
Abstract
Aims/hypothesis
We tested the hypothesis that NEFA concentrations are higher in obese subjects with fatty liver than in obese subjects without fatty liver.
Materials and methods
We recruited 22 obese (BMI>30 kg/m2) men aged 42–64 years, in whom liver fat was assessed by ultrasound and classified into categories of no, mild to moderate and severe fatty liver by two independent radiologists. Regional and visceral abdominal fat were assessed by dual-energy X-ray absorptiometry and magnetic resonance imaging, and endogenous glucose production, whole-body glucose disposal during an insulin clamp, and NEFA concentrations were measured, along with NEFA suppression (percent (%) suppression and insulin sensitivity index for NEFA during an OGTT).
Results
Seven subjects had no evidence of fatty liver, nine had mild or moderate fatty liver and six had severe fatty liver. The amount of visceral fat was not associated with the degree of fatty liver. Whole-body glucose disposal was inversely associated with fatty liver (38.4, 26.5 and 23.9 μmol kg−1 min−1 for the groups with no fatty liver, mild to moderate fatty liver and severe fatty liver, respectively, p=0.004). NEFA suppression during the OGTT was decreased (62.5, 50.8 and 41%, p=0.03, for no, mild to moderate, and severe fatty liver, respectively) and the insulin sensitivity index for NEFA was decreased (0.80, 0.40 and 0.34, p<0.0001). Regression modelling suggested that NEFA concentrations were associated with fatty liver independently of whole-body glucose production and disposal measurements.
Conclusions/interpretation
In obese men, NEFA concentrations during an OGTT are associated with fatty liver independently of classic measures of insulin sensitivity determined by the hyperinsulinaemic clamp. The contribution to this association by factors regulating NEFA concentrations requires further study.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Non-alcoholic steatohepatitis (NASH) forms part of a spectrum of non-alcoholic fatty liver diseases (NAFLD) that ranges from steatosis to cirrhosis and is associated with obesity [1–3]. NAFLD is now thought to be the most common of all liver disorders in developed nations, with important health and resource implications for these countries, as up to 50% of subjects develop liver fibrosis, 15–17% develop cirrhosis and 3% develop end-stage liver failure requiring transplantation [3–5].
The precise mechanism linking the quantity and location of body fat with NAFLD is uncertain, but because of the strong association between NAFLD, obesity and insulin resistance, it has been suggested that NAFLD is part of the metabolic syndrome [2, 6, 7]. Establishing whether obesity or insulin resistance is the more important factor for the development of fatty liver is difficult because of their strong co-linear association [2, 6–9]. Recent literature has suggested that insulin resistance is more closely associated with the development of NAFLD than with obesity [7, 8, 10]. However, it remains uncertain whether insulin resistance itself is the most important factor for development of fatty liver, or whether other factor(s) produced by fat, namely adipocytokines, inflammatory cytokines and NEFA, are more important. The role of overnight NEFA concentrations (when insulin concentrations are low) and the role of insulin-mediated NEFA suppression in contributing to fatty liver are uncertain. A previous study in slightly overweight men with a BMI of 25–26 kg/m2, stratified by high and low liver fat, showed that insulin-stimulated NEFA suppression was impaired after 2 h, during a low-dose insulin clamp lasting 3 h, in the high-fat group [11]. However, obese individuals have markedly increased NEFA concentrations compared with slightly overweight subjects, while obesity itself is associated with impaired suppression of plasma NEFA concentrations [12–14]. Moreover, obese individuals have increased quantities of visceral fat and the contribution of visceral fat to the pathogenesis of fatty liver in obesity is uncertain. To our knowledge, none of the studies to date that have compared insulin sensitivity in skeletal muscle and liver of obese individuals with and without fatty liver have included measurement of visceral fat and NEFA concentrations. Ultrasonography can be used to identify subjects with fatty liver, and in obese subjects ultrasound has a sensitivity of 94%, a specificity of 88% and a positive predictive value of 94% when compared with histological findings [15, 16]. Therefore, ultrasound is an appropriate tool to identify fatty liver in obese subjects.
The aim of our study was to test the hypothesis that NEFA concentrations are increased and NEFA suppression impaired in subjects with obesity and fatty liver, compared with obese subjects without fatty liver.
Subjects, materials and methods
This cross-sectional study was approved by the local ethics committee. Healthy obese men aged 42–64 years were recruited from the general population. Subjects were asked to avoid strenuous exercise or alcohol on the day prior to attending. An OGTT was undertaken. Blood was then taken at 10, 20, 30, 40, 60, 90 and 120 min. Glucose, total cholesterol, HDL cholesterol and triglycerides, alanine aminotransferase and γ-glutamyl transferase were measured in fasting serum using commercially available kits according to the manufacturers' instructions. Adiponectin, IL-1β, IL-6 and TNF-α were analysed using an ELISA kit (R and D Systems, Abingdon, UK). Suppression of NEFA was measured during the OGTT [17–19]. To assess insulin sensitivity in skeletal muscle and liver, we used a hyperinsulinaemic–euglycaemic clamp technique [20] with a labelled glucose tracer to determine insulin-mediated suppression of hepatic glucose output during the early phase of the clamp [7, 8, 21]. We used anthropometry, dual-energy X-ray absorptiometry (DEXA), bioelectrical impedance and horizontal five-slice cross-sectional magnetic resonance imaging (MRI) to evaluate in detail body fat, truncal fat and visceral fat.
BMI was calculated as height (m) squared divided by weight (kg). Skinfold measurements were made using skinfold calipers (Harpenden; Assist creative resources, Wrexham, UK) at four standard sites. Three measurements were taken at each site and the mean value was used to calculate percentage body fat using the methods described by Durnin and Womersley [22].
The waist was measured over bare skin midway between the costal margin and the iliac crest. Hip circumference was measured at the widest part between the greater trochanter and lower buttock level. Bioelectrical impedance was measured (Bodystat 1500; Bodystat, Isle of Man, UK). DEXA scanning was undertaken with a Delphi W instrument (Hologic, Bedford, MA, USA) using a standard visual method to divide images into trunk, limb and head.
Blood pressure was measured using a Marquette Dash 3,000 monitor (GE Healthcare, Little Chalfont, Bucks, UK) on the non-dominant arm after subjects had become acclimatised to the Clinical Research Facility and had rested for at least 60 min.
Liver ultrasonography was performed by one of two experienced radiologists (with one exception, in which the scan was undertaken by a radiographer). Scoring of the degree of hepatic steatosis for all scans was undertaken independently by both radiologists, by visual assessment of the degree of echogenicity. One of three grades, none= 0, mild to moderate= 1, or severe fatty liver= 2, was assigned. The criteria used for assessment of the liver scans were: (1) comparison of reflectivity with the right kidney; (2) visualisation of the portal tracts; and (3) attenuation of beam. The measure of agreement of scoring between the two radiologists was analysed and a kappa statistic is presented.
Subjects had MRI of the abdominal region. It is uncertain whether single-slice abdominal MRI can be used as a proxy measure of visceral fat. Ross described good correlation between a five-slice technique and a single image at L4–L5 [23], whereas others have not demonstrated good correlation between a single image and multiple images [24]. Although the single-slice technique has been used frequently [25–28], our intention was to obtain images throughout the abdomen. Therefore, we acquired images from five non-contiguous slices extending from 5 cm below to 15 cm above L4–L5, to obtain more detailed information about visceral fat than is available from a single slice. Axial scans were taken with subjects in the supine position (with the exception of one very obese subject in whom better images were obtained by scanning prone). Two subjects could not be scanned because of claustrophobia. Subjects were scanned on an MR scanner (1.5 T Symphony, software release 4VA15A; Siemens, Munich, Germany). A gradient echo 2D FLASH (fast low-angle shot) sequence (TR=111 ms, TE=4.18 ms, flip angle= 70°, slice width=10 mm, slice spacing=50 mm) was used to obtain T 1-weighted images. In order to accommodate the circumference of the individual being scanned within the image, the field of view was varied.
The MRIs were analysed using a proprietary software package (Mimics; Materialise, Leuven, Belgium) to identify regions of subcutaneous and visceral fat within the cross-sectional abdominal MRIs. By examining the histogram of pixel values in each image, a threshold level was set for fat pixels. Three different masks were created, one comprising the whole cross-section of the body, one containing the visceral fat region and one containing the subcutaneous fat region. It was possible to determine the number of pixels contained within each of these masks, and hence calculate the areas of subcutaneous fat and visceral fat, and compare them with the total cross-sectional area. Adipose tissue volume was converted to mass in kg using a density of 0.92 for adipose tissue [23].
Insulin sensitivity was measured using a hyperinsulinaemic–euglycaemic clamp technique with deuterated glucose infusion. Hepatic insulin sensitivity was determined by insulin-mediated suppression of endogenous glucose production [20, 29]. Subjects attended the Clinical Research Facility at 08:00 hours, having abstained from vigorous exercise for 48 h and having fasted from 22:00 hours the night before. Deuterium-labelled glucose was used as a steady-state infusion for 150 min prior to commencing the insulin infusion and was continued throughout the clamp. The insulin infusion was commenced at 1.5 mU kg−1 min−1 and blood was taken at 5-min intervals. A 20% glucose infusion was commenced at a variable rate, adjusted to keep whole-blood glucose at 4.5–5 mmol/l. The clamp continued for 3 h, with blood samples taken at half-hour intervals until the final 30 min, when samples were taken at intervals of 5–10 min for determination of whole-body glucose disposal. A modified version of the equations formulated by Steele [29] was used to calculate total rate of appearance (R a) of glucose, endogenous glucose production and rate of disappearance (R d) of glucose. The tracer to tracee ratio (TTR) was calculated as the ratio of the two areas from the mass spectrometry data. TTR and plasma glucose values were smoothed using the method of optical segments [30]. For the Steele equations, 65% was used as the effective fraction and 0.22 l/kg as the distribution volume of glucose to calculate R a and R d [29, 31]. Endogenous glucose production was measured as insulin-mediated suppression of glucose output in the early part of the clamp and was expressed as percentage suppression 60 min after commencing the insulin infusion, when suppression was linear to this time point.
NEFA were measured fasting at 0, 10, 20, 30, 40, 60, 90 and 120 min during the OGTT to allow measurement of insulin-mediated suppression of NEFA concentration [17–19].
NEFA concentrations were measured in heparinised plasma samples with an autoanalyser (Konelab 20; Thermo Electron, Waltham, MA, USA) using reagents from Wako Chemicals (Richmond, VA, USA). Total cholesterol, HDL cholesterol and triglycerides were measured in fasting serum using in vitro commercial kit methods according to the manufacturer's instructions (Bayer). Adiponectin, TNF-α, IL1-β and IL-6 concentrations were analysed using ELISA kits (R and D Systems Europe). The mean coefficient of variation for the ELISAs was <3% in our hands. The areas under the NEFA curve from 0 to 30 min and from 30 to 120 min were determined and percentage NEFA suppression from 0 to 60 min was calculated. This index of suppression was chosen because most suppression has occurred by 60 min in subjects without fatty liver (as the comparator group). The gradient of NEFA suppression was markedly different in subjects without fatty liver (and almost flat) from 60 to 120 min in this group of subjects. The NEFA area was measured by summing the area of each trapezoid. Trapezoids were formed by generating a straight line between two time-point NEFA measurements and dropping perpendiculars from these time points to the x axis. Insulin sensitivity indices for glycaemia and NEFA were estimated using data from fasting, 1- and 2-h samples for insulin, glucose and NEFA concentrations, according to the method proposed for calculating insulin sensitivity from data obtained during an OGTT [32].
Cardiorespiratory fitness was determined after subjects had avoided strenuous exercise for 24 h and fasted for 8 h. Subjects were asked to perform an incremental treadmill test (Woodmill treadmill) commencing at 1.3 m/s (3 mi/h) at a gradient of 0%; every 2 min, the gradient was increased by 2% or the speed was increased by 0.25 m/s (0.5 mi/h) (i.e. stage 1, 1.3 m/s, 0% gradient; stage 2, 1.3 m/s, 2% gradient; stage 3, 1.55 m/s, 2% gradient) [33]. Physical activity level was assessed by heart rate (HR) monitoring [34] and completion of the Baecke Questionnaire of habitual activity.
All statistical analyses were performed using SPSS for Windows version 11.5. Comparison of mean values of continuous variables from the three groups was undertaken using ANOVA. Where variables were not normally distributed, log transformation was undertaken to normalise the distribution. Pearson correlation coefficients are presented for univariate regression analyses of normally distributed data. Regression analysis was used to determine the measures that were independently associated with fatty liver. A p-value of <0.05 was considered to be statistically significant for all analyses. A kappa statistic was calculated for agreement of independent scoring of liver scans between the two radiologists.
Results
We studied 22 obese men (mean BMI 35.1 kg/m2). Seven subjects (32%) had no fatty liver, nine had mild to moderate fatty liver (41%) and six had severe fatty liver (27%) according to ultrasonography. Agreement between the two radiologists independently scoring the scans was 96% (kappa statistic=0.93 [SE 0.14], p<0.0001). There was disagreement between radiologists on the scoring of only one scan, radiologist 1 scoring this scan as mild fatty liver and radiologist 2 scoring it as moderate fatty liver. (Importantly, this discrepant scoring did not affect the classification of this scan, which was placed in the mild to moderate fatty liver group).
No statistically significant difference in reported alcohol consumption was ascertained between the three groups, and plasma adiponectin was highest among the group with no evidence of fatty liver (6.72, 3.67 and 4.11 ng/l, p=0.04) (Table 1).
There was a trend towards increasing total and regional body fat with the degree of fatty liver but this did not reach statistical significance (Table 2). The correlation between all measures used to determine percentage body fat was close (Pearson correlation coefficients ranged from 0.77 to 0.93; p<0.001). There were no marked differences in the amount of either abdominal visceral fat (p=0.63) or subcutaneous fat (p=0.63) as measured by five-slice MRI (Table 3) in subjects in each of the three groups. Because this finding was somewhat unexpected and since most other investigators have assessed visceral fat by single horizontal MRI scanning, we also examined correlations between each of the single slices and the composite measure obtained from all five horizontal slices. The R 2 values varied considerably, from 0.47 to 0.85, depending on which single slice was chosen. Given that the calculated value from the composite of five slices is most likely to reflect the quantity of total visceral fat, the relatively poor correlation coefficients obtained with a single slice can be interpreted to mean that the use of only a single slice may result in an imprecise value for total visceral fat.
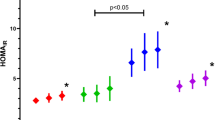
We found that markedly impaired whole-body glucose disposal was associated with fatty liver, as shown by comparing the groups with no fatty liver, mild to moderate fatty liver and severe fatty liver. Whole-body glucose disposal in the hyperinsulinaemic-euglycaemic clamp decreased with increasing severity of fatty liver (38.4, 26.5 and 23.9 μmol kg−1 min−1 for the groups with no fatty liver, mild to moderate fatty liver and severe fatty liver, respectively; p=0.004). Percentage suppression of NEFA concentration after 60 min during the OGTT (Fig. 1) showed a similar pattern for the three groups (suppression at 60 min, 62.5, 50.8 and 41.0%; p=0.03). In contrast, no statistically significant difference was demonstrated in endogenous glucose production between subjects with no fatty liver, mild to moderate fatty liver and severe fatty liver (percentage suppression of R a at 60 min, 64.9, 60.2 and 56.6%, respectively; p=0.78) (Table 4).

NEFA concentrations during the OGTT. Key: diamonds, no fatty liver; squares, mild-to-moderate fatty liver; triangles, severe fatty liver. The three groups were compared using ANOVA. *p<0.05 for comparison between the three groups
NEFA concentrations were increased with fatty liver (for groups with no, mild to moderate, and severe fatty liver respectively: AUC 0–30 min, 224.6, 235.8 and 322.0 h μmol−1 l−1, p=0.008; AUC 30–120 min, 271.6, 357.3 and 511 h μmol−1 l−1; p=0.02) (Fig. 1). Fasting NEFA concentrations were also increased with fatty liver (p=0.04) (Table 1). Since fasting NEFA concentrations and overnight lipolysis are influenced specifically by factors such as cortisol and cytokines (e.g. TNF-α), we measured 9 a.m. cortisol concentrations, undertook a short synacthen test (data not shown) and measured TNF-α, IL-1-β and IL-6 concentrations. Table 1 shows no differences in these measurements for men in the different groups. We determined whether the association between fatty liver and increased NEFA area during the OGTT was independent of the fasting NEFA concentration, as fasting NEFA concentrations were increased in men with fatty liver. Multiple regression modelling was undertaken with fatty liver as the dependent variable. Independent variables were selected from univariate analyses that were statistically significantly different in the three groups and included in the multivariate models. In a regression model that contained whole-body glucose disposal, adiponectin, NEFA area and percentage body fat as independent variables, NEFA area under the curve after an OGTT was associated with fatty liver independent of whole-body glucose disposal (B coefficient 5.02, p=0.025).
Discussion
No previous studies of obesity and fatty liver have included measurements of body composition, visceral fat, insulin sensitivity and NEFA concentrations. Although it is well known that obesity is associated with increased NEFA concentrations, our data emphasise that there are markedly different NEFA concentrations in obese individuals with fatty liver compared with similarly obese individuals who do not have fatty liver. We also show markedly impaired glucose disposal during the hyperinsulinaemic clamp, predominantly reflecting impaired insulin sensitivity in skeletal muscle, in individuals with severe fatty liver, but surprisingly no parallel impairment in insulin-stimulated inhibition of endogenous glucose production (largely reflecting suppression of hepatic glucose production) was found. Our data show increased NEFA concentrations, decreased NEFA suppression and an altered NEFA/insulin sensitivity index in subjects with fatty liver, suggesting abnormality of regulation of lipolysis, re-esterification or uptake of NEFA. To address this issue, isotope labelling studies are needed. Since lipolysis is regulated not only by insulin but also by the hypothalamic pituitary adrenal axis and cortisol concentrations, we measured 9 a.m. cortisol concentrations and undertook a short synacthen test to examine the adrenal response to ACTH. These results showed no differences between subjects according to the presence or absence of fatty liver (data not shown).
We chose to undertake a hyperinsulinaemic–euglycaemic clamp to assess insulin resistance in skeletal muscle in subjects in the three groups. Because it was not feasible to also undertake a low-dose insulin clamp to assess insulin-stimulated suppression of endogenous glucose production, we assessed suppression of endogenous deuterated glucose during the first 60 min of the insulin infusion, when suppression of glucose production was linear, during this relatively high-dose insulin clamp. As this method is not validated against the measurement of low-dose insulin-induced suppression of endogenous glucose production, our hepatic insulin sensitivity findings should be treated with caution. However, we reason from the data that there was no marked difference in hepatic insulin sensitivity across the three groups.
We show marked differences in NEFA concentrations with fatty liver in our obese subjects, and these data suggest two possible defects affecting the regulation of NEFA concentrations that relate to (1) the regulation of overnight NEFA concentrations and (2) insulin-induced suppression of NEFA concentrations, which are relevant to fatty liver. The effect of increased NEFA concentrations on the pathogenesis of fatty liver in humans in vivo is uncertain, but increasing evidence now suggests that increased NEFA supply to the liver may increase hepatic fat accumulation and link hepatic fat accumulation to hepatic inflammation. NEFA are a potent stimulus for hepatic triglyceride synthesis and hepatic fatty acid metabolism is closely linked to inflammation and hepatic steatosis. It has recently been shown that mice overexpressing suppressors of cytokine signalling (SOCS)-1 and SOCS-3 in liver have increased hepatic fatty acid synthesis. Interestingly, inhibition of these signalling proteins normalised fatty acid synthesis and dramatically ameliorated hepatic steatosis [35]. Taken together with our clinical data, these results emphasise the importance of fatty acids in the pathogenesis of fatty liver, regardless of whether the source of fatty acids is exogenous, such as adipose, or from endogenous hepatic production.
We studied obese men without fatty liver and with mild to moderate fatty liver and severe fatty liver, and surprisingly found minimal differences in the amount of visceral fat. Because fatness is often assessed imprecisely, we measured body adiposity by different methods in order to characterise body composition. We demonstrated that there was no significant difference in total fat between the groups and, more importantly, little difference in the amount of visceral fat between groups. We deliberately chose to use a five-slice MRI method, as described by Ross [23], to allow detailed evaluation of visceral fat. We were concerned that an image taken using a single-slice method, as used in many studies [25–27, 36], was inadequate to evaluate the precise amount of visceral fat. In support of our approach, it has previously been shown that an individual's visceral fat content cannot be reliably estimated from a single-slice MRI when compared with contiguous, whole body multislice MRI as the gold standard [24].
In obese subjects the evidence suggests that liver ultrasonography is a useful tool to detect liver fat where magnetic resonance spectroscopy and/or biopsy are not available or not feasible. In obese subjects, liver ultrasound has 88% sensitivity and 94% specificity in detecting fatty liver compared with histology [37], and all of our subjects were obese. Moreover, the kappa statistic shows excellent agreement between the two radiologists who independently scored the ultrasound scans. Our two radiologists disagreed on one scan and for that scan one radiologist scored the degree of fat as mild and the other scored it as moderate. This discrepant result did not affect the placing of this scan in the mild to moderate group of liver fat. Thus, in this group of obese patients, with appropriately experienced radiologists, ultrasound was a useful, reproducible technique for the semiquantitative assessment of liver fat, supporting published results. Our finding that obese subjects with no fatty liver, mild to moderate fatty liver and severe fatty liver have similar amounts of visceral fat suggests that the quantity of visceral fat is not particularly important for fatty liver. Although some studies have reported an increased quantity of visceral fat in association with fatty liver [8, 38, 39], it has been argued convincingly that a quantitative increase in visceral fat is unproven, even in association with the metabolic syndrome [40]. Others have queried the link between the amount of visceral fat and fatty liver, showing no increase in visceral fat in association with fatty liver [41, 42].
Data obtained from the hormone sensitive lipase knock out mouse indirectly support a causal role for NEFA in the pathogenesis of fatty liver [43]. This mouse has decreased NEFA concentrations and also decreased liver fat. The glitazone group of drugs that act primarily as peroxisome proliferator-activated receptor γ ligands, and also decrease NEFA concentrations, may also be effective in the treatment of fatty liver [44]. Thus recently, 18 subjects with biopsy-proven NASH were treated with pioglitazone (30 mg daily) for 48 weeks [44]. NEFA concentrations improved and hepatic fat content and size decreased. Histological features of steatosis, cellular injury, parenchymal inflammation, Mallory bodies and fibrosis were significantly improved from baseline. The results of this study lend further support to the pivotal role of insulin resistance combined with increased concentrations of NEFA in the pathogenesis of hepatic steatosis. Our results support the notion that there is a qualitative defect in adipose tissue function that is important in the pathogenesis of fatty liver. Further support for this notion is provided by the observation that patients with lipodystrophy, in whom adipocyte function is perturbed, often have fatty liver. Disturbed adipocyte function might result in NASH through a contribution to the development of fatty liver. Hepatic steatosis has been described as the first hit in a two-hit model for the progression of NAFLD, the second hit being a proinflammatory phenotype induced by cytokines and reactive oxygen species [45]. Fatty liver may develop as a result of either increased delivery of NEFA from insulin-resistant adipocytes or from increased hepatic fatty acid synthesis. Increased fatty acid synthesis may occur in insulin-resistant states, in which plasma insulin concentrations are high and dietary substrate is provided in excess. Thus, in postabsorptive states, when excess dietary glucose cannot be converted to glycogen or completely oxidised, glucose provides a substrate for fatty acid synthesis. Fatty acids may lead indirectly to hepatic lipotoxicity by promoting hepatic inflammation via stimulating TNF-α, or may directly promote hepatic toxicity via an effect on fat accumulation [46].
We have shown that increased NEFA concentrations were associated with fatty liver, independently of a classical measure of insulin sensitivity that predominantly measures insulin-stimulated glucose uptake in muscle (R d). However, insulin-stimulated glucose disposal was also markedly reduced in our obese men with severe fatty liver. It has been demonstrated previously that increased insulin resistance in muscle occurs in obese diabetic individuals with fatty liver compared with those without fatty liver [8], but in that study subjects with fatty liver were significantly more obese than those without fatty liver, and obesity in itself is associated with insulin resistance in muscle. Because our study compared obese healthy subjects with similar levels of total body adiposity and quantity of visceral fat, any confounding effects of the degree of total fat or the contribution of visceral fat are minimised. The DEXA measurements in our study showed that a trend towards increased truncal fat in subjects with fatty liver (Table 3). Increasing evidence suggests that truncal fat, in particular upper body fat, may contribute an important proportion of plasma fatty acids compared with visceral fat [47]. An increase in NEFA concentrations in subjects with truncal fat and fatty liver may then impair insulin-stimulated whole-body glucose disposal via a direct effect of NEFA on skeletal muscle glucose uptake [48].
In summary, our study has demonstrated for the first time that, in obese individuals with fatty liver, NEFA concentrations are markedly increased compared with similarly obese men without fatty liver. Our results were obtained in a small study and need to be reproduced in a larger study. We suggest more research is needed to elucidate the factors regulating NEFA concentrations in people with fatty liver, as this information may also provide key insights for the development of better treatments for fatty liver and NASH.
Abbreviations
- DEXA:
-
Dual-energy X-ray absorptiometry
- MRI:
-
Magnetic resonance imaging image
- NASH:
-
Non-alcoholic steatohepatitis
- NAFLD:
-
Non-alcoholic fatty liver disease
- Ra:
-
Rate of appearance
- Rd:
-
Rate of disappearance
- TTR:
-
Tracer to tracee ratio
References
Gupte P, Amarapurkar D, Agal S et al. (2004) Non-alcoholic steatohepatitis in type 2 diabetes mellitus. J Gastroenterol Hepatol 19:854–858
Cortez-Pinto H, Camilo ME, Baptista A, De Oliveira AG, De Moura MC (1999) Non-alcoholic fatty liver: another feature of the metabolic syndrome? Clin Nutr 18:353–358
Angulo P, Keach JC, Batts KP, Lindor KD (1999) Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 30:1356–1362
Sheth SG, Gordon FD, Chopra S (1997) Nonalcoholic steatohepatitis. Ann Intern Med 126:137–145
Clark JM, Diehl AM (2003) Nonalcoholic fatty liver disease: an underrecognized cause of cryptogenic cirrhosis. JAMA 289:3000–3004
Marceau P, Biron S, Hould FS et al. (1999) Liver pathology and the metabolic syndrome X in severe obesity. J Clin Endocrinol Metab 84:1513–1517
Marchesini G, Brizi M, Bianchi G et al. (2001) Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 50:1844–1850
Kelley DE, McKolanis TM, Hegazi RA, Kuller LH, Kalhan SC (2003) Fatty liver in type 2 diabetes mellitus: relation to regional adiposity, fatty acids, and insulin resistance. Am J Physiol Endocrinol Metab 285:E906–E916
Westerbacka J, Yki-Jarvinen H, Vehkavaara S et al. (2003) Body fat distribution and cortisol metabolism in healthy men: enhanced 5beta-reductase and lower cortisol/cortisone metabolite ratios in men with fatty liver. J Clin Endocrinol Metab 88:4924–4931
Johanson EH, Jansson PA, Lonn L et al. (2003) Fat distribution, lipid accumulation in the liver, and exercise capacity do not explain the insulin resistance in healthy males with a family history for type 2 diabetes. J Clin Endocrinol Metab 88:4232–4238
Seppala-Lindroos A, Vehkavaara S, Hakkinen AM et al. (2002) Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab 87:3023–3028
Boden G, Shulman GI (2002) Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest 32 (Suppl 3):14–23
Boden G (1999) Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc Assoc Am Phys 111:241–248
Boden G (2002) Interaction between free fatty acids and glucose metabolism. Curr Opin Clin Nutr Metab Care 5:545–549
Saverymuttu SH, Joseph AE, Maxwell JD (1986) Ultrasound scanning in the detection of hepatic fibrosis and steatosis. Br Med J (Clin Res Ed) 292:13–15
Mottin CC, Moretto M, Padoin AV et al. (2004) The role of ultrasound in the diagnosis of hepatic steatosis in morbidly obese patients. Obes Surg 14:635–637
Maison P, Byrne CD, Hales CN, Wareham NJ (2000) Hypertension and its treatment influence changes in fasting nonesterified fatty acid concentrations: a link between the sympathetic nervous system and the metabolic syndrome? Metabolism 49:81–87
Byrne CD, Wareham NJ, Day NE, McLeish R, Williams DR, Hales CN (1995) Decreased non-esterified fatty acid suppression and features of the insulin resistance syndrome occur in a sub-group of individuals with normal glucose tolerance. Diabetologia 38:1358–1366
Zhang J, Holt RI, Wild SH, Poole RB, Holt H, Byrne CD (2005) Plasma adiponectin concentrations are independently predicted by fat insulin sensitivity in women and by muscle insulin sensitivity in men. Diabetes Care 28:755–756
DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237:E214–E223
Staehr P, Hother-Nielsen O, Levin K, Holst JJ, Beck-Nielsen H (2001) Assessment of hepatic insulin action in obese type 2 diabetic patients. Diabetes 50:1363–1370
Durnin JV, Womersley J (1974) Body fat assessed from total body density and its estimation from skinfold thickness: measurements on 481 men and women aged from 16 to 72 years. Br J Nutr 32:77–97
Ross R, Aru J, Freeman J, Hudson R, Janssen I (2002) Abdominal adiposity and insulin resistance in obese men. Am J Physiol Endocrinol Metab 282:E657–E663
Thomas EL, Saeed N, Hajnal JV et al. (1998) Magnetic resonance imaging of total body fat. J Appl Physiol 85:1778–1785
Basu A, Basu R, Shah P, Vella A, Rizza RA, Jensen MD (2001) Systemic and regional free fatty acid metabolism in type 2 diabetes. Am J Physiol Endocrinol Metab 280:E1000–E1006
Bacha F, Saad R, Gungor N, Janosky J, Arslanian SA (2003) Obesity, regional fat distribution, and syndrome X in obese black versus white adolescents: race differential in diabetogenic and atherogenic risk factors. J Clin Endocrinol Metab 88:2534–2540
Ribeiro-Filho FF, Faria AN, Kohlmann NE, Zanella MT, Ferreira SR (2003) Two-hour insulin determination improves the ability of abdominal fat measurement to identify risk for the metabolic syndrome. Diabetes Care 26:1725–1730
Gautier JF, Mourier A, de Kerviler E et al. (1998) Evaluation of abdominal fat distribution in noninsulin-dependent diabetes mellitus: relationship to insulin resistance. J Clin Endocrinol Metab 83:1306–1311
Powrie JK, Smith GD, Hennessy TR et al. (1992) Incomplete suppression of hepatic glucose production in non-insulin dependent diabetes mellitus measured with [6,6-2H2]glucose enriched glucose infusion during hyperinsulinaemic euglycaemic clamps. Eur J Clin Invest 22:244–253
Finegood DT, Bergman RN (1983) Optimal segments: a method for smoothing tracer data to calculate metabolic fluxes. Am J Physiol 244:E472–E479
Finegood DT, Bergman RN, Vranic M (1988) Modeling error and apparent isotope discrimination confound estimation of endogenous glucose production during euglycemic glucose clamps. Diabetes 37:1025–1034
Belfiore F, Iannello S, Volpicelli G (1998) Insulin sensitivity indices calculated from basal and OGTT-induced insulin, glucose, and FFA levels. Mol Genet Metab 63:134–141
Irwin ML, Yasui Y, Ulrich CM et al. (2003) Effect of exercise on total and intra-abdominal body fat in postmenopausal women: a randomized controlled trial. JAMA 289:323–330
Wareham NJ, Hennings SJ, Byrne CD, Hales CN, Prentice AM, Day NE (1998) A quantitative analysis of the relationship between habitual energy expenditure, fitness and the metabolic cardiovascular syndrome. Br J Nutr 80:235–241
Ueki K, Kondo T, Tseng YH, Kahn CR (2004) Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci USA 101:10422–10427
Gautier JF, Mourier A, de Kerviler E, et al. (1998) Evaluation of abdominal fat distribution in noninsulin-dependent diabetes mellitus: relationship to insulin resistance. J Clin Endocrinol Metab 83:1306–1311
Saverymuttu SH, Joseph AE, Maxwell JD (1986) Ultrasound scanning in the detection of hepatic fibrosis and steatosis. Br Med J (Clin Res Ed) 292:13–15
Westerbacka J, Corner A, Tiikkainen M et al. (2004) Women and men have similar amounts of liver and intra-abdominal fat, despite more subcutaneous fat in women: implications for sex differences in markers of cardiovascular risk. Diabetologia 47:1360–1369
Chalasani N, Deeg MA, Persohn S, Crabb DW (2003) Metabolic and anthropometric evaluation of insulin resistance in nondiabetic patients with nonalcoholic steatohepatitis. Am J Gastroenterol 98:1849–1855
Frayn KN (2000) Visceral fat and insulin resistance—causative or correlative? Br J Nutr 83 (Suppl 1):S71–S77
Ryysy L, Hakkinen AM, Goto T, Vehkavaara S, Westerbacka J, Halavaara J, Yki-Jarvinen H (2000) Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes 49:749–758
Tiikkainen M, Tamminen M, Hakkinen AM et al. (2002) Liver-fat accumulation and insulin resistance in obese women with previous gestational diabetes. Obes Res 10:859–867
Voshol PJ, Haemmerle G, Ouwens DM et al. (2003) Increased hepatic insulin sensitivity together with decreased hepatic triglyceride stores in hormone-sensitive lipase-deficient mice. Endocrinology 144:3456–3462
Promrat K, Lutchman G, Uwaifo GI et al. (2004) A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 39:188–196
Diehl AM (1999) Nonalcoholic steatohepatitis. Semin Liver Dis 19:221–229
Byrne CD, Wang TW, Hales CN (1992) Control of Hep G2-cell triacylglycerol and apolipoprotein B synthesis and secretion by polyunsaturated non-esterified fatty acids and insulin. Biochem J 288:101–107
Horowitz JF, Klein S (2000) Whole body and abdominal lipolytic sensitivity to epinephrine is suppressed in upper body obese women. Am J Physiol Endocrinol Metab 278:E1144–E1152
Boden G, Shulman GI (2002) Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest 32 (Suppl 3):14–23
Acknowledgements
We would like to thank the staff of the Wellcome Trust Clinical Research Facility for their help with this study. We would like to thank C. Wang for her help with the assays, L. England for her help with this manuscript and N. Englyst and A. Ward for their advice. We are grateful to M. Hanna for performing the ultrasonography and scoring the liver scans. The work was funded by grant support from the Wellcome Trust to C. Byrne, from Wessex Deanery, and from NHS Research and Development funding.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Holt, H.B., Wild, S.H., Wood, P.J. et al. Non-esterified fatty acid concentrations are independently associated with hepatic steatosis in obese subjects. Diabetologia 49, 141–148 (2006). https://doi.org/10.1007/s00125-005-0070-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-0070-x