes Association (2010) Diagnosis and classification of diabetes mellitus. Diabetes Care 33(Suppl 1):S62–S69
Yamagata K, Oda N, Kaisaki PJ et al (1996) Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature 384:455–458
Yamagata K, Furuta H, Oda N et al (1996) Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature 384:458–460
Froguel P, Zouali H, Vionnet N et al (1993) Familial hyperglycemia due to mutations in glucokinase: definition of a subtype of diabetes mellitus. N Engl J Med 328:697–702
Chèvre JC, Hani EH, Stoffers DA, Habener JF, Froguel P (1998) Insulin promoter factor 1 gene is not a major cause of maturity-onset diabetes of the young in French Caucasians. Diabetes 47:843–844
Beards F, Frayling T, Bulman M et al (1998) Mutations in hepatocyte nuclear factor 1beta are not a common cause of maturity-onset diabetes of the young in the U.K. Diabetes 47:1152–1154
Malecki MT, Jhala US, Antonellis A et al (1999) Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet 23:323–328
The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus (2003) Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus (position statement). Diabetes Care 26(Suppl 1):S5–S20
Pegan S, Arrabit C, Zhou W et al (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci 8:279–287
Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM (2005) Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J 24:229–239
Nagano N, Urakami T, Mine Y et al (2012) Diabetes caused by Kir6.2 mutation: successful treatment with oral glibenclamide switched from continuous subcutaneous insulin infusion in the early phase of the disease. Pediatr Int 54:277–279
Pearson ER, Flechtner I, Njølstad PR et al (2006) Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 355:467–477
Bonellie S, Chalmers J, Gray R, Greer I, Jarvis S, Williams C (2008) Centile charts for birthweight for gestational age for Scottish singleton births. BMC Pregnancy Childbirth 8:5
Ng MC, Lee SC, Ko GT et al (2001) Familial early-onset type 2 diabetes in Chinese patients: obesity and genetics have more significant roles than autoimmunity. Diabetes Care 24:663–671
Xu JY, Dan QH, Chan V et al (2005) Genetic and clinical characteristics of maturity-onset diabetes of the young in Chinese patients. Eur J Hum Genet 13:422–427
Liu L, Furuta H, Minami A et al (2007) A novel mutation, Ser159Pro in the NeuroD1/BETA2 gene contributes to the development of diabetes in a Chinese potential MODY family. Mol Cell Biochem 303:115–120
Villareal DT, Koster JC, Robertson H et al (2009) Kir6.2 variant E23K increases ATP-sensitive K+ channel activity and is associated with impaired insulin release and enhanced insulin sensitivity in adults with normal glucose tolerance. Diabetes 58:1869–1878
Schwanstecher C, Meyer U, Schwanstecher M (2002) Kir6.2 polymorphism predisposes to type 2 diabetes by inducing overactivity of pancreatic beta-cell ATP-sensitive K+ channels. Diabetes 51:875–879
Klupa T, Edghill EL, Nazim J et al (2005) The identification of a R201H mutation in KCNJ11, which encodes Kir6.2, and successful transfer to sustained-release sulphonylurea therapy in a subject with neonatal diabetes: evidence for heterogeneity of beta cell function among carriers of the R201H mutation. Diabetologia 8:1029–1031
Girard CA, Shimomura K, Proks P et al (2006) Functional analysis of six Kir6.2 (KCNJ11) mutations causing neonatal diabetes. Pflugers Arch 453:323–332
Flanagan SE, Patch AM, Mackay DJ et al (2007) Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 56:1930–1937
Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley-Green A, Lindley KJ (2004) Hyperinsulinism in infancy: from basic science to clinical disease. Physiol Rev 84:239–275
Loechner KJ, Akrouh A, Kurata HT et al (2011) Congenital hyperinsulinism and glucose hypersensitivity in homozygous and heterozygous carriers of Kir6.2 (KCNJ11) mutation V290M mutation: K(ATP) channel inactivation mechanism and clinical management. Diabetes 60:209–217
Huopio H, Otonkoski T, Vauhkonen I, Reimann F, Ashcroft FM, Laakso M (2003) A new subtype of autosomal dominant diabetes attributable to a mutation in the gene for sulfonylurea receptor 1. Lancet 361:301–307
Nichols CG, Koster JC, Remedi MS (2007) β-cell hyperexcitability: from hyperinsulinism to diabetes. Diabetes Obes Metab 9(Suppl 2):81–88
Suchi M, MacMullen CM, Thornton PS et al (2006) Molecular and immunohistochemical analyses of the focal form of congenital hyperinsulinism. Mod Pathol 19:122–129
Oyama K, Minami K, Ishizaki K, Fuse M, Miki T, Seino S (2006) Spontaneous recovery from hyperglycemia by regeneration of pancreatic beta-cells in Kir6.2G132S transgenic mice. Diabetes 55:1930–1938
Hwang JS, Shin CH, Yang SW, Jung SY, Huh N (2006) Genetic and clinical characteristics of Korean maturity-onset diabetes of the young (MODY) patients. Diabetes Res Clin Pract 74:75–81
Plengvidhya N, Boonyasrisawat W, Chongjaroen N et al (2009) Mutations of maturity-onset diabetes of the young (MODY) genes in Thais with early-onset type 2 diabetes mellitus. Clin Endocrinol (Oxf) 70:847–853
Acknowledgements
We thank Susumu Seino and Tadao Shibasaki (Division of Cellular and Molecular Medicine, Department of Physiology and Cell Biology, Kobe University Graduate School of Medicine, Japan), Nobuya Inagaki (Department of Diabetes and Clinical Nutrition, Kyoto University, Japan) and Long Yu (State Key Laboratory of Genetic Engineering, School of Life Sciences, Fudan University, China) for their cooperation. We also thank Weiping Jia, Xiaojing Ma, Ming Lu, Can Li, Weijing Zhao, Rong Zhang, Jun Yin, Yuqian Bao and Jing Xu (Shanghai Diabetes Institute, Department of Endocrinology & Metabolism, Shanghai Jiaotong University Affiliated Sixth People’s Hospital, Shanghai, China) for their technical support and cooperation.
Funding
This research was supported by grants from the Project of National Natural Science Foundation of China (nos. 81270876, 30771022 and 30971384) and the Shanghai Scientific & Technical Committee Foundation (nos. 10XD1403400 and 06ZR14051). YL is supported by NIH grant SC1DK087655.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors contributed to the conception and design, or analysis and interpretation of data, drafted the article or revised it critically for important intellectual content, and approved the final version to be published.
Author information
Authors and Affiliations
Corresponding author
Additional information
Limei Liu, Kazuaki Nagashima, Takao Yasuda, Yanjun Liu, Hai-rong Hu and Guang He contributed equally to this study.
Rights and permissions
About this article
Cite this article
Liu, L., Nagashima, K., Yasuda, T. et al. Mutations in KCNJ11 are associated with the development of autosomal dominant, early-onset type 2 diabetes. Diabetologia 56, 2609–2618 (2013). https://doi.org/10.1007/s00125-013-3031-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-3031-9