
Abstract
Key message
A Citrullus amarus mapping population segregating for resistance to Fusarium oxysporum f. sp. niveum race 2 and Papaya ringspot virus was used to identify novel QTL, important for the improvement in watermelon disease resistance.
Abstract
Multiple disease screens of the USDA Citrullus spp. germplasm collection have highlighted the value of Citrullus amarus (citron melon or wild watermelon) as a resource for enhancing modern watermelon cultivars (Citrullus lanatus) with resistance to a broad range of fungal, bacterial and viral diseases of watermelon. We have generated a genetic population of C. amarus segregating for resistance to two important watermelon diseases: Fusarium wilt (caused by the fungus Fusarium oxysporum f. sp. niveum; Fon race 2) and Papaya ringspot virus-watermelon strain (PRSV-W). QTL mapping of Fon race 2 resistance identified seven significant QTLs, with the major QTL representing a novel genetic source of resistance and an opportunity for gene pyramiding. A single QTL was associated with resistance to PRSV-W, which adhered to expectations of a prior study indicating a single-gene recessive inheritance in watermelon. The resistance loci identified here provide valuable genetic resources for introgression into cultivated watermelon for the improvement in disease resistance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Modern watermelon cultivars share a narrow genetic base and exhibit a general lack of resistance to diseases and pests resulting from many years of cultivation and selection for desirable fruit qualities (Levi et al. 2001). Due to the loss of methyl bromide as a soil fumigant in the USA, soilborne pathogens are becoming a more severe problem for watermelon growers. The watermelon crop routinely suffers significant losses due to the soilborne fungal disease Fusarium wilt [caused by Fusarium oxysporum f. sp. niveum (Fon)]. Fusarium wilt is considered the most serious soilborne disease worldwide in watermelon (Wechter et al. 2012; Branham et al. 2017, 2018). It is extremely difficult to entirely eradicate the fungus from a field once established, as the chlamydospores can survive for many years in a field even without the presence of a susceptible host (Martyn 1987). Fon infects the roots of the host and then travels through the xylem vessels while exuding compounds that reduce or prevent the flow of water and nutrients, resulting in wilting of the plant (Zhang et al. 2015).
There are four known pathogenic races of Fon identified as 0, 1, 2 and 3 (Zhou et al. 2010). Several watermelon heirlooms and commercial cultivars possess resistance to Fon races 0 and 1, but not to races 2 or 3. Fon race 2 has been reported in numerous growing regions of the USA (Keinath and DuBose 2009), as well as in watermelon production areas throughout the world. A screen of the US Department of Agriculture (USDA) collection of C. amarus plant introductions (PIs) identified multiple accessions with moderate resistance to Fon race 2 (Wechter et al. 2012). Two of those PIs were selected and self-pollinated for several generations to develop two Fon race 2-resistant C. amarus lines, USVL246-FR2 and USVL252-FR2 (Wechter et al. 2016). Quantitative trait loci (QTL) mapping of the resistance in USVL246-FR2 indicated a complex genetic basis, including polygenic inheritance and moderate heritability (Branham et al. 2017). The genetic architecture of Fon race 2 resistance in USVL252-FR2 has not yet been identified.
In addition to soilborne diseases, potyviruses are a major limiting factor to watermelon production in many regions in the USA (Ali et al. 2012). In recent years, the number of aphid or whitefly-transmitted potyviruses has increased to near epidemic levels, causing significant losses to the watermelon crop across the USA (Ali et al. 2012). The aphid-transmitted virus, Papaya ringspot virus (PRSV-W, formerly Watermelon mosaic virus 1), is among the most damaging potyviruses of watermelon (Gonsalves et al. 2010). PRSV-W is efficiently transmitted by numerous aphids in a nonpersistent manner. The virus particles localize mainly in the feeding mouthparts of the aphid and do not circulate within its viruliferous digestive system. Thus, it is transmitted rapidly and easily among host plants. PRSV-W causes serious plant stunting, along with green mosaic or mottled patterns and malformations of leaves, leaf wrinkling, blisters, distortion and narrow leaf blades. The watermelon fruits from PRSV-infected plants may be malformed, with distinct ringspot patterns and variation in internal flesh texture (Gonsalves et al. 2010). PRSV-W has frequently been identified in watermelon fields in the southern United States (Ali et al. 2012).
Constant pressure from new and emerging plant pathogens has created a critical need to generate watermelon cultivars with increased resistance to diseases, as well as identifying new or additional genetic sources of resistance to the major diseases. In addition, there is a need to conduct genetic studies to identify gene loci associated with resistance, develop molecular markers useful in breeding programs and incorporate the resistance into the genetic background of elite watermelon cultivars.
Watermelon belongs to the genus Citrullus, a xerophytic group that thrives primarily in dry areas of the African tropics (Wehner 2008). The genus Citrullus comprises several known diploid (n = 11) species, including Citrullus amarus (previously known as C. lanatus var. citroides) (Chomicki and Renner 2015). C. amarus exists in diverse regions throughout southern Africa and is considered a valuable resource for enhancing modern watermelon cultivars with resistance to a broad range of fungal diseases such as Fusarium wilt and potyviruses of watermelon (Ling et al. 2009; Wechter et al. 2012; Levi et al. 2013).
The C. amarus PI 248252 and PI 244019 exhibit high levels of resistance to Fon race 2 and PRSV-W, respectively (Strange et al. 2002; Wechter et al. 2012; Guner et al. 2018), and were used for the development of the resistant inbred lines USVL252-FR2 (Wechter et al. 2016) and PI 244019PRSV−R. We developed genetic populations (F2 and F2:3) derived from a cross of USVL252-FR2 x PI 244019PRSV-R to identify QTLs associated with resistance. In the present study, we phenotyped these genetic populations for resistance to Fon race 2 and PRSV-W and genotyped them using genotyping-by-sequencing (GBS) technology (Elshire et al. 2011), which generated thousands of single-nucleotide polymorphisms (SNPs). We constructed a high-density genetic map for the F2 population and identified QTLs associated with resistance to these two major diseases of watermelon.
Materials and methods
Plant materials and growth conditions
Genetic populations (F2 and F2:3) that segregated for both Fon race 2 and PRSV-W resistance were generated by hand-pollination from a cross of USVL252-FR2 and PI 244019PRSV-R. USVL252-FR2 is a C. amarus breeding line, derived from PI 482252, that is tolerant to Fon race 2 (Wechter et al. 2016) and susceptible to PRSV-W (Strange et al. 2002). PI 244019PRSV-R, derived from C. amarus PI 244019, is resistant to PRSV-W (Strange et al. 2002; Guner et al. 2018) and susceptible to Fon race 2 (Wechter et al. 2012).
Fungal inoculum preparation
Fon race 2 culture preparation followed the optimized procedures described in Wechter et al. (2012) using isolate B05-30 (kindly provided by Anthony Keinath, Clemson University). Race designation was validated by inoculating and evaluating the standard differentials for Fon, which included ‘Sugar Baby,’ ‘AllSweet’ and PI 296341-FR. Single-spore isolations were grown on Difco™ Potato Dextrose Agar (Becton, Dickinson & Co. Sparks, MD) for two weeks at 25 °C under a 12-h diurnal lighting cycle of fluorescent light. Five sections of 1 cm2 were cut from the agar and added to 250 mL of Difco™ Potato Dextrose Broth (Becton, Dickinson & Co., Sparks, MD). The suspension was agitated for 14 days with a rotary shaker at 200 rpm at 25 °C and then filtered through two layers of cheese cloth. Conidial concentration (both microconidia and macroconidia) was assessed with a hemocytometer under a microscope and adjusted to 1 × 106 conidia per mL with sterile tap water. An electric cement mixer was used to saturate and homogenize a 1:1:1 tri-soil mix of perlite–vermiculite–Metromix 360 potting soil (Sun Gro Horticulture, Agawam, MA) with the spore suspension (~ 1 L of spore suspension to 1 kg soil mix). F3 seeds from each F2:3 family were planted into the saturated conidial tri-mix in 50-cell propagation trays (Hummert International, Earth City, MO).
Virus culture and inoculation
The PRSV-W inoculum (kindly provided by Sakata Seeds, Fort Myers, FL) was maintained on the squash cultivar ‘Gray Zucchini.’ Inoculum was prepared by macerating virus-infected leaf tissue (1:5 w/v) with a mortar and pestle in 0.01 M phosphate-buffered saline (pH 7.0). After lightly dusting the seedlings (1-2 true leaf stage) with carborundum (320 mesh grit powder, Fisher Scientific, Hampton, NH), the virus inoculum was gently rubbed on leaves with a cotton swab, as described by Ling et al. (2009). Following inoculation, the seedlings were placed under shade in a greenhouse at the US Vegetable Laboratory (USDA, Charleston, SC) for a few hours to minimize direct sunlight damage to the virus and newly inoculated leaves. The greenhouse temperature was 26 °C and 18 °C during the day (14 h) and night (10 h), respectively. The second inoculation was performed 7 days after the first inoculation to prevent any potential escape from the inoculation (Ling et al. 2009). Plants were monitored daily for the presence of viral symptoms, and the final disease severity evaluation was performed 30 days after the first inoculation.
Experimental design and disease evaluations
Fon race 2
Population type and experimental design were chosen based upon the complexity of the genetic architecture of each trait. Inheritance of Fon race 2 resistance is complex and polygenic with moderate heritability (Ren et al. 2015; Branham et al. 2017). Therefore, we evaluated 205 F2:3 families and their parents for Fon race 2 resistance using a randomized complete block design with two independent greenhouse studies of two replicates of 10 plants each (i.e., a total of 40 F3 plants per family). Seedlings were maintained as described in Branham et al. (2017). The first test was planted on June 20, 2017, and the second on August 1, 2017. Disease response of each F2:3 family was evaluated at 28 days post-inoculation (dpi) as the percentage of F3 plants that were completely healthy (i.e., no chlorosis or wilting). Variance components were estimated with ASReml-R version 3.0 (Gilmour et al. 2009) to fit a linear mixed model using a restricted maximum likelihood algorithm. All factors within the model were fit as random effects, which included family, test, the interaction between family and test, replicate and tray. Correlation of disease response between the two tests was assessed with Pearson’s correlation coefficient (r).
PRSV-w
Inheritance studies of PRSV-W resistance in watermelon have indicated single-gene-mediated resistance (Guner et al. 2018). Thus, 151 F2 individuals and 12 individuals from each parental line were screened for resistance to PRSV-W in a greenhouse trial. Only five plants of PI 244019PRSV-R were evaluated for disease response due to low germination rate in this trial. In addition, five plants of the cultivars ‘Charleston Gray’ and ‘Sugar Baby’ were used as susceptible controls. Two plants of each of these cultivars were mock-inoculated (dusted with carborundum and gently rubbed) and used as negative controls. Seeds were germinated in a commercial soil mix (MetroMix 360) in a greenhouse at the US Vegetable Laboratory (USDA) in Charleston, South Carolina, with 26 °C day temperature for 14 h under natural lighting and 18 °C night temperature for 10 h.
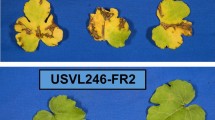
Disease severity (DS) of each plant was evaluated on a five-point rating scale as follows: 1—no symptoms, 2—slight leaf yellowing or very slight mosaic on inoculated leaves with normal appearance on new leaves, 3—deformed leaves with yellow mosaic, 4—stunted plant with severely deformed leaves with mosaic appearance and 5—stunted plant with extensive mosaic appearance and severe leaf deformation, mostly narrow leaves and reduced plant growth or plant death (Fig. 1). Fit of recessive inheritance of resistance to PRSV-W in this population was evaluated with a Chi-squared test with a DS of 1 considered resistant and > 1 susceptible.

Photographs of F2 individuals inoculated with Papaya ringspot virus-watermelon strain, depicting symptoms representative of resistant (disease severity [DS] of 1) and susceptible (DS = 3) responses
Population genotyping
Genomic DNA of 356 F2 plants, the F1 hybrid and parents was extracted from young leaf tissue and sent for genotyping at the Genomic Diversity Facility of Cornell University (Ithaca, NY, USA) using GBS technology. Genomic DNA samples (96-plex) were digested with ApeKI to construct GBS libraries, which were sequenced on an Illumina HiSeq 2000/2500 to generate 100-bp, single-end reads (Elshire et al. 2011). Reads were grouped into tags with the GBSv2 pipeline of TASSEL 5.2.30 (Glaubitz et al. 2014). Tags were aligned to the C. amarus reference genome of USVL246-FR2 (unpublished) using the Burrows–Wheeler aligner (BWA) version 0.7.12-5 (Li and Durbin 2009). SNPs were called with GBSv2 using the default parameters. Many of the most likely sequencing errors were removed by filtering with VCFtools version 0.1.15 (Danecek et al. 2011) to remove SNPs with more than 90% missing data or a minor allele frequency (MAF) < 0.01. Missing data were imputed with the FSFHap plug-in (Swarts et al. 2014) of TASSEL using the windowLD algorithm. Consensus parental genotypes (A or B), after removing ambiguous or heterozygous loci, were identified with the ABH plug-in of TASSEL from 10 sequencing samples of each parental line. Population genotypes (A, C, G or T) were then converted to the consensus parental genotypes (A or B).
Genetic map construction
All remaining analyses, including data quality control, genetic map construction and QTL mapping, were completed in the qtl package (Broman et al. 2003) of R (R core team 2018). SNPs with more than 20% missing data were removed. SNPs were binned with a single representative SNP chosen at random from each set with identical segregation patterns. Single SNPs deviating from the segregation pattern of the surrounding region (as determined by Chi-squared tests) were removed as they were likely residual sequencing errors. Linkage groups were formed using a maximum recombination frequency of 0.35 and a minimum logarithm of odds (LOD) score of 20. Markers were ordered by the minimum number of obligate crossovers with a greedy algorithm. The Lander–Green algorithm (Lander and Green 1987) was used to construct a multi-locus genetic linkage map with a maximum likelihood approach, and the Kosambi mapping function (Kosambi 1943) was used to convert recombination fractions to genetic distances.
QTL mapping of disease resistance
QTLs associated with each trait (PRSV-W disease severity rating of the F2 population and F2:3 family mean disease incidence for Fon race 2 resistance) were mapped with Haley–Knott regression (Haley and Knott 1992). The optimal QTL model with the highest penalized LOD score (Manichaikul et al. 2009) for each trait was chosen using an automated forward and backward selection algorithm, implemented with the stepwiseqtl function (Broman and Sen 2009; Broman and Speed 2002; Zeng et al. 1999) from the qtl package of R (Broman et al. 2003). One thousand permutations of a two QTL scan (scantwo function) were used to determine the model selection penalties and the genome-wide significance threshold (α = 0.05). The final QTL model for each trait was visualized with standard interval mapping (scanone function) followed by forward selection. The unpublished draft genome annotation of USVL246-FR2 was used to identify the location and predicted function of candidate genes from the 1.5-LOD intervals of significant QTLs. Comparison of homologs of candidate genes of other cucurbit species was explored using tools available at CuGenDB (Zheng et al. 2019).
Results
Population segregation of disease response
Fon race 2
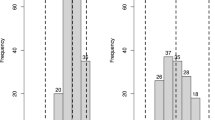
The Fon race 2-resistant parent, USVL252-FR2, had a high mean percentage of unaffected individuals (71.3%), while the susceptible parent, PI 244019PRSV-R, showed an intermediate level of resistance with a mean of 48.8% unaffected (Table 1). PI 244019PRSV-R was developed from a few rounds of selfing from the highly susceptible PI 244019 (6.3% survival in Wechter et al. 2012), so the intermediate disease response was unexpected. Despite the lower than expected difference between the parents, the F2:3 population displayed transgressive segregation with eight families more resistant than USVL252-FR2 and 134 families more susceptible than PI 244019PRSV-R (Online resource 1). Disease response followed a normal distribution with a mean of 43.6% and a range of 4.6–80.4% unaffected across the F2:3 families (Fig. 2a). Family and the interaction of family and test contributed 13.1 and 5.8 percent of the total phenotypic variation (%VP), respectively (Table 2). Almost a third of the variation in Fon race 2 resistance was explained by test, evident by lower mean percent of healthy plants for the parents, F1 and F2:3 families in test 2 (Table 1). Correlation of disease response between tests was highly significant (p = 4.3 × 10−7), but moderate in magnitude (r = 0.34).

Histograms of a mean percent unaffected of each F3 family in the Citrullus amarusF2:3 population after inoculation with Fusarium oxysporum f. sp. niveum race 2 and b disease severity of F2 individuals after inoculation with Papaya ringspot virus-watermelon strain. Means of the parents (USVL252-FR2 and PI 244019PRSV-R) are indicated by vertical dashed lines
PRSV-w
Population segregation of resistance to PRSV-W followed a single-gene recessive model of inheritance (p = 0.12), with a DS of 1 considered resistant and 2–4 considered susceptible (Fig. 2b). All F2 individuals survived; therefore, the maximum DS in the population was 4. Mean DS for the population was 2.8 with a standard deviation of 1.3. Plants of the PRSV-W-resistant parent (PI 244019PRSV-R) were asymptomatic with the exception of one plant (DS = 2), which had a mean DS of 1.2. All replicates of USVL252-FR2 had DS scores of 3 or 4, with an overall mean DS of 3.6.
Genotyping and map construction
GBS of ten samples of each parent and 356 F2 individuals produced over 1.8 billion good bar-coded reads. The reads grouped into 288,957 tags, with 79% mapped to the USVL246-FR2 reference genome. TASSEL pipeline identified 53,143 SNPs, which was reduced to 11,367 SNPs after removal of those with more than 90% missing data or a MAF < 0.01. FSFHap imputation reduced the average missing data across all loci from 31.7 to 17.5%. The ABH plug-in filtered the genotype set to 4,208 SNPs by removing loci where the parental genotypes were missing, heterozygous or ambiguous. SNPs with more than 20% missing data were dropped, and the remaining high-quality SNPs were grouped into 1874 bins, with SNPs in each bin having identical segregation patterns across the population. The binned SNPs formed eleven linkage groups with a total map distance of 1326.7 centiMorgans (cM). Each linkage group (named according to chromosome) had high coverage with 123–258 SNPs, and a mean and maximum distance between SNPs of 0.7 and 16.8 cM, respectively (Online resource 1).
QTL mapping of resistance
Fon race 2
The optimal QTL model of mean percent asymptomatic families across tests consisted of a single major QTL (qFon2-1; LOD score 12.8) explaining 18.9% of the variation in disease response with additive and dominance effects of 9.6 and − 1.3, respectively. An additional 23.8 percent of the variation in disease response was explained by three minor QTLs (qFon2-5, qFon2-6 and qFon2-8.2), ranging from 7.1 to 9.4%VP each (Table 3). The major QTL (qFon2-1) was consistently identified in each test, but association of the minor QTLs with disease response varied (Fig. 3). QTL mapping of each test independently found three additional minor QTLs (qFon2-2, qFon2-8.1 and qFon2-11) that were not associated with resistance to Fon race 2 across tests (Fig. 3; Table 3). As expected under transgressive segregation, resistance was inherited from both parents with resistance alleles for qFon2-1, qFon2-5 and qFon2-8.2 provided by USVL252-FR2 and qFon2-2, qFon2-6 and qFon2-11 by PI 244019PRSV-R (Table 3). Interaction of the alleles at qFon2-8.1 resulted in underdominance with the mean percent of unaffected individuals higher for families homozygous for either allele than for families heterozygous at this locus. The genome-wide significance thresholds for Fon race 2 resistance across tests, within test 1 and within test 2 were 3.7, 3.8 and 3.9, respectively. The 1.5-LOD intervals of the QTLs associated with Fon race 2 resistance were wide, extending from 6 to 28 cM (Table 3). These intervals corresponded to 1.6 to 15 Mb in the C. amarus genome (Table 3) and contained 159 to 640 potential candidate genes each (Online resource 2).

Logarithm of odds (LOD) scores for forward model selection of up to seven QTL associated with mean asymptomatic F3 individuals per F2:3 family: a across two greenhouse tests, b within test 1 and c within test 2, after inoculation with Fusarium oxysporum f. sp. niveum race 2. The initial scan shows the likelihood of the first QTL being located at each SNP in the genome (linkage group = chromosome) with subsequent scans showing the LOD of an additional QTL with the effects of the previous QTL(s) controlled for in the model. The dashed line marks the genome-wide significance threshold
PRSV-w
A single QTL (qPRSV-3) was significantly associated (LOD score > 3.9) with PRSV-W resistance in the F2 population with a LOD score of 9.1. qPRSV-3 explained 24.2%VP and had additive and dominance effects of 0.77 and − 0.97, respectively (Fig. 4; Table 3). PRSV-W resistance was recessively inherited with the resistance alleles contributed by PI 244019PRSV-R (the PRSV-W-resistant parent). The 1.5-LOD interval covered 8.5 cM which mapped to 2.2 Mb of the C. amarus genome, a region with 171 potential candidate genes, including the eukaryotic elongation factor eIF4E at the QTL peak (Online resource 2).

Logarithm of odds (LOD) scores for forward model selection of up to seven QTL associated with disease severity in the F2 population after inoculation with Papaya ringspot virus-watermelon strain. The initial scan shows the likelihood of the first QTL being located at each SNP in the genome (linkage group = chromosome) with subsequent scans showing the LOD of an additional QTL with the effects of the previous QTL(s) controlled for in the model. The dashed line marks the genome-wide significance threshold
Discussion
The distribution of Fon race 2 resistance varied widely across the F2:3 population with both phenotypic and genetic data confirming transgressive segregation. Sixty-five percent of the F2:3 families were more susceptible than either parent. Although epistasis was not detected, there could potentially be deleterious interactions between the alleles of multiple single loci causing both the higher than expected number of susceptible F2:3 families in the population and the higher susceptibility of the F1 as compared to either parent or the population mean. Environmental factors substantially impacted the variation in Fon race 2 resistance, noticeable in the lower mean percent of asymptomatic individuals of the parents and population in the second test and the higher association of QTL from the first test. Test 2 was inoculated in a greenhouse in Charleston, SC, in the hottest month of the year (August). High temperatures in greenhouse settings in Charleston have been noted to cause extremes in Fusarium wilt phenotyping, especially in terms of plant survival. Multiple QTL studies have detected a strong environmental component in disease response of Citrullus species to inoculation with Fon (Meru and McGregor 2016a, b; Branham et al. 2019). Resistance means of the F2:3 families were highly significantly correlated between the tests despite the large difference in disease incidence.
Segregation of PRSV-W resistance in the F2 population confirmed the action of a single recessive gene as reported in a previous inheritance study of PRSV-W resistance in three C. amarus PIs, including the progenitor of the resistant parent, PI 244019PRSV-R (Guner et al. 2018). The resistant and susceptible parental means for DS fell in the tails of the population distribution as expected.
GBS (Elshire et al. 2011) of the population provided sufficient marker density to saturate the genetic map as more than half the filtered markers had identical segregation patterns and were binned. The high-density genetic map constructed here is comparable to all previously reported intraspecific C. amarus genetic maps (Branham et al. 2017, 2019) with intermediate values in terms of numbers of SNPs, mean distance between SNPs and total map length.
Four QTL were associated with Fon race 2 resistance means across tests, and three additional QTL were associated with the means from a single test. Five of the seven unique QTL identified here collocate with QTL previously reported for Fon races 1 and/or 2. The 1.5-LOD interval of the major QTL associated with Fon race 2 resistance in this population, qFon2-1, overlaps the major QTL associated with Fon race 1 resistance in multiple germplasm sources of C. lanatus (Lambel et al. 2014; Branham et al. 2018) and in the C. amarus PI 296341 (Ren et al. 2015). Given the resolution of the current data, we are unable to determine whether the resistance to the two races provided by this locus is conveyed by different alleles of the same gene or different causal genes that are linked. QTL intervals for qFon2-2, qFon2-5 and qFon2-8.1 overlapped with those associated with Fon race 2 resistance in the C. amarus line USVL246-FR2 (Branham et al. 2017). qFon2-11 collocated with previously reported QTL in C. lanatus for Fon races 1 (Branham et al. 2018) and 2 (Meru and McGregor 2016b). Two of the QTL associated with Fon race 2 resistance in this population (qFon2-6 and qFon2-8.2) have not been reported previously and may represent novel resistance sources. No QTL studies of PRSV-W resistance have been reported in watermelon, but the identification of a single recessive QTL (qPRSV-3) confirms the findings of prior inheritance studies in C. amarus (Guner et al. 2018).
The 1.5-LOD intervals of the QTL associated with resistance to Fon race 2 and PRSV-W extended across large physical distances (i.e., megabases) of the genome, which correspond to regions covering hundreds of genes each. Future research will therefore focus on only the most relevant candidate genes based upon functional annotation. Nucleotide binding site–leucine-rich repeats (NBS-LRR) and receptor-like protein kinases (RLKs) are two of the main types of dominant resistance (R) genes and have both been shown to provide resistance to different formae speciales of F. oxysporum in multiple host species.
Two of the Fon race 2 resistance QTL collocated with NBS-LRR genes, with one in the interval of the major QTL qFon2-1. Resistance to F. oxysporum f. sp. conglutinans in Chinese cabbage was narrowed to a region of two tandem NBS-LRR genes through differential expression in an RNA-seq study and population segregation analysis (Shimizu et al. 2014). Positional cloning and transgenic complementation identified an NBS-LRR as the causal gene for resistance to F. oxysporum f. sp. lycopersici race 2 in tomato (Simons et al. 1998). Resistance to F. oxysporum f. sp. melonis (Fom) races 0 and 2, and races 0 and 1 in melon was shown through map-based cloning to be conferred by the NBS-LRR genes, Fom-1 and Fom-2, respectively (Joobeur et al. 2004; Brotman et al. 2013). The Fom-1 gene (conferring resistance to Fom race 2 in melon) is within a block of R genes on chromosome 9 (Brotman et al. 2013). A syntenic block of homologous R genes (> 10) was located in the 1.5-LOD interval of qFon2-8.2, including a homolog of Fom-1.
A loss-of-function mutation in an RLK gene in A. thaliana provided broad-spectrum resistance to multiple formae speciales of F. oxysporum, including f. sp. matthioli, f. sp. conglutinans and f. sp. raphanin in the mutant as compared to the wild type (Diener and Ausubel 2005). Cloning and transgenic complementation in A. thaliana and tomato validated RLKs as the causal genes conferring resistance to F. oxysporum f. sp. matthioli (Cole and Diener 2013) and F. oxysporum f. sp. lycopersici race 3 (Catanzariti et al. 2015), respectively. RLK genes were located in the 1.5-LOD intervals of all seven QTL associated with Fon race 2 resistance in this population.
Eukaryotic translation initiation factor 4E (eIF4E) has been shown to provide recessive resistance to potyviruses through map-based cloning followed by transgenic complementation in A. thaliana (Lellis et al. 2002), pepper (Ruffel et al. 2002) and lettuce (Nicaise et al. 2003). The function of eIF4E was disrupted in cucumber using gene editing with clustered regularly interspaced short palindromic repeats (CRISPR/Cas9) [Chandrasekaran et al. 2016]. Homozygous mutant plants were resistant to multiple viruses, including PRSV-W, while heterozygous mutants and non-mutants were highly susceptible. A non-synonymous SNP in the eIF4E gene in C. lanatus was associated with resistance to Zucchini yellow mosaic virus (Ling et al. 2009). The homologous eIF4E gene in C. amarus, CaU03G008250, is located at the QTL peak of qPRSV-3 and may be the recessive source of resistance to PRSV-W in PI 244019PRSV-R.
Using a biparental mapping population of C. amarus, we were able to identify QTL associated with resistance to two of the most devastating pathogens of watermelon, Fon race 2 and PRSV-W. The major QTL for Fon race 2 resistance identified on chromosome 1 provides a novel source of resistance that can be combined with the previously identified QTL on chromosome 9 through gene pyramiding to provide durable resistance for watermelon improvement. Mapping of the PRSV-W resistance QTL to chromosome 3 will allow the development of markers to speed up the process of incorporating this recessive resistance into an elite watermelon background.
References
Ali A, Abdalla O, Bruton B, Fish W, Sikora E, Zhang S, Taylor M (2012) Occurrence of viruses infecting watermelon, other cucurbits, and weeds in the parts of southern United States. Plant Health Progress 13:9. https://doi.org/10.1094/php-2012-0824-01-rs
Branham SE, Levi A, Farnham MW, Patrick Wechter W (2017) A GBS-SNP-based linkage map and quantitative trait loci (QTL) associated with resistance to Fusarium oxysporum f. sp. niveum race 2 identified in Citrullus lanatus var. citroides. Theor Appl Genet 130:319–330. https://doi.org/10.1007/s00122-016-2813-0
Branham SE, Wechter WP, Lambel S et al (2018) QTL-seq and marker development for resistance to Fusarium oxysporum f. sp. niveum race 1 in cultivated watermelon. Mol Breed 38:1–9. https://doi.org/10.1007/s11032-018-0896-9
Branham S, Levi A, Wechter P (2019) QTL mapping identifies novel source of resistance to Fusarium wilt race 1 in Citrullus amarus. Plant Dis. https://doi.org/10.1094/pdis-09-18-1677-re
Broman KW, Sen S (2009) A guide to QTL mapping with R/qtl, vol 46. Springer, New York
Broman KW, Speed T (2002) A model selection approach for the identification of quantitative trait loci in experimental crosses (with discussion). J R Stat Soc B 64(641–656):731–775
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890. https://doi.org/10.1093/bioinformatics/btg112
Brotman Y, Normantovich M, Goldenberg Z et al (2013) Dual resistance of melon to fusarium oxysporum races 0 and 2 and to papaya ring-spot virus is controlled by a pair of head-to-head-oriented nb-lrr genes of unusual architecture. Mol Plant 6:235–238. https://doi.org/10.1093/mp/sss121
Catanzariti A-M, Lim GTT, Jones DA (2015) The tomato I-3 gene: a novel gene for resistance to Fusarium wilt disease. New Phytol 207:106–118. https://doi.org/10.1111/nph.13348
Chandrasekaran J, Brumin M, Wolf D et al (2016) Development of broad virus resistance in non-transgenic cucumber using CRISPR/Cas9 technology. Mol Plant Pathol 17:1140–1153. https://doi.org/10.1111/mpp.12375
Chomicki G, Renner SS (2015) Watermelon origin solved with molecular phylogenetics including Linnaean material: another example of museomics. New Phytol 205:526–532. https://doi.org/10.1111/nph.13163
Cole SJ, Diener AC (2013) Diversity in receptor-like kinase genes is a major determinant of quantitative resistance to Fusarium oxysporum f. sp matthioli. New Phytol 200:172–184. https://doi.org/10.1111/nph.12368
Core Team R (2018) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Gilmour AR, Gogel BJ, Cullis BR, et al. (2009) ASReml user guide release 3.0. VSN International Ltd, Hemel Hemp-stead
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Diener AC, Ausubel FM (2005) Resistance to Fusarium oxysporum 1, a dominant Arabidopsis disease-resistance gene, is not race specific. Genetics 171:305–321. https://doi.org/10.1534/genetics.105.042218
Elshire RJ, Glaubitz JC, Sun Q et al (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. https://doi.org/10.1371/journal.pone.0019379
Glaubitz JC, Casstevens TM, Lu F et al (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9:e90346. https://doi.org/10.1371/journal.pone.0090346
Gonsalves D, Tripathi S, Carr JB, Suzuki JY (2010) Papaya ringspot virus. Plant Health Instructor. https://doi.org/10.1094/PHI-I-2010-1004-01
Guner N, Pesic-VanEsbroeck Z, Rivera-Burgos LA, Wehner TC (2018) Inheritance of resistance to Papaya ringspot virus-watermelon strain in watermelon. HortScience 53:624–627. https://doi.org/10.21273/hortsci12944-18
Haley CS, Knott SA (1992) A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity (Edinb) 69:315–324. https://doi.org/10.1038/hdy.1992.131
Joobeur T, King JJ, Nolin SJ et al (2004) The fusarium wilt resistance locus Fom-2 of melon contains a single resistance gene with complex features. Plant J 39:283–297. https://doi.org/10.1111/j.1365-313X.2004.02134.x
Keinath AP, DuBose V (2009) First report of Fusarium oxysporum f. sp niveum race 2 in South Carolina watermelon fields. Phytopathology 99(6):S63
Kosambi DD (1943) The estimation of map distances from recombination values. Ann Eugen 12:172–175. https://doi.org/10.1111/j.1469-1809.1943.tb02321.x
Lambel S, Lanini B, Vivoda E et al (2014) A major QTL associated with Fusarium oxysporum race 1 resistance identified in genetic populations derived from closely related watermelon lines using selective genotyping and genotyping-by-sequencing for SNP discovery. Theor Appl Genet 127:2105–2115. https://doi.org/10.1007/s00122-014-2363-2
Lander ES, Green P (1987) Construction of multilocus genetic linkage maps in humans. Proc Natl Acad Sci USA 84:2363–2367. https://doi.org/10.1073/pnas.84.8.2363
Lellis AD, Kasschau KD, Whitham SA, Carrington JC (2002) Loss-of-susceptibility mutants of Arabidopsis thaliana reveal an essential role for eIF(iso)4E during potyvirus infection. Curr Biol 12:1046–1051. https://doi.org/10.1016/S0960-9822(02)00898-9
Levi A, Thomas CE, Wehner TC, Zhang X (2001) Low genetic diversity indicates the need to broaden the genetic base of cultivated watermelon. HortScience 36:1096–1101
Levi A, Thies JA, Wechter WP et al (2013) High frequency oligonucleotides: targeting active gene (HFO-TAG) markers revealed wide genetic diversity among Citrullus spp. accessions useful for enhancing disease or pest resistance in watermelon cultivars. Genet Resour Crop Evol 60:427–440. https://doi.org/10.1007/s10722-012-9845-3
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Ling KS, Harris KR, Meyer JD, Levi A, Guner N, Wehner TC, Bendahmane A, Havey MJ (2009) Non-synonymous single nucleotide polymorphisms in the watermelon eIF4E gene are closely associated with resistance to Zucchini yellow mosaic virus. Theor Appl Genet 120(1):191–200
Manichaikul A, Moon JY, Sen Ś et al (2009) A model selection approach for the identification of quantitative trait loci in experimental crosses, allowing epistasis. Genetics 181:1077–1086. https://doi.org/10.1534/genetics.108.094565
Martyn RD (1987) Fusarium oxysporum f. sp. niveum race 2: a highly aggressive race new to the United States. Plant Dis 71:233–236
Meru G, McGregor C (2016a) Genotyping by sequencing for SNP discovery and genetic mapping of resistance to race 1 of Fusarium oxysporum in watermelon. Sci Hortic (Amsterdam) 209:31–40. https://doi.org/10.1016/j.scienta.2016.06.005
Meru G, McGregor CE (2016b) A genetic locus associated with resistance to Fusarium oxysporum f. sp. niveum Race 2 in Citrullus lanatus-type Watermelon. J Am Soc Hortic Sci 141:617–622. https://doi.org/10.21273/JASHS03890-16
Nicaise V, German-Retana S, Sanjuan R et al (2003) The Eukaryotic translation initiation factor 4E controls lettuce susceptibility to the potyvirus lettuce mosaic virus. Plant Physiol 132:1272–1282. https://doi.org/10.1104/pp.102.017855
Ren Y, Jiao Di, Gong G et al (2015) Genetic analysis and chromosome mapping of resistance to Fusarium oxysporum f. sp. niveum (FON) race 1 and race 2 in watermelon (Citrullus lanatus L.). Mol Breed 35:1–9. https://doi.org/10.1007/s11032-015-0375-5
Ruffel S, Dussault MH, Palloix A et al (2002) A natural recessive resistance gene against potato virus Y in pepper corresponds to the eukaryotic initiation factor 4E (elF4E). Plant J 32:1067–1075. https://doi.org/10.1046/j.1365-313X.2002.01499.x
Shimizu M, Fujimoto R, Ying H et al (2014) Identification of candidate genes for fusarium yellows resistance in Chinese cabbage by differential expression analysis. Plant Mol Biol 85:247–257. https://doi.org/10.1007/s11103-014-0182-0
Simons G, Groenendijk J, Wijbrandi J et al (1998) Dissection of the fusarium I2 gene cluster in tomato reveals six homologs and one active gene copy. Plant Cell 10:1055–1068. https://doi.org/10.1105/tpc.10.6.1055
Strange EB, Guner N, Pesic-Vanesbroeck Z, Wehner TC (2002) Plant genetic resources screening the watermelon germplasm collection for resistance to Papaya ringspot virus type-W. Publ Crop Sci 42:1324–1330
Swarts K, Li H, Romero Navarro JA et al (2014) Novel methods to optimize genotypic imputation for low-coverage, next-generation sequence data in crop plants. Plant Genome. https://doi.org/10.3835/plantgenome2014.05.0023
Wechter WP, Kousik C, McMillan M, Levi A (2012) Identification of resistance to Fusarium oxysporum f. sp. niveum race 2 in citrullus lanatus var. citroides plant introductions. HortScience 47:334–338. https://doi.org/10.1002/ird.1717
Wechter WP, McMillan MM, Farnham MW, Levi A (2016) Watermelon germplasm lines USVL246-FR2 and USVL252-FR2 tolerant to Fusarium oxysporum f. sp. niveum race 2. HortScience 51:1065–1067
Wehner TC (2008) Watermelon. In Vegetables I:381–418. Springer, New York
Zeng ZB, Kao CH, Basten CJ (1999) Estimating the genetic architecture of quantitative traits. Genet Res 74:279–289. https://doi.org/10.1017/S0016672399004255
Zhang M, Xu JH, Liu G, Yao XF, Li PF, Yang XP (2015) Characterization of the watermelon seedling infection process by Fusarium oxysporum f. sp. niveum. Plant Pathol 64(5):1076–1084
Zheng Y, Wu S, Bai Y et al (2019) Cucurbit genomics database (CuGenDB): a central portal for comparative and functional genomics of cucurbit crops. Nucleic Acids Res 47:D1128–D1136. https://doi.org/10.1093/nar/gky944
Zhou XG, Everts KL, Bruton BD (2010) Race 3, a new and highly virulent race of Fusarium oxysporum f. sp. niveum causing fusarium wilt in watermelon. Plant Dis 94:92–98. https://doi.org/10.1094/PDIS-94-1-0092
Acknowledgements
This research is a part of the Cucurbit Coordinated Agricultural Project (CucCAP) supported by the USDA, National Institute of Food and Agriculture (NIFA), Specialty Crop Research Initiative (SCRI) Grant # 2015-51181-24285, and used resources provided by the SCINet project of the USDA Agricultural Research Service, ARS project number 0500-00093-001-00-D.
Funding
This study was funded, in part, by the US Department of Agriculture (USDA) project number 6080-22000-028-00 and the National Institute of Food and Agriculture, Specialty Crops Research Initiative project number 6080-21000-019-08.
Author information
Authors and Affiliations
Contributions
AL, WPW, SB and KL designed and implemented the experiments. NG, MB and EK developed F2 and F3 population and maintained and supplied PRSV inoculum, WPW, SB, KL and AL phenotyped the population. LM and BC assisted in increasing genetic populations, inoculating and maintaining plants in greenhouse, and in conducting DNA isolation, genotyping and ELISA tests. SB analyzed the data. SB, WPW, KL and AL wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
The experiment conducted complies with the laws of the USA.
Additional information
Communicated by Albrecht E. Melchinger.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Branham, S.E., Patrick Wechter, W., Ling, KS. et al. QTL mapping of resistance to Fusarium oxysporum f. sp. niveum race 2 and Papaya ringspot virus in Citrullus amarus. Theor Appl Genet 133, 677–687 (2020). https://doi.org/10.1007/s00122-019-03500-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03500-3