
Abstract
Aims/hypothesis
Although thiazolidinediones are now widely used to treat type 2 diabetes, their mechanism of action remains largely unknown. They are agonists for the transcription factor PPARγ, and in addition to their insulin-sensitising effects, they can promote adipogenesis and control gene expression in adipose tissues. We have explored the effect of rosiglitazone on insulin-mediated induction of pivotal genes involved in lipid metabolism and thermogenesis in brown fat. The genes studied were: (1) lipoprotein lipase (lpl), which is involved in lipid uptake; (2) hormone-sensitive lipase (hsl), which mobilises fatty acids from stored triglycerides; (3) fatty acid synthase (fas), which regulates de novo lipogenesis; and (4) the uncoupling proteins (ucp) 1 and 3, which control thermogenesis.
Methods
We used fetal rat primary brown adipocytes cultured with insulin, rosiglitazone or both combined. Then, we studied gene expression by northern and western blotting, as well as ‘run-on’ and gel-shift assays to identify binding of potential transcription factors to the fas promoter.
Results
Exposure to rosiglitazone for 24 h induced ucp-1, lpl and hsl gene expression and when rosiglitazone was combined with insulin a synergistic effect on lpl and ucp-3 mRNA expression was produced. These effects were consistent with increased LPL and HSL activities as well as respiration rates, mainly in response to exogenous palmitate. In contrast, treatment with rosiglitazone did not alter FAS mRNA basal levels but prevented the induction elicited by insulin in a time- and dose-dependent manner. Correspondingly diminished FAS protein levels and activity, as well as cellular lipid content, were observed, indicating an antilipogenic action of rosiglitazone in brown adipocytes. Furthermore, rosiglitazone impaired insulin increase in the FAS transcription rate by antagonising insulin-induced binding of upstream stimulatory factors to the E-box consensus sequence in the FAS promoter and insulin-induced binding of activating protein-1.
Conclusions/interpretation
Rosiglitazone prevents insulin-induced up-regulation of the main lipogenic enzyme but increases the expression of those enzymes involved in lipid uptake and mobilisation, favouring fatty acid utilisation through uncoupled respiration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thiazolidinediones (TZDs) are insulin-sensitising drugs that improve insulin action in vivo across a wide spectrum of insulin-resistant states, and have recently been introduced as therapeutic agents for the treatment of type 2 diabetes [1]. Although TZDs were developed without knowledge of their molecular targets, they are agonists for the nuclear receptor peroxisome proliferating activated receptor (PPAR)γ, and the relative ability of different TZDs to bind and activate PPARγ in vitro correlates with the antidiabetic action of these drugs in vivo [1, 2]. Moreover, PPARγ are ligand-activated transcription factors that play a critical role in the adipogenic differentiation process. Accordingly, either TZDs or ectopic expression of PPARγ dramatically promoted adipogenic differentiation from pre-adipocytes or non-adipogenic cells [3, 4]. Besides increasing insulin sensitivity and adipogenesis, TZDs control specific gene expression in white adipose tissue, a tissue in which there is high expression of PPARγ. In this regard, the TZD rosiglitazone up-regulated hormone-sensitive lipase (HSL) and lipoprotein lipase (LPL) gene expression and increased insulin responsiveness in lipolysis and lipogenesis in human subcutaneous adipocytes [5]. Moreover, induction of glycerol kinase gene expression by TZDs on murine mature adipocytes has been described [6]. All these effects have been proposed as the mechanism of action for TZDs to reduce circulating NEFA, which cause insulin resistance in skeletal muscle and other insulin target tissues. The increase in fat mass due to LPL expression was consistent with the weight gain observed in humans during treatment with rosiglitazone [2]. Furthermore, recent studies have reported attenuated insulin resistance (due to increased expression of adiponectin, PPARγ and PGC-1, and decreased expression of IL-6) without changes in the fat content of the subcutaneous adipose tissue from patients with highly active antiretroviral therapy-associated lipodystrophy treated with rosiglitazone [7].
Although white adipose tissue is the main adipose tissue, brown adipose tissue is also present in small mammals. Brown adipose tissue is specialised in non-shivering thermogenesis and is responsible for the heat production associated with expression of the mitochondrial uncoupling protein (UCP)-1 [8]. Brown adipocytes are also a target for TZD action, since they have high expression of PPARγ; TZDs were found to promote adipogenic and thermogenic differentiation in these cells [9, 10]. Moreover, we have recently shown that rosiglitazone produces insulin sensitisation under physiological and insulin-resistance conditions in cultured brown fat cells [11, 12]. Regarding gene expression, rosiglitazone up-regulated ucp-1 through binding to the PPAR response element present in the enhancer of this gene [13–15]. Fuels for thermogenesis are provided as fatty acids, which can be obtained: (1) via uptake from lipoproteins by the action of LPL; (2) via lipolysis from stored triglycerides by HSL; and (3) via de novo lipid synthesis by fatty acid synthase (FAS). Brown adipocytes express LPL and are also responsible for prominent de novo lipid synthesis, since glucose is an essential lipogenic substrate and insulin one of the most potent signals in inducing the expression of FAS [10, 16]. Therefore, this cell model provides an ideal system for investigating regulation of the expression of pivotal genes involved in lipid metabolism and thermogenesis by rosiglitazone and/or insulin.
Materials and methods
Materials
Insulin and BSA (fraction V, essentially NEFA-free) were from Sigma Chemical (St. Louis, MO, USA). Wy14643 was purchased from Biomol Research Laboratories (Plymouth, UK). Rosiglitazone was kindly provided by Dr S. A. Smith (Glaxo SmithKline, Harlow, UK). Fetal calf serum, PBS, culture media and Trizol were from Invitrogen (Paisley, UK). Autoradiographic films were Kodak X-O-MAT/AR (Eastman Kodak, Rochester, NY, USA). (α-32P)dCTP, (α-32P)UTP and the multiprimer DNA-labelling system kit were purchased from Amersham Biosciences (Little Chalfont, UK). The mouse anti-FAS antibody was from BD Biosciences Pharmingen (San Diego, CA, USA). The antibody anti-UCP-1 was from Alexis (Lausanne, Switzerland). The antibodies for supershift experiments were anti-sterol regulatory element binding protein (SREBP)1 ‘sc-8984’ from Santa Cruz (Palo Alto, CA, USA), and anti-upstream stimulatory factor (USF) 1 and 2 was kindly provided by Dr Viollet (Institut Cochin de Genetique Moleculaire, Paris, France) [17]. All other reagents used were of the purest grade available.
Cell culture
Fetal brown adipocytes were obtained from interscapular brown adipose tissue of 20-day-old Wistar rat fetuses and isolated by collagenase dispersion as described previously [16]. Isolated cells were plated in tissue culture dishes (4×106 cells per 100-mm diameter dish) in minimal essential medium with Earle’s salts supplemented with 10% fetal calf serum, the presence of serum being essential for the cells to attach to the plastic surface of the dishes. After 4–6 h of culture at 37°C, cells were rinsed twice with PBS and a 70% confluent monolayer was observed under inverse light microscopy. Cells were maintained for 20 h in a serum-free medium supplemented with 0.2% (w/v) BSA and then cultured for different times (from 3 to 48 h) with or without insulin (10 nmol/l), rosiglitazone (10 μmol/l) or Wy14643 (10 μmol/l) alone or in combination. Wistar rats were obtained from the Complutense University Animal House (Madrid, Spain). The principles of laboratory animal care were followed and the study was approved by the local ethics committee and carried out in accordance with the Declaration of Helsinki.
Northern blotting
RNA was isolated and submitted to northern blot analysis as described previously [14]. For serial hybridisation with different probes, such as FAS, LPL, HSL, UCP-1 and UCP-3 cDNAs, the blots were stripped and then rehybridised as needed in each case. A final hybridisation with the 18 S rRNA cDNA was performed for normalisation. The membranes were subjected to autoradiography and the relative densities of the hybridisation signals were measured by densitometric scanning of the autoradiograms in a laser densitometer (Molecular Dynamics, Sunnyvale, CA, USA).
Western blotting
Proteins were isolated and submitted to western blot analysis as described previously [11]. Immunoreactive bands were visualised using the enhanced chemiluminescence (ECL-Plus) western blotting protocol (Amersham).
Brown adipocyte respiration
After different treatments, cells were detached from the plates and suspended at 4×105 cells/ml in PBS containing 5 mmol/l pyruvate, 10 mmol/l d-glucose, 10 mmol/l fructose and 32 μmol/l BSA, pH 7.4, 37°C, in a Hansa-Tech oxygen electrode chamber [18]. Other additions are described in the figure legends.
FAS activity
Cells were sonicated for 30 s at 1.5 mA in 0.1 mmol/l EDTA, 1 mmol/l dithiothreitol, 1 mm/l MgCl2, 10% (v/v) glycerol, 0.1 mmol/l NaH2P04 (pH 7.2), and centrifuged at 12,000×g for 4 min. The cytosolic supernatant was used for the spectrophotometric determination of FAS activity, as described previously [19]. Enzyme activity was expressed as nanomoles of NADPH consumed per minute per milligram of protein.
LPL activity
Cellular pellets were homogenised in 0.2 mol/l Tris-HCl (pH 8.2) at 4°C and delipidated with acetone–diethyl ether as described previously [20]. LPL activity was assayed in triplicate using an egg lecithin-stabilised emulsion of 14C-fatty acid-labelled triolein as substrate containing: 2.5 mmol/l triolein, 2.4% BSA, 0.2 mol/l Tris, 0.1 mol/l NaCl, and 0.8% heated rat serum (pH 8.2), in the absence or presence of 1 mol/l NaCl (high saline concentrations). LPL activity was determined by subtracting the non-LPL-dependent activity (high salt) from the total lipolytic activity.
HSL activity
Cellular pellets were homogenised in three volumes of 0.25 mol/l sucrose containing 1 mmol/l EDTA, 1 mmol/l dithiothreitol, 10 μg/ml antipain (pH 7.4) as described previously [20]. Infranatants were obtained by centrifugation at 110,000×g in a Beckman centrifuge (model TL-100) for 45 min at 4°C. A phospholipid-stabilised emulsion of a dioleoylglycerol ether analogue, 1(3)-mono[3H]oleyl-2-O-oleoylglycerol, was used to assay the hormone-sensitive diacylglycerol lipase activity. Inhibition experiments were performed by pre-incubation with 100 mmol/l NaF.
Lipid content
Cytoplasmic lipid content was determined by Nile Red fluorescence emission in a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) as described previously [16]. Results represent mean intensities of fluorescence, obtained from the histograms of number of cells vs intensity of fluorescence, and are expressed in arbitrary units.
Nuclear run-on transcription assay
Nuclei were prepared and the run-on transcription assay was performed using (α-32P)UTP following the protocol described previously [16]. The incorporation of (α-32P)UTP into RNA was determined by scintillation counting, and equal counts were hybridised to filter-immobilised plasmids as described for northern blotting. Quantification of the signals was performed by densitometric scanning of the slots and signals were standardised using the β-actin signal.
Gel-shift assay
Nuclear extracts were prepared and incubated with labelled oligonucleotides following the gel mobility shift assay described previously [21]. The double-stranded oligonucleotide used as the AP-1 probe was composed of the sequence 5′-AGCTTGATGAGTCAGGGGCCGGATC-3′, and for the −65-bp E-box of the FAS promoter the sequence used was 5′-AGCTGTCAGCCCATGTGGCGTGGCCGC-3′. The AP-1 and E-box double-stranded oligonucleotides were chemically synthesised by Sigma-Genosys (Haverhill, UK). Oligonucleotides were labelled using Klenow polymerase and (α-32P)dCTP. The binding reaction was performed at 4°C for 15 min in a buffer containing 0.5 ng of doubled-stranded oligonucleotide probe, 2 μg of poly(dIdC), and 10 μg of protein in buffer C, supplemented with 35 mmol/l MgCl2. For supershift and competition assays, nuclear extracts were previously incubated for 15 min at 4°C with 1 μl of the corresponding antibodies or 100-fold excess of cold probe. DNA–protein complexes were resolved on a 6% polyacrylamide gel. The gel was dried and exposed to film at −70°C.
Data analysis
Results are presented as means±SEM (n=4) for duplicate dishes from four independent experiments. Statistical significance was tested with ANOVA, followed by the protected least-significant different test. A p value of less than 0.05 was considered significant. In experiments using X-ray films (Hyperfim), different exposure times were used to ensure that bands were not saturated.
Results
Rosiglitazone up-regulates the expression of UCP-1, UCP-3, LPL and HSL in brown adipocytes
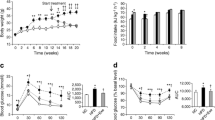
Rat fetal brown adipocytes were maintained for 20 h in a serum-free medium and further cultured for 24 h in the absence or presence of rosiglitazone (10 μmol/l) or insulin (10 nmol/l), individually or combined. The expression of the thermogenic genes ucp-1 and ucp-3 was studied by northern and western blotting, respectively (Fig. 1). Rosiglitazone significantly increased UCP-1 mRNA and protein levels (5-fold), insulin caused a 50% increase in these levels, and the combined presence of insulin and rosiglitazone produced a lower effect (4-fold) than rosiglitazone alone (Fig. 1a,b). The UCP-3 mRNA accumulation induced by rosiglitazone or insulin individually increased 2-fold but the result was additive when both compounds were combined (Fig. 1a). In order to determine whether changes in UCP expression were associated with changes in the thermogenic capacity of the cells, respiratory rates were calculated. As shown in Fig. 1c, the basal respiratory rates showed a good correlation with the UCP-1 levels in all the treatments studied. However, the most striking feature was the stimulation of respiration by exogenous palmitate, which should activate UCP-1 in situ. In the presence of oligomycin (absence of ATP synthesis), palmitate increased respiration by 36% in insulin-treated cells, by 148% in cells stimulated with rosiglitazone, and by 86% under both treatments.

Rosiglitazone induces UCP-1 expression and respiration in brown adipocytes. Brown adipocytes were serum-deprived for 20 h and further cultured for 24 h in the absence (control, C) or presence of 10 nmol/l insulin (I), 10 μmol/l rosiglitazone (R) or both (IR). a Total RNA was submitted to northern blotting with labelled UCP-1, UCP-3 and 18 S rRNA cDNAs. b Total protein was submitted to western blotting with anti-UCP-1 antibody and developed with ECL. Representative autoradiograms out of four are shown. Histograms from densitometric analysis of the autoradiograms are expressed in arbitrary units and represent means±SEM (n=4). *p<0.05 and ***p<0.001 for differences vs C; ♦ p<0.05 for differences in the presence of (IR) vs (R). c Traces of the respiration of cells after the following additions were made: 2.5 μg/ml oligomycin (Oligo); 64 μmol/l palmitate (Palm) (2:1 molar ratio to albumin); and 10 μmol/l FCCP. Rates are expressed in nanomoles of O2 consumed per minute and per 106 cells. A representative experiment of four independent experiments is shown
Since rosiglitazone appeared to induce UCP-1 expression and respiration rates in brown adipocytes, we investigated the possibility that it might enhance the availability of NEFA for oxidation. In consequence, we studied LPL and HSL gene expression under identical culture conditions as described above. As shown by northern blot analysis in Fig. 2a, the presence of either insulin or rosiglitazone doubled the LPL mRNA content; this effect was higher (5-fold) when both compounds were added together. Lower doses of rosiglitazone also had a synergistic effect with insulin on LPL mRNA accumulation (Fig. 3b). LPL activity was also measured in lysates from cells treated as described above. Both insulin and rosiglitazone individually had a positive effect on LPL activity, and combined produced an additive effect on LPL activity (Fig. 2b), in a similar fashion to mRNA accumulation. Rosiglitazone doubled HSL mRNA and protein content regardless of the absence or presence of insulin (Fig. 2c, d). Moreover, rosiglitazone increased HSL activity by 2-fold; this effect was partially precluded when insulin was present in the culture medium (Fig. 2e).

Rosiglitazone up-regulates the expression of LPL and HSL. Cells were cultured as in Fig. 1. a Total RNA was analysed by northern blotting with labelled LPL and 18 S rRNA cDNAs. A representative example of four autoradiograms is shown. Histograms from densitometric analysis represent means±SEM (n=4). b LPL activity was measured in cellular homogenates and was expressed as picokatals per milligram of protein (picokatals, picomoles of substrate transformed per second). Results are shown as means±SEM (n=4). *p<0.05 and ***p<0.001 for differences vs control (C); ♦ p<0.001 for differences between values in the presence of IR vs I and vs R. c Total RNA was analysed by northern blotting with labelled HSL and 18 S rRNA cDNAs. d Total protein was submitted to western blotting with antibodies against HSL or against β-actin and developed with ECL. A representative example of four autoradiograms is shown. e HSL activity in cellular homogenates was measured as described in Materials and methods and is expressed as milliunits per milligram protein. Results are expressed as means±SEM (n=4). ***p<0.05 for differences vs control (C); Δp<0.05 for differences between values in the presence of I vs its absence

Rosiglitazone down-regulates insulin-induced FAS gene expression in a dose- and time-dependent manner, and also down-regulates cellular lipid content. Brown adipocytes were serum-deprived for 20 h and further cultured for 24 h in the absence (control, C) or presence of 10 nmol/l insulin (I) or 10 μmol/l rosiglitazone (R) or 10 μmol/l Wy14643 (Wy) alone or in combination (a); or with insulin with or without increasing concentrations of rosiglitazone (b); or for 6, 24 or 48 h in the absence or presence of insulin with or without rosiglitazone (c). Total RNA was extracted under the different conditions and submitted to northern blotting with labelled probes. A representative of four autoradiograms is shown. Densitometric analysis of FAS mRNA levels after standardisation using the 18 S rRNA signal is shown. Results (arbitrary densitometric units) are presented as means±SEM (n=4). d Cells were cultured for 48 h as in Fig. 1. Total protein was submitted to western blotting with anti-FAS antibody and developed with ECL. A representative example of four autoradiograms is shown. Histograms represent means±SEM (n=4). e Cell extracts were used to determine FAS activity. Results are shown as means±SEM (n=4) and are expressed as nanomoles of NADPH used per minute per milligram of protein. f Cytoplasmic lipid content was determined by Nile Red fluorescence as described in Materials and methods. Results (arbitrary densitometric units) are expressed as means±SEM (n=4). *p<0.05 and ***p<0.001 for differences between values in the presence of I vs control (C); Δp<0.001 for differences between values in the presence of IR vs I
Rosiglitazone impairs insulin-induced FAS gene expression by antagonising the binding of insulin-induced USFs to the FAS promoter and insulin-induced AP-1 binding activity
Fatty acids can be synthesised de novo by the lipogenic enzyme FAS. In this regard, treatment for 24 h with insulin, a potent lipogenic signal, produced a significant 9-fold) FAS mRNA accumulation compared with untreated cells (Fig. 3a). However, rosiglitazone alone failed to have any positive effect on FAS mRNA accumulation and, surprisingly, significantly decreased the response to insulin. In contrast to the effect of rosiglitazone, the PPARα agonist Wy14643 (10 μmol/l) did not alter the insulin effect on FAS mRNA levels. To further explore the mechanisms for the inhibition of insulin-induced FAS gene expression by rosiglitazone, we carried out dose-response and time-course experiments. When insulin-treated brown adipocytes were incubated in the presence of various doses of this compound, the insulin-induced increase in FAS mRNA accumulation was precluded in a concentration-dependent manner (Fig. 3b). The expression of FAS was detectable after 6 h of treatment with insulin, reaching maximal values at 24 h which were maintained for 48 h. The presence of rosiglitazone inhibited this insulin effect at all the time points studied (Fig. 3c). Rosiglitazone not only precluded an insulin-induced increase in FAS mRNA accumulation, but also impaired insulin induction of FAS protein content in cells after treatment for 48 h as determined by western blot analysis (Fig. 3d). Accordingly, insulin stimulated FAS activity after 48 h of treatment, but rosiglitazone totally precluded the increase elicited by insulin at this time (Fig. 3e). In consequence, the increase in cytoplasmic lipid content produced by treatment with insulin for 48 h and detected by Nile Red fluorescence was reduced by the presence of rosiglitazone (Fig. 3f).
Since TZDs are insulin sensitisers, the striking effect of rosiglitazone, i.e. antagonising insulin action on FAS expression, prompted us to investigate this phenomenon further. Accordingly, nuclear ‘run-on’ assays were performed in transcriptionally active nuclei isolated from cells treated for 3 h in the absence or presence of insulin or rosiglitazone or both combined (Fig. 4a). The transcription rate of the FAS gene increased 5-fold under insulin treatment relative to untreated cells. Rosiglitazone alone did not produce any effect, but significantly reduced the insulin induction of FAS transcription (Fig. 4a). We considered the possibility that the inhibitory effect of rosiglitazone on insulin-induced FAS gene expression could be mediated by interfering with transcription factors needed for insulin action. In this regard, the promoter region of the FAS gene contains a proximal insulin response sequence containing an E-box motif at the position −65 bp, a potential binding site for two transcription factors: USFs and SREBP [22–24]. We carried out electrophoretic mobility shift assays with a labelled oligonucleotide corresponding to the −65-bp E-box sequence and were able to observe an increase in specific protein–DNA complexes in extracts from insulin-stimulated cells, which was abolished in cells co-treated with rosiglitazone (Fig. 4b). This band disappeared in competition experiments with unlabelled probe. Specific antibodies against USFs but not against SREBP eliminated most of the binding to the E-box sequence. Furthermore, the promoter region of FAS has been reported to possess an inverted CCAAT box at positions −106 to −85 bp, a potential sequence for binding the transcription factor AP-1, a factor that was activated by insulin in brown adipocytes [25, 26]. We therefore carried out electrophoretic mobility shift assays with oligonucleotide probes corresponding to the AP-1 consensus sites in nuclear extracts from cells cultured for 24 h either in the absence or presence of insulin, rosiglitazone or both combined (Fig. 4c). Insulin treatment doubled nuclear protein binding to DNA corresponding to the AP-1 consensus site as compared to untreated cells. However, rosiglitazone treatment completely impaired insulin-enhanced AP-1 DNA binding activity (Fig. 4c).

Rosiglitazone impairs insulin-induced FAS transcription by antagonising insulin-induced USFs binding to the FAS promoter and insulin-induced AP-1 binding activity. Cells were treated for 3 h (a) or 24 h (b, c) as in Fig. 1. Nuclei (a) were prepared and used for a run-on transcription assay as described in Materials and methods. Representative autoradiograms of labelled transcripts representing FAS, β-actin and the vector PUC12 are shown in addition to histograms from the densitometric analysis after standardisation with the β-actin signal. b Nuclear extracts were incubated with labelled doubled-stranded oligonucleotide corresponding to the −65-bp E-box of the FAS promoter used as the probe. Some reactions included 15 min of previous incubation with USF or SREBP antibodies or a 100-fold molar excess of unlabelled probe used as a competitor (Comp), with the specific band that disappeared marked by an arrow. c Nuclear extracts were subjected to electrophoretic mobility shift assay using an AP-1 consensus site-labelled probe. A representative example of four autoradiograms is shown. Histograms from densitometric analysis are expressed in arbitrary units and represent means±SEM (n=4). Statistical significance, see Fig. 3
Discussion
In this paper we investigated the role of rosiglitazone on insulin-mediated induction of pivotal genes involved in lipid metabolism and thermogenesis in primary cultures of rat fetal brown adipocytes. Our results show that insulin produces a small induction of UCP-1 and UCP-3 expression while rosiglitazone induction of both UCPs, mainly UCP-1, is robust and seems to be directly due to the presence of the PPAR response element present in their promoters [13–15]. The concentration of rosiglitazone used in our experiments was 10 μmol/l, the optimal dose determined in previous studies for use in brown adipocytes [13], and close to the peak plasma concentration reached in humans, 2 μmol/l, after oral administration at the doses used in clinical practice [27]. The positive effect of insulin on UCP-3 gene expression observed in brown adipocytes is in contrast to the inhibitory effect described in skeletal muscle, indicating tissue-specific regulation of this gene [28]. Moreover, rosiglitazone also increases basal respiration rates and uncoupled respiration in response to palmitate, which clearly indicates that UCP-1 is functional and that rosiglitazone induces thermogenesis in these cells. Respiratory rates may appear low when compared to those observed in mature brown adipocytes [18], but a low respiratory capacity is characteristic of immature adipocytes [29].
In parallel with UCP-1, rosiglitazone induces LPL expression in brown adipocytes, in agreement with the PPAR response element found in the human LPL promoter [30]. When combined with insulin, rosiglitazone produces a synergistic effect on LPL mRNA accumulation and enzymatic activity, as found in brown adipose tissue of rats treated with a non-TZD PPARγ agonist [31]. These results suggest a likely increase in NEFA uptake from lipoproteins in response to insulin and/or rosiglitazone, in a manner similar to that detected in human subcutaneous adipocytes [5]. Brown adipocytes treated with rosiglitazone showed an increased basal respiration rate and LPL activity although they were cultured in serum-free medium with no lipoprotein availability. This would imply that rosiglitazone induces the mechanism necessary for obtaining NEFA as fuel for thermogenesis from lipoproteins, although under our experimental conditions NEFA probably originated from the hydrolysis of stored triglycerides through the activation of HSL. With regard to this possibility, we observed an induction of HSL mRNA and protein accumulation, resulting in increased HSL enzymatic activity in the presence of rosiglitazone, in agreement with reported data on human adipose tissue [5]. However, insulin exerts its antilipolytic action on HSL activity in the presence of rosiglitazone, probably by affecting the phosphorylation of the enzyme which is necessary for its full activation [32].
Lipid synthesis in murine adipocytes is largely a consequence of de novo fatty acid synthesis from glucose, although this metabolic pathway is minimal or absent in humans, as recently reported [33]. Previous findings have shown that insulin is a potent activator of the transcription of the lipogenic gene FAS in brown adipocytes [16] as well as in 3T3-L1 adipocytes [22, 24]. In the present study, we describe for the first time the negative regulation by rosiglitazone of the insulin-mediated induction of FAS mRNA, protein, activity and lipid content in brown adipocytes. This inhibition seems to be a transcriptionally mediated event, as indicated by the ‘run-on’ experiments, and dependent on PPARγ activation since the PPARα agonist Wy14643 does not behave in the same way. Dose-response experiments with rosiglitazone indicated that half-maximal inhibition occurred at a concentration similar to the Kd for PPARγ, 40 nmol/l, which supports the hypothesis of a PPARγ-mediated mechanism [34]. Our results are in agreement with the inhibitory effect of troglitazone (1 μmol/l) on insulin-induced FAS expression reported in 3T3-L1 adipocytes [35].
The fact that rosiglitazone does not produce any effect, either positive or negative, on FAS basal expression, and that so far there is no evidence of the presence of a PPAR response element in the FAS promoter, suggests that direct binding of PPARγ to the promoter is not involved in the inhibitory mechanism, and competition for limiting amounts of general co-activators could be the explanation. As a matter of fact, rosiglitazone per se does not modify protein–DNA complexes bound to E-box or AP-1 sequences but prevents insulin-induced binding. This mechanism, known as transrepression, has been related to some anti-inflammatory and anti-proliferative actions of PPARγ agonists. Thus, PPARγ ligands blocked PMA-mediated induction of the cox-2 promoter by inhibiting AP-1 activity as a consequence of the inhibition of c-JUN expression and competition for limiting amounts of the general co-activator p300/CBP [36]. Competition for the same co-activator was involved in the inhibition of transcription of genes induced by interferon-γ and/or lipopolysaccharide, including inducible NO synthase gene [37] and also in cyclin D1 repression by PPARγ [38]. Recently, rosiglitazone has also been shown to prevent the binding of transcription factors of the fos/jun family to the AP-1 site by inhibition of jun kinase in heart, resulting in protection from ischaemia [39]. Therefore, the mechanism for transcriptional repression by PPARs is either competition for limiting amounts of general co-activators or direct inhibition of the transcription factors that bind to response elements. We found that rosiglitazone inhibits AP-1 binding activity induced by insulin in brown adipocytes, and although the implication of this transcription factor for FAS gene expression has not been documented, the presence of an inverted CCAAT box in the −106 to −85-bp section of the FAS promoter has been reported [25]. Moreover, it has been reported that insulin activates the FAS promoter through the proximal insulin-response sequence containing an E-box motif at the −65-bp position [22]. We found that insulin induces protein binding to that sequence and supershift analysis has demonstrated that USFs are the proteins bound. Therefore, we have shown that rosiglitazone precludes insulin-induced USF protein binding to specific sequences in the FAS promoter, and that this correlates with the inhibition observed in FAS mRNA and protein accumulation. USFs have been reported to interact with PGC-1 [40, 41] and with CBP/p300 [42, 43], co-activators for many nuclear receptors including PPARγ [44, 45]. Competition between USFs and PPARγ for the same co-activator, for which PPARγ bound to rosiglitazone would have a higher affinity, is a possibility to explore in the future.
Our results on TZD amelioration of the insulin effects on brown adipose tissue fat content in rodents differ from those regarding weight gain due to an increase in white adipose tissue mass detected in humans during rosiglitazone therapy [2]. These effects, however, are less prominent in rodents, and so far no changes in brown adipose tissue adiposity in humans have been reported. Moreover, no changes in fat content but attenuation of insulin resistance were observed after treatment with rosiglitazone in patients with highly active antiretroviral therapy-associated lipodystrophy [7]. Furthermore, fat hypertrophy was detected in adipose-specific PPARγ knock-out mice which also exhibit fat cell loss [46]. Nevertheless, the actions of rosiglitazone in vivo, such as decreasing circulating NEFA and increasing glucose utilisation in skeletal muscle, may be the result of the insulin-sensitising effect observed in response to the primary action of TZDs on white adipose tissue and brown adipose tissue. Therefore, the antagonistic effects of rosiglitazone on insulin action—including (1) impairment of FAS expression and lipid accumulation in brown adipocytes together with (2) the increment in the uptake and mobilisation of lipids through the expression and activation of LPL and HSL and (3) the effective waste of energy as a result of the induction of UCPs and oxidation of NEFA in these cells—would result in a decrease in the levels of circulating NEFA and contribute to the alleviation of insulin resistance.
Abbreviations
- AP-1:
-
activating protein 1
- ECL:
-
enhanced chemiluminescence
- FAS:
-
fatty acid synthase
- HSL:
-
hormone-sensitive lipase
- LPL:
-
lipoprotein lipase
- PGC-1:
-
PPARγ co-activator-1
- PPAR:
-
peroxisome proliferating activated receptor
- SREBP:
-
sterol regulatory element binding protein
- TZD:
-
thiazolidinediones
- UCP:
-
uncoupling protein
- USF:
-
upstream stimulatory factor
References
Olefsky JM, Saltiel AR (2000) PPAR gamma and the treatment of insulin resistance. Trends Endocrinol Metab 11:362–368
Olefsky JM (2000) Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J Clin Invest 106:467–472
Lowell BB (1999) PPARgamma: an essential regulator of adipogenesis and modulator of fat cell function. Cell 99:239–242
Rosen ED, Spiegelman BM (2001) PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem 276:37731–37734
McTernan PG, Harte AL, Anderson LA et al (2002) Insulin and rosiglitazone regulation of lipolysis and lipogenesis in human adipose tissue in vitro. Diabetes 51:1493–1498
Guan HP, Li Y, Jensen MV, Newgard CB, Steppan CM, Lazar MA (2002) A futile metabolic cycle activated in adipocytes by antidiabetic agents. Nat Med 8:1122–1128
Sutinen J, Kannisto K, Korsheninnikova E et al (2004) Effects of rosiglitazone on gene expression in subcutaneous adipose tissue in highly active antiretroviral therapy-associated lipodystrophy. Am J Physiol Endocrinol Metab 286:E941–E949
Cannon B, Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359
Tai TA, Jennermann C, Brown KK et al (1996) Activation of the nuclear receptor peroxisome proliferator-activated receptor gamma promotes brown adipocyte differentiation. J Biol Chem 271:29909–29914
Rabelo R, Camirand A, Silva JE (1997) 3′,5′-cyclic adenosine monophosphate-response sequences of the uncoupling protein gene are sequentially recruited during darglitazone-induced brown adipocyte differentiation. Endocrinology 138:5325–5332
Hernandez R, Teruel T, Lorenzo M (2003) Rosiglitazone produces insulin sensitisation by increasing expression of the insulin receptor and its tyrosine kinase activity in brown adipocytes. Diabetologia 46:1618–1628
Hernandez R, Teruel T, de Alvaro C, Lorenzo M (2004) Rosiglitazone ameliorates insulin resistance in brown adipocytes of Wistar rats by impairing TNF-alpha induction of p38 and p42/p44 mitogen-activated protein kinases. Diabetologia 47:1615–1624
Teruel T, Smith SA, Peterson J, Clapham JC (2000) Synergistic activation of UCP-3 expression in cultured fetal rat brown adipocytes by PPARalpha and PPARgamma ligands. Biochem Biophys Res Commun 273:560–564
Teruel T, Clapham JC, Smith SA (1999) PPARalpha activation by Wy 14643 induces transactivation of the rat UCP-1 promoter without increasing UCP-1 mRNA levels and attenuates PPARgamma-mediated increases in UCP-1 mRNA levels induced by rosiglitazone in fetal rat brown adipocytes. Biochem Biophys Res Commun 264:311–315
Teruel T, Hernandez R, Benito M, Lorenzo M (2003) Rosiglitazone and retinoic acid induce uncoupling protein-1 (UCP-1) in a p38 mitogen-activated protein kinase-dependent manner in fetal primary brown adipocytes. J Biol Chem 278:263–269
Teruel T, Valverde AM, Benito M, Lorenzo M (1996) Insulin-like growth factor I and insulin induce adipogenic-related gene expression in fetal brown adipocyte primary cultures. Biochem J 319:627–632
Viollet B, Lefrancois-Martinez AM, Henrion A, Kahn A, Raymondjean M, Martinez A (1996) Immunochemical characterization and transacting properties of upstream stimulatory factor isoforms. J Biol Chem 271:1405–1415
Cunningham SA, Nicholls DG (1987) Induction of functional uncoupling protein in guinea pigs infused with noradrenaline. Studies with isolated brown adipocytes. Biochem J 245:485–491
Teruel T, Valverde AM, Alvarez A, Benito M, Lorenzo M (1995) Differentiation of rat brown adipocytes during late foetal development: role of insulin-like growth factor I. Biochem J 310:771–776
Martin-Hidalgo A, Holm C, Belfrage P, Schotz MC, Herrera E (1994) Lipoprotein lipase and hormone-sensitive lipase activity and mRNA in rat adipose tissue during pregnancy. Am J Physiol 266:E930–E935
Hernandez R, Teruel T, Lorenzo M (2003) Insulin and dexamethasone induce GLUT4 gene expression in foetal brown adipocytes: synergistic effect through CCAAT/enhancer-binding protein alpha. Biochem J 372:617–624
Wang D, Sul HS (1997) Upstream stimulatory factor binding to the E-box at −65 is required for insulin regulation of the fatty acid synthase promoter. J Biol Chem 272:26367–26374
Casado M, Vallet VS, Kahn A, Vaulont S (1999) Essential role in vivo of upstream stimulatory factors for a normal dietary response of the fatty acid synthase gene in the liver. J Biol Chem 274:2009–2013
Kim JB, Sarraf P, Wright M et al (1998) Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest 101:1–9
Rangan VS, Oskouian B, Smith S (1996) Identification of an inverted CCAAT box motif in the fatty-acid synthase gene as an essential element for modification of transcriptional regulation by cAMP. J Biol Chem 271:2307–2312
Teruel T, Valverde AM, Navarro P, Benito M, Lorenzo M (1998) Inhibition of PI 3-kinase and RAS blocks IGF-I and insulin-induced uncoupling protein 1 gene expression in brown adipocytes. J Cell Physiol 176:99–109
Park JY, Kim KA, Kang MH, Kim SL, Shin JG (2004) Effect of rifampin on the pharmacokinetics of rosiglitazone in healthy subjects. Clin Pharmacol Ther 75:157–162
Guillet-Deniau I, Mieulet V, Le Lay S et al (2002) Sterol regulatory element binding protein-1c expression and action in rat muscles: insulin-like effects on the control of glycolytic and lipogenic enzymes and UCP3 gene expression. Diabetes 51:1722–1728
Klaus S, Munzberg H, Truloff C, Heldmaier G (1998) Physiology of transgenic mice with brown fat ablation: obesity is due to lowered body temperature. Am J Physiol 274:R287–R293
Schoonjans K, Peinado-Onsurbe J, Lefebvre AM et al (1996) PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J 15:5336–5348
Laplante M, Sell H, MacNaul KL, Richard D, Berger JP, Deshaies Y (2003) PPAR-gamma activation mediates adipose depot-specific effects on gene expression and lipoprotein lipase activity: mechanisms for modulation of postprandial lipemia and differential adipose accretion. Diabetes 52:291–299
Holm C (2003) Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem Soc Trans 31:1120–1124
Arner P (2003) The adipocyte in insulin resistance: key molecules and the impact of the thiazolidinediones. Trends Endocrinol Metab 14:137–145
Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA (1995) An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 270:12953–12956
Barthel A, Kruger KD, Roth RA, Joost HG (2002) Concentration-dependent stimulatory and inhibitory effect of troglitazone on insulin-induced fatty acid synthase expression and protein kinase B activity in 3T3-L1 adipocytes. Naunyn-Schmiedebergs Arch Pharmacol 365:290–295
Subbaramaiah K, Lin DT, Hart JC, Dannenberg AJ (2001) Peroxisome proliferator-activated receptor gamma ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence for involvement of activator protein-1 and CREB-binding protein/p300. J Biol Chem 276:12440–12448
Li M, Pascual G, Glass CK (2000) Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol 20:4699–4707
Wang C, Fu M, D’Amico M et al (2001) Inhibition of cellular proliferation through IkappaB kinase-independent and peroxisome proliferator-activated receptor gamma-dependent repression of cyclin D1. Mol Cell Biol 21:3057–3070
Khandoudi N, Delerive P, Berrebi-Bertrand I, Buckingham RE, Staels B, Bril A (2002) Rosiglitazone, a peroxisome proliferator-activated receptor-gamma, inhibits the Jun NH(2)-terminal kinase/activating protein 1 pathway and protects the heart from ischemia/reperfusion injury. Diabetes 51:1507–1514
Michael LF, Wu Z, Cheatham RB et al (2001) Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci U S A 98:3820–3825
Moore ML, Park EA, McMillin JB (2003) Upstream stimulatory factor represses the induction of carnitine palmitoyltransferase-Ibeta expression by PGC-1. J Biol Chem 278:17263–17268
De Luca A, Severino A, De Paolis P et al (2003) p300/cAMP-response-element-binding-protein (‘CREB’)-binding protein (CBP) modulates co-operation between myocyte enhancer factor 2A (MEF2A) and thyroid hormone receptor–retinoid X receptor. Biochem J 369:477–484
Breen GA, Jordan EM (1999) Transcriptional activation of the F(1)F(0) ATP synthase alpha-subunit initiator element by USF2 is mediated by p300. Biochim Biophys Acta 1428:169–176
Gelman L, Zhou G, Fajas L, Raspe E, Fruchart JC, Auwerx J (1999) p300 interacts with the N- and C-terminal part of PPARgamma2 in a ligand-independent and -dependent manner, respectively. J Biol Chem 274:7681–7688
Kodera Y, Takeyama K, Murayama A, Suzawa M, Masuhiro Y, Kato S (2000) Ligand type-specific interactions of peroxisome proliferator-activated receptor gamma with transcriptional coactivators. J Biol Chem 275:33201–33204
He W, Barak Y, Hevener A et al (2003) Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A 100:15712–15717
Acknowledgements
T. Teruel was the recipient of a postdoctoral fellowship from the Comunidad Autonoma de Madrid. R. Hernandez was the recipient of a postgraduate fellowship from the Ministerio de Educacion y Cultura. This work was supported by Grant BMC2002-01322 (Direccion General de Investigacion Cientifica, MCYT, Spain). We are also grateful for the support of COST B17 Action on ‘Insulin resistance, obesity and diabetes mellitus in the elderly’ (European Commission), and of Red de grupos de Diabetes Mellitus G03/212 (Instituto de Salud Carlos III, MSC, Spain). T. Teruel and R. Hernandez contributed equally to the experimental work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Teruel, T., Hernandez, R., Rial, E. et al. Rosiglitazone up-regulates lipoprotein lipase, hormone-sensitive lipase and uncoupling protein-1, and down-regulates insulin-induced fatty acid synthase gene expression in brown adipocytes of Wistar rats. Diabetologia 48, 1180–1188 (2005). https://doi.org/10.1007/s00125-005-1744-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-1744-0