
Abstract
Aims/hypothesis
Arctigenin is a natural compound that had never been previously demonstrated to have a glucose-lowering effect. Here it was found to activate AMP-activated protein kinase (AMPK), and the mechanism by which this occurred, as well as the effects on glucose and lipid metabolism were investigated.
Methods
2-Deoxyglucose uptake and AMPK phosphorylation were examined in L6 myotubes and isolated skeletal muscle. Gluconeogenesis and lipid synthesis were evaluated in rat primary hepatocytes. The acute and chronic effects of arctigenin on metabolic abnormalities were observed in C57BL/6J and ob/ob mice. Changes in mitochondrial membrane potential were measured using the J-aggregate-forming dye, JC-1. Analysis of respiration of L6 myotubes or isolated mitochondria was conducted in a channel oxygen system.
Results
Arctigenin increased AMPK phosphorylation and stimulated glucose uptake in L6 myotubes and isolated skeletal muscles. In primary hepatocytes, it decreased gluconeogenesis and lipid synthesis. The enhancement of glucose uptake and suppression of hepatic gluconeogenesis and lipid synthesis by arctigenin were prevented by blockade of AMPK activation. The respiration of L6 myotubes or isolated mitochondria was inhibited by arctigenin with a specific effect on respiratory complex I. A single oral dose of arctigenin reduced gluconeogenesis in C57BL/6J mice. Chronic oral administration of arctigenin lowered blood glucose and improved lipid metabolism in ob/ob mice.
Conclusions/interpretation
This study demonstrates a new role for arctigenin as a potent indirect activator of AMPK via inhibition of respiratory complex I, with beneficial effects on metabolic disorders in ob/ob mice. This highlights the potential value of arctigenin as a possible treatment of type 2 diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes is characterised by abnormal glucose and lipid metabolism due to insulin resistance and impaired insulin secretion. Skeletal muscle is the primary tissue in glucose homeostasis, accounting for approximately 80% of whole-body glucose disposal under insulin-stimulated conditions [1]. Glucose transport is the first rate-limiting step in glucose metabolism. Reduced glucose transport in skeletal muscle leads to impaired whole-body glucose uptake and contributes to the pathogenesis of type 2 diabetes mellitus [2–4].
Glucose transport in skeletal muscle can be activated by two separate pathways (insulin-dependent and insulin-independent). In type 2 diabetic patients, insulin-mediated glucose transport in skeletal muscle is impaired due to defects in insulin signalling pathways [5]. Thus, therapies that target insulin-independent pathways in the regulation of glucose transport are likely to be more beneficial in the treatment of type 2 diabetes mellitus. Several factors, such as muscle contraction (exercise), hypoxia and cellular stress have been demonstrated to increase glucose transport independently of insulin in skeletal muscle, effects to which the activation of AMP-activated protein kinase (AMPK) has been suggested to contribute [6–8]. Moreover, it has been shown that activation of AMPK by known AMPK activators restored glucose transport in skeletal muscle from patients with type 2 diabetes mellitus, while insulin-induced glucose transport was impaired [9]. As such, activation of AMPK in skeletal muscle could be an attractive way to increase glucose transport independently of insulin, thus bypassing the defective insulin signalling in type 2 diabetic patients.
AMPK is a heterotrimeric protein composed of a catalytic α subunit and regulatory β and γ subunits. It is activated in response to an increase of the intracellular AMP/ATP ratio due to several stimuli, e.g. hypoxia, glucose deprivation or the action of various hormones, cytokines and adipokines. AMPK thus acts as an efficient sensor of cellular energy states and plays an important role in the regulation of whole-body energy homeostasis [9]. Liver kinase B1 (LKB1) and Ca2+/calmodulin-dependent protein kinase kinase (CaMKK) are two distinct upstream kinases that activate AMPK by phosphorylation of Thr172. LKB1 is activated by an increase of the AMP/ATP ratio, while CaMKK is activated by an increase in intracellular Ca2+ [10, 11]. Once activated, AMPK leads to a concomitant activation of energy-producing processes (e.g. fatty acid oxidation and glucose uptake) and inhibition of energy-consuming biosynthetic processes (e.g. fatty acid and sterol synthesis) to restore energy balance [12]. Genetic and pharmacological studies have demonstrated that AMPK controls whole-body glucose homeostasis by regulating metabolism in various peripheral tissues, such as skeletal muscle, liver and adipose tissues [13]. Metformin, one of the most commonly used drugs for the treatment of type 2 diabetes, has been demonstrated to exert pleiotropic beneficial effects by activation of AMPK [14], suggesting that AMPK could be a very attractive drug target for the treatment of type 2 diabetes.
Recently, several small molecules have been reported to activate AMPK. Direct activators of AMPK, such as the thienopyridone family, act without increasing cellular AMP/ATP ratios [15], while various pharmacological agents have been reported to activate AMPK indirectly by modes of action that have not been fully delineated. Natural products have played a significant role in new drug discovery and development processes [16], with some reported to possess glucose-lowering effects [17, 18]. Moreover, natural products such as alkaloids, bitter melon extracts and epigallocatechin-3-gallate (EGCG) have been found to activate AMPK and improve metabolic disease [17, 19]. However, most of these agents displayed inadequate efficacy in vivo, leading to the demand to develop more effective AMPK activators.
Given the importance of skeletal muscle’s capacity for glucose transport for whole-body glucose homeostasis and that glucose transport could be induced by activation of AMPK, we conducted a primary screening of our natural products library for 2-deoxyglucose uptake in L6 myotubes, followed by a secondary screening based on the evaluation of AMPK phosphorylation. During these screenings, arctigenin, a natural compound that had never been previously shown to have a glucose-lowering effect, was found to increase glucose uptake and AMPK phosphorylation in L6 myotubes. In the present study, the mechanisms involved in the activation of AMPK by arctigenin were explored, and the effects of arctigenin on glucose and lipid metabolism investigated in vitro and in vivo.
Methods
Animals
C57BL/6J male mice and Sprague–Dawley male rats were purchased from SLAC Laboratory Animals (Shanghai, China). B6.V-Lep ob/Lep ob mice (Jackson Laboratory, Bar Harbor, ME, USA) were bred at the Shanghai Institute of Materia Medica (Chinese Academy of Sciences, Shanghai, People’s Republic of China). The animals were maintained under a 12 h light–dark cycle with free access to water and food. Animal experiments were approved by the Animal Care and Use Committee, Shanghai Institute of Materia Medica.
2-Deoxyglucose uptake, GLUT4 translocation and protein phosphorylation in L6 myotubes
L6 myoblasts were differentiated to myotubes and treated with or without arctigenin, followed by 2-deoxyglucose uptake measurement and analysis of GLUT4 translocation or protein phosphorylation by western blot (electronic supplementary material [ESM] Methods).
2-Deoxyglucose uptake and protein phosphorylation in isolated muscle
Soleus and extensor digitorum longus (EDL) muscles were isolated for analysis of 2-deoxyglucose uptake and protein phosphorylation (see ESM Methods).
Gluconeogenesis, lipid synthesis and protein phosphorylation in primary cultured rat hepatocytes
Rat hepatocytes were isolated by collagenase digestion from rats that had been fasted for 24 h (gluconeogenesis analysis) or from fed rats (protein phosphorylation or lipid synthesis analysis). Gluconeogenesis, fatty acid and sterol synthesis, and protein phosphorylation were measured as described in ESM Methods.
Transfection of small interfering RNA
L6 myotubes were transfected with small interfering RNA (siRNA) using siLentFect lipid (catalogue number 170–3360; Bio-Rad Laboratories, Hercules, CA, USA) as described in ESM Methods.
Western blot analysis
Protein preparation and western blot analysis were performed as described in ESM Methods.
Acute arctigenin administration in normal mice
The effects of acute administration of arctigenin on gluconeogenesis and AMPKα phosphorylation were observed in male C57BL/6J mice (see ESM Methods).
AMPK acitivity assay
AMPK activity was measured by monitoring phosphorylation of the AMARA peptide substrate (see ESM Methods).
Hela cells culture
Full details are provided in ESM Methods.
Mitochondrial membrane potential assay
This assay was performed according to a previous report [20], with modifications (see ESM Methods).
Adenine nucleotide extraction and measurement
Adenine nucleotides were extracted from L6 myotubes and measured by HPLC as described in ESM Methods.
Measurement of respiration in L6 myotubes and isolated mitochondria
Liver mitochondria were prepared from Sprague–Dawley male rats. Isolated liver was finely chopped, homogenised (Polytron Homogeniser; Kinematica, Lucerne, Switzerland) and centrifuged (300 g) as described in a previous report [21]. Mitochondria were resuspended in respiration medium at a concentration of 60 mg/ml. Respiration measurements in L6 myotubes and isolated mitochondria were performed in a 782 2-channel oxygen system (Strathkelvin Instruments, Motherwell, UK) (see ESM Methods).
Determination of lactate content
L6 myotubes were cultured in a 24 well plate and treated for 1 or 4 h with 3 μmol/l arctigenin or 50 μmol/l rosiglitazone (positive control) in FBS-free DMEM. Lactate in the medium was measured with a lactate assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, People’s Republic of China).
Arctigenin treatment in ob/ob mice
Study 1
Female ob/ob mice (6–7 weeks old) were assigned to three groups (n = 8) based on blood glucose and body weight, and subjected to gavage treatment with vehicle (0.5% carboxymethyl cellulose, wt/vol.), 200 mg/kg arctigenin or 250 mg/kg metformin, respectively, for 23 days. The effects of arctigenin on metabolic disorders of ob/ob mice were investigated. HOMA-IR was calculated (see ESM Methods).
Study 2
Female ob/ob mice (6–7 weeks) were assigned to two groups (n = 18) based on blood glucose and body weight, and subjected to twice daily intraperitoneal injection of 25 mg/kg arctigenin or vehicle (0.5% Tween 80, vol./vol.) for 22 days. AKT phosphorylation in skeletal muscle, liver and fat isolated from arctigenin-treated or vehicle-treated (control) mice was evaluated under basal and insulin-stimulated conditions (see ESM Methods).
RNA isolation and real-time PCR
Gene expression was analysed by real-time PCR as described in ESM Methods. For details on primer sequences, see ESM Table 1.
Statistical analysis
Results are expressed as mean±SEM. The statistical analysis was performed with a two-tailed unpaired t test. Values of p < 0.05 were considered to be statistically significant.
Results
Arctigenin increases glucose uptake and AMPK phosphorylation in L6 myotubes
To identify new glucose-lowering lead compounds from natural products, we screened our phytocompounds collection for 2-deoxyglucose uptake in L6 myotubes. Arctigenin, a natural product (Fig. 1a), was found to increase glucose uptake under basal and insulin-stimulated conditions. Incubation of L6 myotubes with 1 μmol/l arctigenin resulted in a 1.49-fold increase in glucose uptake, which is comparable with the effect of 100 nmol/l insulin (Fig. 1b). Arctigenin increased basal glucose uptake in a dose- and time-dependent manner. Glucose uptake increased to 1.83-fold of control after treatment with 3 μmol/l arctigenin for 2 h, whereas the same dose of berberine only caused a 1.49-fold enhancement (Fig. 1c, d). Moreover, arctigenin treatment increased GLUT4 protein levels in the plasma membrane, whereas no change of total GLUT4 abundance was observed, suggesting that arctigenin stimulated GLUT4 translocation in L6 myotubes (Fig. 1e). Arctigenin dose-dependently increased AMPK and acetyl coenzyme A synthetase (ACC) phosphorylation in L6 myotubes (Fig. 1f, g). Significant increases of AMPK and ACC phosphorylation were observed within 15 min of arctigenin treatment (3 μmol/l) and were maintained for up to 120 min (Fig. 1h, i).

Arctigenin (ARC-G) increases glucose uptake and AMPK phosphorylation in L6 myotubes. a Chemical structure of arctigenin. b Increased basal and insulin-stimulated (100 nmol/l) glucose uptake in L6 myotubes after 2 h of treatment with 1 μmol/l arctigenin. c Dose-dependent increase in glucose uptake by L6 myotubes exposed to arctigenin at the indicated concentrations for 2 h. Berberine (3 μmol/l) was set as a positive control. d Time-dependent stimulation of glucose uptake by 3 μmol/l arctigenin. e GLUT4 translocation in L6 myotubes treated by arctigenin or insulin for 2 h. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PM, plasma membrane. f Dose-dependent increase in AMPK and ACC phosphorylation (p-AMPK, p-ACC) in L6 myotubes by 2 h treatment with arctigenin at the indicated concentrations, quantified (g) as relative optical density. White bars, p-AMPKα/AMPKα; black bars, p-ACC/GAPDH. h Time-dependent stimulation of AMPK and ACC phosphorylation by 3 μmol/l arctigenin, quantified (i) as relative optical density. Key as above (g). Results are shown as means±SEM from three independent experiments; *p < 0.05 and **p < 0.01 vs corresponding controls (as indicated)
Arctigenin increases glucose uptake and AMPK phosphorylation in isolated muscles
Measurement of 2-deoxyglucose uptake was performed in isolated EDL and soleus muscles from C57BL/6J mice. Incubation of EDL or soleus muscles with 3 μmol/l arctigenin resulted in an increase of glucose uptake by 31.7% (p = 0.1) and 18% (p < 0.05), or 42% (p < 0.05) and 20.6% (p < 0.01) under basal and insulin-stimulated conditions, respectively (Fig. 2a, b). Arctigenin also dose-dependently increased AMPK and ACC phosphorylation in isolated EDL and soleus muscles (Fig. 2c–f).

Arctigenin (ARC-G) increases glucose uptake and AMPK phosphorylation in isolated muscles of C57BL/6J mice. a Increased basal and insulin-stimulated (12 nmol/l) glucose uptake in EDL and (b) soleus muscles by 2 h of treatment with 3 μmol/l arctigenin. White bars, control; black bars, arctigenin. c Dose-dependent increase in AMPK and ACC phosphorylation (p-AMPK, p-ACC) in EDL muscles by 1 h of treatment with arctigenin at the indicated concentrations. AICAR (2 mmol/l) was set as a positive control. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. d Blot findings (c), quantified as relative optical density. White bars, p-AMPKα/AMPKα; black bars, p-ACC/GAPDH. e Dose-dependent increase in AMPK and ACC phosphorylation in soleus muscles with treatment as above (c, d), quantified (f) as relative optical density. Key as above (d). Results are means±SEM; n = 10–14 (a, b), n = 4–6 (c–f); *p < 0.05 and **p < 0.01 vs corresponding controls (as indicated)
Arctigenin-stimulated glucose uptake is AMPK-dependent
To determine whether arctigenin-stimulated glucose uptake involves either the phosphoinositide-3 kinase (PI3 kinase)/AKT pathway or activation of AMPK, the effect of the PI3 kinase inhibitor, wortmannin, or the AMPK inhibitor, compound C, on arctigenin-stimulated glucose uptake was observed in L6 myotubes. As shown in Fig. 3a, wortmannin blocked insulin-stimulated glucose uptake, but had no effect on arctigenin stimulation, whereas arctigenin-stimulated glucose uptake was fully blocked by compound C. Furthermore, arctigenin-stimulated glucose uptake was completely lost upon Ampkα1 (also known as Prkaa1) and Ampkα2 (also known as Prkaa2) silencing in L6 myotubes (Fig. 3c, d). These data indicate that arctigenin-stimulated glucose uptake was mediated by activation of AMPK.

a Effect of arctigenin (ARC-G) on glucose uptake and (b) AMPK phosphorylation (p-AMPK) in L6 myotubes in the presence of compound C and wortmannin. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. c Effect of arctigenin in L6 myotubes on glucose uptake and (d) Ampkα1/α2 silencing. c White bars, control; black bars, arctigenin. a, b L6 myotubes were preincubated for 30 min in the absence or presence of wortmannin (100 nmol/l) or compound C (20 μmol/l), and then treated with 3 μmol/l arctigenin for 2 h, followed by 2-deoxyglucose uptake measurement (a), or p-AMPK, and ACC and AKT phosphorylation (p-ACC, p-AKT) analysis by western blot (b). Insulin (100 nmol/l) was set as positive control. c, d In siRNA studies, L6 myotubes were transfected for 48 h with siLentFect lipid (Blank), scrambled siRNA (negative control) or a combination of two siRNA sequences directed against the α1 and α2 isoforms of AMPK (Ampk siRNA), followed by 2-deoxyglucose uptake measurement (c) or analysis of AMPK phosphorylation and protein abundance by western blot (d). Results (a, c) are shown as means±SEM from three independent experiments; *p < 0.05 and **p < 0.01 vs corresponding controls (as indicated)
Arctigenin increases AMPK phosphorylation, and inhibits gluconeogenesis and lipid synthesis in primary hepatocytes
Primary rat hepatocytes were incubated for 1 h in the absence or presence of 2.5, 5, 10, 20 or 40 μmol/l arctigenin. AMPK and ACC phosphorylation were increased dose-dependently, suggesting that arctigenin activated the AMPK signalling pathway in hepatocytes (Fig. 4a). Since activation of AMPK is known to regulate glucose and lipid metabolism in liver, the effects of arctigenin on gluconeogenesis and lipid synthesis were evaluated in primary rat hepatocytes. As shown in Fig. 4b–d, glucagon-induced hepatic gluconeogenesis was significantly suppressed by arctigenin treatment in a dose-dependent manner; arctigenin also significantly decreased the insulin-stimulated synthesis of fatty acid and sterol. Moreover, the suppression of gluconeogenesis and lipid synthesis by arctigenin was completely blocked by compound C pretreatment (Fig. 4e–h), suggesting that arctigenin inhibited hepatic gluconeogenesis and lipid synthesis by activating AMPK.

Effects of arctigenin (ARC-G) on AMPK phosphorylation (p-AMPK), gluconeogenesis and lipid synthesis in rat primary hepatocytes. a Dose-dependent increase in p-AMPK and ACC phosphorylation (p-ACC) by 1 h of treatment with arctigenin at the indicated concentrations. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. b Dose-dependent inhibition of glucagon-stimulated (10 nmol/l) gluconeogenesis by 20 h incubation with arctigenin treatment at the indicated concentrations. c Dose-dependent inhibition of insulin-stimulated (10 nmol/l) fatty acid and sterol (d) synthesis by 24 h of exposure to arctigenin at the indicated concentrations. e Rat primary hepatocytes were preincubated for 30 min in the absence or presence of compound C (20 μmol/l) and then treated with 20 μmol/l arctigenin for 20 h, followed by the measurement of glucagon-stimulated (10 nmol/l) gluconeogenesis. f Rat primary hepatocytes were preincubated as above (e) and then treated with 20 μmol/l arctigenin for 24 h, followed by the measurement of insulin-stimulated (10 nmol/l) fatty acid and (g) sterol synthesis. e–g White bars, without compound C; black bars, with compound C. h Compound C (20 μmol/l) treatment inhibited the ACC phosphorylation induced by arctigenin in rat primary hepatocytes. Values (b–g) are means±SEM from three independent experiments; *p < 0.05 and **p < 0.01 vs corresponding controls (as indicated); † p < 0.05 and †† p < 0.01 vs ‘0’ group
Arctigenin inhibits gluconeogenesis and increases AMPK phosphorylation in vivo
To assess the effect of arctigenin on gluconeogenesis in vivo, we performed a pyruvate tolerance test, as administration of the gluconeogenic substrate pyruvate increases blood glucose levels by promoting gluconeogenesis in liver. As shown in Fig. 5a, b, administration of pyruvate significantly enhanced blood glucose levels in control mice, but this effect was attenuated in mice pre-treated with 300 mg/kg arctigenin by gavage, indicating reduced gluconeogenesis. In addition, the phosphorylation of AMPK and ACC in liver isolated from arctigenin-treated mice was significantly increased by 36% and 74%, respectively (Fig. 5c, d), suggesting that arctigenin activated AMPK in vivo.

Acute effect of arctigenin (ARC-G) on gluconeogenesis and hepatic AMPK phosphorylation (p-AMPK) in C57BL/6J mice. Mice were treated as described in Methods. In the pyruvate tolerance test, blood glucose levels (a) were measured and the AUC (b) calculated. White circles, vehicle control; black squares, arctigenin. c Hepatic p-AMPK and ACC phosphorylation (p-ACC) were analysed by western blot and quantified (d) as relative optical density. White bars, vehicle control; black bars, arctigenin. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Values (a, b, d) are means±SEM; n = 8; *p < 0.05 and **p < 0.01 vs vehicle
Arctigenin activates AMPK indirectly, an action that is not dependent on LKB1, CaMKK or peroxynitrite
AMPK enzymatic activity measurement was performed by monitoring phosphorylation of the AMARA peptide. The results showed that arctigenin did not activate AMPK directly (ESM Fig. 1a). Moreover, arctigenin increased AMPK phosphorylation in Hela cells lacking LKB1 (ESM Fig. 1b, c). The increased AMPK phosphorylation and glucose uptake induced by arctigenin was also unaffected by the CaMKK inhibitor, STO-609, and by the nitric oxide synthase (NOS) inhibitor, l-NAME (ESM Fig. 1d–g). These data indicate that arctigenin activated AMPK independently of LKB1, CaMKK, and the peroxynitrite (ONOO−) or NOS pathways.
Arctigenin reduces mitochondrial respiration by inhibiting respiratory complex I
Several studies have linked mitochondrial function with AMPK activation [17, 18, 22, 23]. We observed effects of arctigenin on mitochondrial membrane potential (∆ψm). As shown in Fig. 6a, arctigenin depolarised ∆ψm in a dose-dependent manner, but with relatively mild effect (maximum effect lower than 30%). Arctigenin treatment also dose-dependently increased the AMP/ATP ratio in L6 myotubes (Fig. 6b). To determine whether the change in nucleotide ratio was due to an effect on cellular respiration, we examined oxygen consumption in L6 myotubes and observed a dose-dependent inhibition of respiration (Fig. 6d). Furthermore, the effects of arctigenin on ADP-stimulated respiration in the presence of complex I (glutamate + malate) or complex II (succinate) substrates were examined in isolated liver mitochondria. Arctigenin produced a dose-dependent inhibition of oxygen consumption with complex I-linked substrate, but not with complex II-linked respiration (Fig. 6e). In addition, arctigenin increased lactate production in L6 myotubes, as the reduction of aerobic respiration may lead to compensated elevation of anaerobic respiration (Fig. 6f).

Effect of arctigenin (ARC-G) on the AMP/ATP ratio and respiration. a Dose-dependent depolarisation of ∆ψm in L6 myotubes by 10 min of arctigenin treatment at the indicated concentrations. Berberine (3 μmol/l) and carbonyl cyanide m-chlorophenyl hydrazone (CCCP; 10 μmol/l) were set as positive control. b Dose-dependent increase of the AMP/ATP ratio in L6 myotubes exposed to arctigenin at the indicated concentrations for 2 h. c Time-dependent stimulation of the AMP/ATP ratio in L6 myotubes by 10 μmol/l arctigenin. d Dose-dependent inhibition of the respiration of intact L6 myotubes by arctigenin treatment at the indicated concentrations. e Effect of arctigenin on the respiration of isolated mitochondria from rat liver. Arctigenin dose-dependently inhibited mitochondrial respiration in ADP-stimulated respiration in the presence of complex I (glutamate+malate) substrates, but not of complex II (succinate) substrates. f Effect of arctigenin (3 μmol/l) on lactate release in L6 myotubes after 1 h (white bars) or 4 h (black bars) of treatment. Rosiglitazone (50 μmol/l) was set as a positive control. Values are means±SEM; n = 3 (a), n = 4 (b, c, f), n = 7 (d), n = 6 (e); *p < 0.05, **p < 0.01 vs corresponding controls (as indicated)
Arctigenin ameliorates metabolic disorders in ob/ob mice
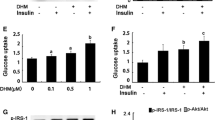
The acute and chronic glucose-lowering potential of arctigenin (200 mg/kg, orally) was evaluated in ob/ob mice, with metformin (250 mg/kg, orally) included as positive control. A single administration of arctigenin resulted in a statistically significant decrease of blood glucose levels by 34.2% after 2 h, although this effect was less pronounced than that of metformin (decrease 62.3%) (Fig. 7a). Chronic administration of arctigenin for 23 days significantly decreased random and fasting blood glucose levels. During the whole treatment period, administration of 200 mg/kg arctigenin reduced random and fasting blood glucose levels, with an average reduction rate of 32.2% and 35.2%; administration of 250 mg/kg metformin caused average reductions of 22.2% and 46.0%, respectively (Fig. 7b, c). After 20 days of treatment with arctigenin, glucose tolerance improved, an effect similar to that seen with metformin (Fig. 7d). No changes in body weight were observed during arctigenin or metformin treatment (Table 1). However, arctigenin treatment caused a significant reduction of subcutaneous, but not of perirenal or mesenteric fat weight by 21% (Table 1). Arctigenin did not alter serum triacylglycerol, NEFA and adiponectin levels, but did significantly decrease cholesterol levels (Table 1). The fasting serum insulin level in arctigenin-treated mice was not significantly changed (Table 1), whereas the HOMA of insulin resistance (HOMA-IR) tended to be decreased (p = 0.08; Table 1). Moreover, arctigenin significantly decreased hepatic and muscle triacylglycerol content by 27% and 19.2%, respectively, but had no substantial effect on hepatic cholesterol content (Table 1). Chronic i.p. administration of arctigenin for 22 days (25 mg/kg, twice daily) significantly increased insulin-stimulated AKT phosphorylation in gastrocnemius muscle and liver, but not in perirenal fat (Fig. 7e–g). Arctigenin treatment showed no effect on levels of GLUT4 in gastrocnemius muscle (Fig. 7h) and had no significant effect on hepatic Pepck (also known as Pck1) expression, but it did significantly decrease G6pase (also known as G6pc) mRNA expression (Fig. 7i).

Effect of arctigenin (ACR-G) on metabolic abnormalities of ob/ob mice. a Arctigenin lowered fasting blood glucose values after a single oral administration. Random-fed blood glucose (b) and fasting blood glucose (c) were determined throughout the treatment. d Glucose tolerance was determined on day 20 of the treatment. White circles, vehicle; black squares, arctigenin (200 mg/kg); black triangles, metformin 250 (mg/kg). e AKT phosphorylation (p-AKT) in gastrocnemius muscle, liver (f) and perirenal fat (g) was evaluated. h Protein abundance of GLUT4 in gastrocnemius muscle was determined by western blot. i Hepatic Pepck and G6pase mRNA levels were determined by real-time PCR. White bars (e–g, i), vehicle control; black bars, arctigenin. Values are means±SEM for n = 8–9 mice; *p < 0.05 and **p < 0.01 vs vehicle-treated mice
Discussion
As a natural product derived from Arctium lappa, arctigenin has been demonstrated to induce various biological activities, such as anti-inflammatory and cortical neuron-protecting effects [24]. A recent study showed that arctigenin served as an anti-tumour agent by blocking the expression of unfolded protein response (UPR) target genes [25]. Although UPR has been reported to be involved in the pathophysiology of insulin resistance and diabetes [26–28], our preliminary work showed that arctigenin had no effects on UPR or the Jun N-terminal kinase (JNK) pathway in liver of ob/ob mice (ESM Fig. 2). In the present study, we found that arctigenin activated AMPK by inhibiting mitochondria complex I. We also showed that arctigenin modulates glucose and lipid metabolism in skeletal muscle and liver, and can ameliorate metabolic disorders in ob/ob mice.
Glucose transport in skeletal muscle is the major component of whole-body glucose uptake and plays a key role in maintaining whole-body glucose homeostasis [2]. Here, a screening of our natural compounds library led to an astonishing discovery, namely that arctigenin significantly stimulated glucose uptake in L6 myotubes, an effect observed to occur in addition to the increased glucose uptake induced by insulin. Since AMPK activation is implicated as an insulin-independent mechanism in stimulation of glucose uptake in skeletal muscle [29], we assessed the effect of arctigenin on AMPK phosphorylation in L6 myotubes. As expected, AMPK phosphorylation was significantly enhanced by arctigenin treatment, a finding that was associated with increased phosphorylation of ACC, a downstream target of AMPK [30], suggesting that activation of AMPK had occurred. In agreement with our studies in L6 myotubes, arctigenin also stimulated glucose uptake, and increased AMPK and ACC phosphorylation in isolated mouse soleus and EDL muscle, providing further evidence of activation of AMPK by arctigenin treatment in skeletal muscle. To investigate the role of AMPK activation in mediating arctigenin-stimulated glucose uptake, inhibitor and siRNA studies were performed in L6 myotubes. Compound C, an AMPK inhibitor, fully blocked arctigenin-stimulated glucose uptake, whereas wortmannin, a PI3 kinase inhibitor, showed no effect. Furthermore, arctigenin-stimulated glucose uptake was completely lost upon silencing of Ampkα1/α2. Thus, arctigenin-stimulated glucose uptake in skeletal muscle was mediated by activation of AMPK, which is similar to the effect of metformin and 5-amino-imidazole carboxamide ribonucleotide (AICAR) [9, 14].
Liver is the major site for storage and release of carbohydrates, and for fatty acid synthesis. Hepatic metabolism plays a key role in the regulation of whole-body energy homeostasis. Elevated hepatic glucose production is a major cause of fasting hyperglycaemia in type 2 diabetic patients [31], while chronic hyperinsulinaemia in the insulin-resistant state results in increased lipid accumulation in the liver [32]. Activation of AMPK in liver leads to the stimulation of fatty acid oxidation, and inhibition of gluconeogenesis and lipogenesis, thus playing important roles in the regulation of glucose and lipid metabolism [15]. Evidence that the activation of AMPK in liver by metformin was required for metformin to exert its inhibitory effect on hepatocyte glucose production further reinforced the importance of AMPK activation in hepatic metabolism [33]. Since arctigenin has been shown to activate AMPK in skeletal muscle, we further studied its effect on liver. As expected, an increase in AMPK and ACC phosphorylation was observed in primary hepatocytes treated with arctigenin, suggesting that activation of AMPK in liver had occurred. Moreover, arctigenin dose-dependently inhibited glucagon-stimulated gluconeogenesis and insulin-stimulated lipid synthesis in rat primary hepatocytes. However, the latter two inhibitory effects of arctigenin were fully abrogated by treatment with compound C, demonstrating that AMPK activation was required for arctigenin-mediated effects to take place. In addition, the activation of hepatic AMPK and suppression of gluconeogenesis by arctigenin treatment was also demonstrated in vivo.
Although arctigenin activated AMPK in skeletal muscle and liver, it showed no effect on the activation of recombinant AMPK kinase, suggesting that arctigenin activated AMPK indirectly. LKB1 and CaMKK are two upstream kinases of AMPK [34], and a recent study showed that ONOO− played a critical role in AMPK activation by metformin in liver and that endothelial NOS was required for metformin action in vitro and in vivo [35]. However, our results showed that arctigenin activated AMPK independently of LKB1, CaMKK, and the ONOO− or NOS pathways.
Since mild perturbation of mitochondrial function has been shown to be associated with activation of AMPK [36–38], the effect of arctigenin on mitochondrial potential was examined. After arctigenin treatment in L6 myotubes, a relatively mild depolarisation of ∆ψm was observed. Several factors may lead to a fall in ∆ψm, including inhibition of mitochondrial respiration, induction of proton leakage (uncoupling) [39] or opening of the mitochondrial permeability transition pore. The action of arctigenin was not related to mitochondrial permeability transition pore opening, as no apoptosis was observed after arctigenin treatment (data not shown), whereas mitochondrial permeability transition pore opening was tightly correlated with the initiation of apoptosis [40]. We also ruled out the possibility that arctigenin acts directly as an uncoupling agent, as it did not accelerate the respiration rate in isolated mitochondria, as typical uncoupling agents would do [41]. Thus, decreased ∆ψm caused by arctigenin is likely to be associated with inhibition of mitochondrial respiration. This hypothesis was further confirmed in L6 myotubes by the dose-dependently inhibited respiration and increase of the AMP/ATP ratio after arctigenin treatment. Moreover, in isolated mitochondria, arctigenin inhibited ADP-stimulated respiration with glutamate and malate as substrates, but had no effect on respiration with succinate as the substrate. This suggests that arctigenin inhibited respiration via a direct effect on mitochondrial respiratory complex I. Therefore, arctigenin activated AMPK by inhibiting mitochondrial respiratory complex I, further leading to an elevated AMP/ATP ratio. This action shares similar mechanisms with several clinically used glucose-lowering drugs such as rosiglitazone and metformin [38, 42, 43], highlighting the prospect of arctigenin as a lead compound for the treatment of type 2 diabetes mellitus.
Whole-body glucose homeostasis is maintained by a balance between glucose production and glucose uptake by peripheral tissues. Activation of AMPK increases glucose uptake independently of insulin signalling [44] in skeletal muscle and suppresses hepatic glucose production, thus producing a glucose-lowering effect in type 2 diabetes [15]. In the present study, a single oral administration of arctigenin significantly decreased blood glucose levels after 2 h in ob/ob mice, but this effect was less pronounced than that of metformin. Our study of chronic administration of arctigenin showed that arctigenin significantly reduced random and fasting blood glucose levels in ob/ob mice. Moreover, unlike the weaker effect of arctigenin in the acute study, the glucose-lowering effect of arctigenin (200 mg/kg orally) on random-fed blood glucose was comparable with that of metformin (250 mg/kg), suggesting that arctigenin worked longer than metformin, as the random-fed blood glucose values were measured at 24 h post-dose. The glucose-lowering effect of arctigenin was not due to insulin secretion, since administration of arctigenin to ob/ob mice didn’t cause insulin secretion upon glucose loading (ESM Fig. 3).
In addition to glucose-lowering, lipid metabolism in ob/ob mice was also improved by arctigenin treatment. A significant decrease in serum cholesterol levels was observed in ob/ob mice after 23 days of treatment with arctigenin. The triacylglycerol content in liver was also reduced, consistent with previous reports on AICAR, metformin or A-769662 [15, 33]. This reduced lipid accumulation in liver can be explained by arctigenin-induced activation of AMPK, and the subsequent inhibition of hepatic lipogenesis and increase of fatty acid oxidation [45]. In addition, chronic treatment with arctigenin in ob/ob mice resulted in a significant reduction of triacylglycerol content in skeletal muscle, possibly due to the activation of AMPK induced by arctigenin, followed by an increase of fatty acid oxidation in skeletal muscle [46]. Since excess lipid accumulation in muscle and liver is associated with insulin resistance in humans [47] and rodents [48], the decrease in hepatic and intramuscular lipid content will subsequently improve insulin sensitivity, further improving glucose metabolism. These findings are further supported by the increased insulin-stimulated AKT phosphorylation in gastrocnemius muscle and liver of ob/ob mice after chronic arctigenin treatment. Moreover, hepatic G6pase mRNA was significantly reduced in arctigenin-treated mice, suggesting that the decreased expression of gluconeogenic genes may also contribute to the glucose-lowering effect of arctigenin. Taken together, our results indicate that arctigenin improves overall glucose and lipid metabolism in ob/ob mice, an effect likely to be mediated by the activation of AMPK in liver and skeletal muscle.
In conclusion, our studies demonstrate the beneficial effects of arctigenin as a potent indirect activator of AMPK on the regulation of glucose and lipid homeostasis. Arctigenin caused inhibition of mitochondrial respiratory complex I, thus leading to activation of AMPK and subsequent beneficial metabolic outcomes, such as enhanced glucose uptake in skeletal muscle, and suppression of hepatic gluconeogenesis and lipid synthesis. Administration of arctigenin decreased blood glucose, improved glucose tolerance and dyslipidaemia, decreased triacylglycerol accumulation and increased insulin-stimulated AKT phosphorylation in skeletal muscle and liver in ob/ob mice. These results highlight the potential value of arctigenin as a potent lead compound for the treatment of type 2 diabetes.
Abbreviations
- ACC:
-
Acetyl coenzyme A synthetase
- AICAR:
-
5-Amino-imidazole carboxamide ribonucleotide
- AMPK:
-
AMP-activated protein kinase
- CaMKK:
-
Ca2+/calmodulin-dependent protein kinase kinase
- EDL:
-
Extensor digitorum longus
- LKB1:
-
Liver kinase B1
- ∆ψm:
-
Mitochondrial membrane potential
- NOS:
-
Nitric oxide synthase
- ONOO– :
-
Peroxynitrite
- PI3-kinase:
-
Phosphoinositide-3 kinase
- siRNA:
-
Small interfering RNA
- UPR:
-
Unfolded protein response
References
DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP (1981) The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30:1000–1007
Ren JM, Marshall BA, Gulve EA et al (1993) Evidence from transgenic mice that glucose transport is rate-limiting for glycogen deposition and glycolysis in skeletal muscle. J Biol Chem 268:16113–16115
Hansell CA, Schiering C, Kinstrie R et al (2011) Universal expression and dual function of the atypical chemokine receptor D6 on innate-like B cells in mice. Blood 117:5413–5424
Kelley DE, Goodpaster B, Wing RR, Simoneau JA (1999) Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol 277:E1130–E1141
Leng Y, Steiler TL, Zierath JR (2004) Effects of insulin, contraction, and phorbol esters on mitogen-activated protein kinase signaling in skeletal muscle from lean and ob/ob mice. Diabetes 53:1436–1444
Zierath JR, Krook A, Wallberg-Henriksson H (2000) Insulin action and insulin resistance in human skeletal muscle. Diabetologia 43:821–835
Long YC, Zierath JR (2006) AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest 116:1776–1783
Winder WW (2001) Energy-sensing and signaling by AMP-activated protein kinase in skeletal muscle. J Appl Physiol 91:1017–1028
Koistinen HA, Galuska D, Chibalin AV et al (2003) 5-amino-imidazole carboxamide riboside increases glucose transport and cell-surface GLUT4 content in skeletal muscle from subjects with type 2 diabetes. Diabetes 52:1066–1072
Shaw RJ, Kosmatka M, Bardeesy N et al (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 101:3329–3335
Woods A, Dickerson K, Heath R et al (2005) Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2:21–33
Towler MC, Hardie DG (2007) AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 100:328–341
Viollet B, Lantier L, Devin-Leclerc J et al (2009) Targeting the AMPK pathway for the treatment of type 2 diabetes. Front Biosci 14:3380–3400
Musi N, Hirshman MF, Nygren J et al (2002) Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51:2074–2081
Cool B, Zinker B, Chiou W et al (2006) Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 3:403–416
Newman DJ, Cragg GM (2007) Natural products as sources of new drugs over the last 25 years. J Nat Prod 70:461–477
Lee YS, Kim WS, Kim KH et al (2006) Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 55:2256–2264
Baur JA, Pearson KJ, Price NL et al (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444:337–342
Collins QF, Liu HY, Pi J, Liu Z, Quon MJ, Cao W (2007) Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, suppresses hepatic gluconeogenesis through 5′-AMP-activated protein kinase. J Biol Chem 282:30143–30149
Wagner BK, Kitami T, Gilbert TJ et al (2008) Large-scale chemical dissection of mitochondrial function. Nat Biotechnol 26:343–351
Turner N, Li JY, Gosby A et al (2008) Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 57:1414–1418
Zou MH, Kirkpatrick SS, Davis BJ et al (2004) Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem 279:43940–43951
Fryer LG, Parbu-Patel A, Carling D (2002) The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 277:25226–25232
Zhao F, Wang L, Liu K (2009) In vitro anti-inflammatory effects of arctigenin, a lignan from Arctium lappa L., through inhibition on iNOS pathway. J Ethnopharmacol 122:457–462
Sun S, Wang X, Wang C et al (2011) Arctigenin suppresses unfolded protein response and sensitizes glucose deprivation-mediated cytotoxicity of cancer cells. Planta Med 77:141–145
Hotamisligil GS (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140:900–917
Deldicque L, Cani PD, Philp A et al (2010) The unfolded protein response is activated in skeletal muscle by high-fat feeding: potential role in the downregulation of protein synthesis. Am J Physiol Endocrinol Metab 299:E695–E705
Ye R, Jung DY, Jun JY et al (2010) Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes 59:6–16
Bergeron R, Russell RR 3rd, Young LH et al (1999) Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol 276:E938–E944
Hardie DG (2003) Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144:5179–5183
DeFronzo RA, Ferrannini E, Simonson DC (1989) Fasting hyperglycemia in non-insulin-dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism 38:387–395
Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M (2004) Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 432:1027–1032
Zhou G, Myers R, Li Y et al (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174
Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D (2007) Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J 403:139–148
Fujita Y, Hosokawa M, Fujimoto S et al (2010) Metformin suppresses hepatic gluconeogenesis and lowers fasting blood glucose levels through reactive nitrogen species in mice. Diabetologia 53:1472–1481
Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ (2000) Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes 49:527–531
Kosaka T, Okuyama R, Sun W et al (2005) Identification of molecular target of AMP-activated protein kinase activator by affinity purification and mass spectrometry. Anal Chem 77:2050–2055
Konrad D, Rudich A, Bilan PJ et al (2005) Troglitazone causes acute mitochondrial membrane depolarisation and an AMPK-mediated increase in glucose phosphorylation in muscle cells. Diabetologia 48:954–966
Hutchinson DS, Csikasz RI, Yamamoto DL et al (2007) Diphenylene iodonium stimulates glucose uptake in skeletal muscle cells through mitochondrial complex I inhibition and activation of AMP-activated protein kinase. Cell Signal 19:1610–1620
Narita M, Shimizu S, Ito T et al (1998) Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci U S A 95:14681–14686
Harper JA, Dickinson K, Brand MD (2001) Mitochondrial uncoupling as a target for drug development for the treatment of obesity. Obes Rev 2:255–265
Brunmair B, Staniek K, Gras F et al (2004) Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes 53:1052–1059
Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348:607–614
Narkar VA, Downes M, Yu RT et al (2008) AMPK and PPARdelta agonists are exercise mimetics. Cell 134:405–415
Viollet B, Guigas B, Leclerc J et al (2009) AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf) 196:81–98
Winder WW, Hardie DG (1996) Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol 270:E299–E304
Pan DA, Lillioja S, Kriketos AD et al (1997) Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46:983–988
Oakes ND, Bell KS, Furler SM et al (1997) Diet-induced muscle insulin resistance in rats is ameliorated by acute dietary lipid withdrawal or a single bout of exercise: parallel relationship between insulin stimulation of glucose uptake and suppression of long-chain fatty acyl-CoA. Diabetes 46:2022–2028
Acknowledgements
This study was supported by the National Basic Research Program of China (973 Program, 2009CB522300) and National Nature Science Foundation of China (Grant 90813029).
Contribution statement
S-LH, R-TY, JG, YF, Y-LD, FH, Y-HH, Y-DT and YL all contributed to the conception and design of the study, and contributed to analysis and interpretation of data. All the authors participated in drafting and revising the article and all have approved the final version of the article.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Arctigenin (ARC-G) actives AMPK indirectly and not dependent on LKB1, CaMKK or ONOO−. a AMPKα1β1γ1 kinase assay. AMPK kinase was treated with arctigenin at the indicated concentrations. AMP (200 μmol/l) was set as a positive control. b, c Hela cells were incubated with arctigenin at the indicated concentrations for 2 h, followed by the AMPK phosphorylation analysis. Metformin (1mmol/l) and AICAR (0.5mmol/l) were set as negative control and positive control, respectively. d, e L6 myotubes were preincubated in the absence or presence of STO-609 (10 μg/ml) for 30 min and then treated with 3 μmol/l arctigenin for 2 h, followed by AMPK and ACC phosphorylation analysis (d) and 2-deoxyglucose uptake measurement (e). f, g L6 myotubes were preincubated in the absence or presence of L-NAME (200 μmol/l) for 30min and then treated with 3 μmol/l arctigenin for 2 h, followed by AMPK and ACC phosphorylation analysis (f) and 2-deoxyglucose uptake measurement (g). Results are shown as means±SEM from three independent experiments. *p<0.05, **p<0.01 vs corresponding controls (as indicated). g white bars, without L-NAME; black bars, with L-NAME (PDF 5,209 kb)
ESM Fig. 2
Arctigenin (ARC-G) showed no effect on the expression or phosphorylation levels of unfolded protein response (UPR) related proteins in livers of ob/ob mice after chronic arctigenin treatment (200 mg/kg, orally, 23 days; PDF 211 kb)
ESM Fig. 3
Acute effect of arctigenin (ARC-G) on glucose tolerance in ob/ob mice. 400 mg/kg arctigenin or vehicle (0.5% carboxymethyl cellulose, wt/vol) was orally administered to 5 h fasting female C57BL/6 mice (9-week-old) 1 h prior to oral glucose challenge (1.5 g/kg). Blood samples were collected at 0, 15, 30, 60 and 120 min after glucose loading via the retroorbital sinus. Serum glucose (a) and insulin (b) concentrations were measured with an enzymatic colorimetric method and insulin ELISA kit, respectively. white circles, vehicle control; black squares, ARC-G (PDF 1429 kb)
ESM Table 1
PDF 123 kb
ESM Methods
PDF 362 kb
Rights and permissions
About this article
Cite this article
Huang, SL., Yu, RT., Gong, J. et al. Arctigenin, a natural compound, activates AMP-activated protein kinase via inhibition of mitochondria complex I and ameliorates metabolic disorders in ob/ob mice. Diabetologia 55, 1469–1481 (2012). https://doi.org/10.1007/s00125-011-2366-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2366-3