
Abstract
Cadmium (Cd) is a widespread soil pollutant and poses a significant threat to human health via the food chain. Large phenotypic variations in Cd concentration of radish roots and shoots have been observed. However, the genetic and molecular mechanisms of Cd accumulation in radish remain to be elucidated. In this study, a genetic linkage map was constructed using an F2 mapping population derived from a cross between a high Cd-accumulating cultivar NAU-Dysx and a low Cd-accumulating cultivar NAU-Yh. The linkage map consisted of 523 SRAP, RAPD, SSR, ISSR, RAMP, and RGA markers and had a total length of 1,678.2 cM with a mean distance of 3.4 cM between two markers. All mapped markers distributed on nine linkage groups (LGs) having sizes between 134.7 and 236.8 cM. Four quantitative trait loci (QTLs) for root Cd accumulation were mapped on LGs 1, 4, 6, and 9, which accounted for 9.86 to 48.64 % of all phenotypic variance. Two QTLs associated with shoot Cd accumulation were detected on LG1 and 3, which accounted for 17.08 and 29.53 % of phenotypic variance, respectively. A major-effect QTL, qRCd9 (QTL for root Cd accumulation on LG9), was identified on LG 9 flanked by NAUrp011_754 and EM5me6_286 markers with a high LOD value of 23.6, which accounted for 48.64 % of the total phenotypic variance in Cd accumulation of F2 lines. The results indicated that qRCd9 is a novel QTL responsible for controlling root Cd accumulation in radish, and the identification of specific molecular markers tightly linked to the major QTL could be further applied for marker-assisted selection (MAS) in low-Cd content radish breeding program.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cadmium (Cd) is a nonessential heavy metal that is highly toxic to living organisms. Contamination of agricultural soils with Cd due to atmospheric deposition and the use of animal manures, phosphate fertilizers, and sewage sludge has become a severe problem worldwide (McGrath et al. 2001; Tanhuanpää et al. 2007). Cd2+ is rapidly absorbed by roots and can be loaded into the xylem for its transport into aerial organs (Grant et al. 1998). Cd2+ inhibits plant growth and development as a consequence of alterations in photosynthesis, respiration, and nitrogen metabolism as well as a decrease in water and mineral nutrient uptake (Deckert 2005; Besson-Bard et al. 2009). Cadmium can accumulate in human bodies over time through ingestion of food containing Cd, leading to a risk of chronic toxicity with excessive intake (Grant et al. 2008; Ueno et al. 2009). In humans, Cd might cause genotoxic and cytotoxic effects, leading finally to the inhibition of cell proliferation and apoptosis. Thus, the reduction of Cd accumulation in vegetable crops for minimizing human dietary intake of Cd has become an important food-safety issue.
Radish (Raphanus sativus L., 2n = 2× = 18), an annual or biennial herb of the Brassicaceae family, is one of the most important root vegetable crops in the world, especially in East Asia (Wang and He 2005). Liu et al. (2008) reported a large phenotypic variation in Cd concentration among different radish genotypes, indicating the possibility of developing cultivars with low Cd accumulation in the edible tissues. Low-Cd content cultivar development is an attractive method for reducing the potential Cd accumulation in crops (Grant et al. 2008). However, because of determination of Cd concentration is laborious, time consuming, and expensive, marker-assisted selection (MAS), the use of molecular markers linked to a desired gene, could be an alternative to phenotyping (Tanhuanpää et al. 2007). In order to minimize human intake of Cd, development of low-Cd radish cultivars has become a major objective in breeding programs.
A genetic linkage map is the pre-requisite for positioning genes/QTLs of interested traits on the chromosome and exploring genetic and molecular mechanisms of genetic variations in natural germplasm and breeding lines. Several molecular marker systems including restriction fragment length polymorphism (RFLP), randomly amplified polymorphic DNA (RAPD), amplified fragment length polymorphisms (AFLP), simple sequence repeats (SSR), and single nucleotide polymorphism (SNP) have been employed to construct linkage maps in radish using interspecific or intraspecific populations with a total coverage from 554 to 1517 cM (Bett and Lydiate, 2003; Tsuro et al. 2005, 2008; Budahn et al. 2009; Kamei et al. 2010; Shirasawa et al. 2011; Li et al. 2011). Target genes/QTLs associated with some important agronomic traits, such as flowering time, root shape and pigmentation, and disease and pest resistance have been assigned on these maps (Tsuro et al. 2008; Budahn et al. 2009; Kamei et al. 2010). However, most of these maps did not have enough markers to completely cover the whole genome of radish. Therefore, to increase the efficiency of QTL mapping and marker-assisted selection (MAS), a high-density linkage map needed to be constructed with a large number of PCR-based markers in radish.
QTL mapping is a powerful tool for understanding the genetic and molecular mechanisms underlying complicated traits that are controlled by multiple genes, many of which are important for agricultural products (Yamamoto et al. 2009; Ishikawa et al. 2010). Using QTL mapping, the chromosomal regions involved in Zn and Cd accumulation in the hyperaccumulator species Thlaspi caerulescens was identified. Two QTLs were identified for Cd accumulation in roots and one QTL was mapped for Cd accumulation in shoots (Deniau et al. 2006). In rice (Oryza sativa L.), several QTLs for cadmium accumulation and translocation have been reported (Kashiwagi et al. 2009; Ishikawa et al. 2005, 2010; Ueno et al. 2009; Xue et al. 2009). Ishikawa et al. (2005) reported that three putative QTLs controlling Cd concentration were mapped on chromosomes 3, 6, and 8 using 39 chromosome segment substitution lines (CSSLs) in brown rice. In soybean (Glycine max L.), one major QTL (Cda1) associated with seed low Cd accumulation has recently been identified (Jegadeesan et al. 2010). In durum wheat, Cd accumulation was found to be governed by a major gene Cdu1 (Clarke et al. 1997), which was mapped to 5BL (Knox et al. 2009). A single QTL for grain Cd accumulation in oat (Avena sativa L.) has been reported (Tanhuanpää et al. 2007). However, the genetic analysis of QTLs associated with Cd accumulation in radish roots and shoots has not yet been reported.
In this study, using an F2 population derived from an inter-varietal cross between the high- and low Cd-accumulating radish lines, ‘Nau-Dysx’ and ‘Nau-Yh’, a genetic linkage map was constructed with RAPD, SRAP (sequence-related amplified polymorphism), SSR, ISSR (inter-simple sequence repeats), RAMP (randomly amplified microsatellite polymorphism), and RGA (resistant gene analog) markers. The aims of this study were to identify putative QTLs associated with Cd accumulation in radish roots and shoots and to identify molecular markers tightly linked to the major QTLs for Cd accumulation. The results from this study would facilitate genetic improvement of root Cd accumulation in radish breeding programs.
Materials and methods
Plant materials
Two radish advanced inbred lines, NAU-Dysx (high Cd accumulation) and NAU-Yh (low Cd accumulation), were used for their contrasting cadmium accumulation ability in roots and shoots. NAU-Dysx with a cylindrical white skin root was used as the female parent. NAU-Yh with a small globular red skin root was used as the pollen donor. The mapping population consisting of 172 F2 individuals was obtained by selfing the F1 progeny derived from the crossing.
Hydroponic culture and Cd treatment
Seeds of two parental lines and 172 F2 individuals were surface sterilized in 1.2 % NaOCl and incubated at 25 °C for 3 days in the dark. The germinating seeds were sown into sand in plastic pots and the seedlings were cultured in a growth cabinet (14 h light at 25 °C, 10 h dark at 18 °C). After 3 weeks, seedlings with similar size were transplanted into a 20-L plastic container with modified half-strength Hoagland’s nutrient solution consisting of 2 mM Ca(NO3)2, 2 mM MgSO4, 0.88 mM K2SO4, 0.25 mM KH2PO4, 10 μM NaCl, 10 μM H3BO3, 1 μM ZnSO4, 0.6 μM MnSO4, 0.1 μM CuSO4, 0.1 μM (NH4)6Mo7O24, and 20 μM Fe(III)-EDTA (pH5.4). The nutrient solution was refreshed every 3 days. One week later, 50 mg L−1 CdCl2·2.5H2O was added to the solution and cultured in the growth cabinet for 21 days.
Cadmium determination and statistical analysis
After the hydroponic culture period, the roots of the seedlings were washed with running deionized water three times to remove Cd2+ from the plant surface. Shoot height (SH) and root length (RL) were measured. The seedlings were divided into root and shoot (leaves and stem) tissues and oven-dried at 105 °C for 1 h and then at 70 °C for 48 h. Root dry weight (RDW) and shoot dry weight (SDW) were weighed, and total dry weight (TDW) was calculated according to SDW + RDW. Dried tissues (0.5 g) were digested in 5 mL of HNO3–HClO4 (2:1 v/v) mix solution for 12 h and resuspended in 25 mL of deionized water. Cadmium concentration was determined by a graphite furnace atomic absorption spectroscopy (AAnalyst 700; PerkinElmer, Waltham, MA, USA). For two parental lines, six repeated samples were prepared and the mean values of Cd concentrations were calculated from these replicates.
Paired-sample t test was conducted to detect the differences in means of all traits between NAU-Dysx and NAU-Yh. One-way ANOVA analysis was conducted using SAS Version 9.0 (SAS Institute Inc., Cary, North Carolina) to verify the significance of variance component for each trait. Pearson’s correlations for all combinations of traits on the basis of F2 means across environments were also calculated.
Molecular marker analysis
Total genomic DNA was extracted from young leaves according to the modified CTAB method of Liu et al. (2003). DNA concentration and quality were estimated on a 1.2 % agarose gel. DNA was diluted to a final concentration of 10 ng/μl and stored at 4 or −20 °C for further use.
Arbitrary RAPD primers from the Operon Technologies (Alameda, CA, USA) and ISSR primers described by Liu et al. (2007) were screened for polymorphism of the two parental lines. The amplification was performed in the PTC-100 Thermal cycle (MJ Research Inc., USA) according to the reported protocol of Williams et al. (1990), with some minor modifications (Liu et al. 2007). PCR products were resolved by electrophoresis on 1.2 % agarose gel run in 1× TAE buffer, stained with ethidium bromide, and visualized under UV light (Liu et al. 2007).
SRAP primer combinations from Li and Quiros (2001) were used for SRAP analysis and the SRAP-PCR procedure was performed as described by Liu et al. (2007). SSR primer pairs derived from B. rapa (Suwabe et al. 2002) and EST-SSR markers developed from radish (Wang et al. 2007a) were used for SSR analysis. SSR amplification was performed as described by Tsuro et al. (2005). PCR products were separated on 8 % non-denaturing polyacrylamide gel (Acr:Bis = 29:1) and visualized by silver staining (Bassam et al. 1991; Liu et al. 2007).
RAMP primers from the combinations with SSR-anchored primers and ten selected RAPD primers were used for RAMP analysis. RAMP-PCR reactions were performed in a total volume of 10 μl containing 10 ng genomic DNA, 2.0 mM MgCl2, 0.20 mM dNTPs, 0.50 μM each primer, and 0.5 U Taq DNA polymerase (TAKARA). RGA primer pairs from Fourmann et al. (2001) were used for RGA analysis. RGA-PCR amplification was performed as described by Saal and Struss (2005) with a small modification. PCR products were separated and visualized as described for SSR analysis.
Linkage map construction
All primers that exhibited stable and scorable polymorphism between the parental lines were further evaluated on the 172 F2 individuals of the mapping population. X2 test was performed to test segregation distortion at each marker locus against the expected 3:1 (dominant) or 1:2:1 (codominant) segregation ratio for the F2 population. The genetic linkage map was constructed using JoinMap ver. 3.0 (Van Ooijen and Voorrips 2001) with a LOD threshold of 3.0, a recombination fractions threshold of 0.4, and a jump threshold of 5.0. The Kosambi’s mapping function (Kosambi 1944) was used to convert recombination fractions into genetic map distances in centiMorgans (cM).
QTL analysis
QTL analysis was conducted with composite interval mapping (CIM) procedure using Windows QTL Cartographer ver. 2.5 (Zeng 1994; Wang et al. 2007b). CIM analysis was performed using the Model 6, with the window size set at 10 cM and a walk speed of 1 cM. The genome-wide LOD score threshold (α = 0.05) for declaring the presence of QTLs was determined using the permutation test (1,000 replications) (Churchill and Doerge 1994). Based on the permutation results, the LOD score threshold was set at 3.0 for the trait to declare the presence of a significant QTL. The additive effect and percentage of phenotypic variance explained by each QTL were estimated at the peak LOD score. Graphic presentation of the linkage groups (LGs) and the QTLs were drawn with MapChart version 2.2 (Voorrips 2002).
Results
Phenotypic variation in Cd concentration of two parental lines and the F2 mapping population
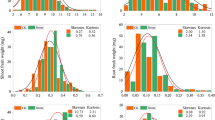
Mean value, standard deviation, range, skewness, and kurtosis for each Cd-related trait measured in the parental lines and the F2 populations were calculated in Table 1. The root Cd concentration (RCd) of the parental lines was significantly different (Table 1; Fig. 1), in which RCd in Nau-Dysx (17.35 mg kg−1) was approximately 3.8 times higher than that of Nau-Yh (4.58 mg kg−1) (P<0.01, F = 6.70). The shoot Cd concentration (SCd) of Nau-Dysx (1.95 mg kg−1) was significantly 2.7 times higher than that of Nau-Yh (0.72 mg kg−1) (P<0.01, F = 5.80) (Table 1).

Phenotypic distribution of Cd concentration in the F2 mapping population derived from the cross Nau-Dysx (high Cd) × Nau-Yh (low Cd). a Frequency distribution of Cd concentration in roots of the F2 lines; b Frequency distribution of Cd concentration in shoots of the F2 lines. Arrow indicates the level of the two parental lines
For the 172 F2 individuals, the frequency distribution of RCd and SCd showed a continuous phenotypic variation with the kurtosis and skewness less than 1, indicating a normal distribution of quantitative traits. The RCd varied from 1.42 to 19.65 mg kg−1 with a mean of 8.52 ± 0.46 mg kg−1, and the SCd ranged from 0.26 to 2.46 mg kg−1 with a mean of 1.17 ± 0.12 mg kg−1 (Table 1; Fig. 1). The transgressive segregation, however, suggests that some additional minor genes may also influence Cd accumulation in the Nau-Dysx × Nau-Yh population, indicating that Cd accumulation in radish is a quantitative trait. Correlation analysis showed that the RCd had significant positive correlations with RL and RDW (r = 0.42 and 0.54, respectively, P < 0.01). In addition, SCd had significant positive correlations with SH and SDW (r = 0.46 and 0.62, respectively, P < 0.01) (Table 2).
Marker polymorphism
Six types of PCR-based markers were initially screened for polymorphism between the two parental lines. Those that showed polymorphism were further screened on the 172 F2 individuals. A total of 1,848 primers (680 SRAP, 600 RAPD, 278 SSR, 120 RAMP, 100 ISSR and 70 RGA) were used for polymorphism screening, in which 226 SRAP, 128 RAPD, 54 SSR, 26 ISSR, 16 RAMP, and 8 RGA primers generated polymorphic bands between the parental lines. The total polymorphism rate of six types of PCR-based markers varied from 11.4 % (RGA) to 33.2 % (SRAP), and the polymorphism for ISSR, RAPD, SSR, and RAMP markers were 26.0, 21.3, 19.4, and 13.3 %, respectively. In total, 645 informative loci (317 SRAP, 152 RAPD, 80 SSR, 51 ISSR, 31 RAMP, and 14 RGA loci) were successfully genotyped within the two parental lines and 172 F2 individuals, and finally subjected to linkage analysis.
Genetic linkage map construction
Markers deviating significantly (P < 0.01) from the expected 3:1 (dominant marker) or 1:2:1 (codominant marker) segregation ratios were excluded from analysis. A total of 592 polymorphic loci (287 SRAP, 135 RAPD, 78 SSR, 49 ISSR, 29 RAMP, and 14 RGA loci) were used for linkage map analysis with LOD score set at 3.0. At last, 523 out of 592 loci (88 %) including 248 SRAP, 114 RAPD, 76 SSR, 47 ISSR, 26 RAMP, and 12 RGA markers were successfully integrated into the linkage analysis with 69 loci remaining unlinked (Table 3).
The linkage map had a total genetic distance of 1,678.2 cM with an average distance of 3.4 cM between adjacent markers and distributed on nine linkage groups (LGs), which is equivalent to the haploid chromosome number in radish (Bett and Lydiate 2003). The average length per linkage group was 186.5 cM, but ranged from 134.7 cM (LG8) to 236.8 cM (LG6). The number of markers in each linkage group varied from 29 (LG8) to 79 (LG5). The average distance between linked markers for each linkage group varied from 2.4 cM (LG5) to 4.8 cM (LG8) (Fig. 2; Table 3).

Genetic linkage map of radish (Raphanus sativus L.) based on an F2 population derived from the intervarietal cross Nau-Dysx (high Cd) × Nau-Yh (low Cd).The marker name and map distances in centiMorgans (cM) are indicated on the right and left side of the linkage groups (LGs), respectively. Significant QTL for root length (RL), shoot height (SH), root dry weight (RDW), shoot dry weight (SDW), total dry weight (TDW), root Cd concentration (RCd) and shoot Cd concentration (SCd) were indicated by bars and whiskers on the right side of the linkage group, which are 1- and 2-LOD likelihood intervals, respectively. The putative QTL region along the linkage groups is highlighted by bar segment and marker interval to the putative QTL is shown in bold
QTL mapping for Cd-related agronomic traits
A total of 19 putative QTLs were identified to be associated with the five Cd-related agronomic traits. These QTLs were mapped onto radish linkage groups (LGs) 1, 2, 3, 4, 5, 6, 7, and 9, and individual QTL could explain 4.73–31.42 % of the phenotypic variance (PV). The main effects and graphical locations of the 19 QTLs are shown in Table 4.
Five putative QTLs associated with root length (RL) were detected on LGs 1 (qRL1), 3 (qRL3.1 and qRL3.2), 5 (qRL5), and 7 (qRL7), respectively. The accumulated contribution of these QTLs explained 68.72 % of the total phenotypic variance, and individual QTL explained 5.82–27.83 % of the PV. Three of these QTLs (qRL3.1, qRL5 and qRL7) showed overdominance and the other two QTLs (qRL1 and qRL3.2) showed dominance. Moreover, three of these QTLs (qRL1, qRL3.1 and qRL7) increased RL by the presence of Nau-Dysx alleles, whereas the other two QTLs (qRL3.2 and qRL5) increased RL by the presence of Nau-Yh alleles.
Two QTLs associated with shoot height (SH) were identified on LGs 2 (qSH2) and 5 (qSH5), which explained 12.46 and 6.52 % of the PV, respectively. Together the two QTLs accounted for 18.98 % of the total phenotypic variance. The QTL qSH2 showed dominance and increased SH by the presence of the Nau-Dysx allele, whereas qSH5 showed partial dominance and increased SH by the presence of the Nau-Yh allele.
Three QTLs for root dry weight (RDW) were detected on LGs 5 (qRDW5), 6 (qRDW6), and 9 (qRDW9), respectively. Together these QTLs accounted for 45.69 % of the total phenotypic variance, and individual QTL accounted for 4.73–31.42 % of the PV. The QTL qRDW5 showed partial dominance and increased RDW by the presence of the Nau-Yh allele, whereas the other two QTLs (qRDW6 and qRDW9) showed overdominance and increased RDW by the presence of Nau-Dysx alleles.
Three QTLs for shoot dry weight (SDW) were identified on LGs 2 (qSDW2), 6 (qSDW6), and 9 (qSDW9), respectively. Together these three QTLs accounted for 48.18 % of the total phenotypic variance, and individual QTL accounted for 8.68–26.64 % of the PV. The QTL qSDW6 showed dominance and increased SDW by the presence of the Nau-Dysx allelle, whereas the other two QTLs (qSDW2 and qSDW9) showed overdominance and increased SDW by the presence of Nau-Yh alleles.
A total of six QTLs for total dry weight (TDW) were identified on LGs 1 (qTDW1.1 and qTDW1.2), 5 (qTDW5), 6 (qTDW6), 7 (qTDW7), and 9 (qTDW9), respectively. The accumulated contribution of these six QTLs accounted for 86.3 % of the total phenotypic variance, and individual QTL explained 6.18–22.85 % of the PV. Three of these QTLs (qTDW1.1, qTDW6 and qTDW9) showed overdominance and the other three QTLs (qTDW1.2, qTDW5 and qTDW7) showed partial to complete dominance. Moreover, four of these QTLs (qTDW1.1, qTDW5, qTDW6 and qTDW9) increased TDW by the presence of Nau-Dysx alleles, whereas the other two QTLs (qTDW1.2 and qTDW7) increased TDW by the presence of Nau-Yh alleles.
QTL mapping for Cd accumulation in roots and shoots
The QTLs for Cd accumulation in roots and shoots of radish are summarized in Table 4. Four putative QTLs for root Cd accumulation (RCd) were identified on LGs 1 (qRCd1), 4 (qRCd4), 6 (qRCd6), and 9 (qRCd9), respectively. The percentage of PV explained by these QTLs varied from 9.86 to 48.64 %, and all together the four QTLs accounted for 94.3 % of the total phenotypic variance. A major QTL qRCd9 with an LOD value of 23.6 was mapped on LG9 flanked by NAUrp011_754 and EM5me6_286, which accounted for 48.64 % of the PV (Table 4; Fig. 3). The QTL qRCd6 showed dominance and the other three QTLs (qRCd1, qRCd4 and qRCd9) showed overdominance. The RCd was increased by the presence of Nau-Yh alleles at qRCd1 and qRCd6 and by the presence of Nau-Dysx alleles at qRCd4 and qRCd9.

A major QTL for root Cd accumulation on LG 9 in the F2 population derived from the cross Nau-Dysx (high Cd) × Nau-Yh (low Cd) was identified using composite interval mapping. The LOD values from each centiMorgan of the linkage groups (LGs) were plotted against the LGs
Within a 10.0-cM region around the major QTL qRCd9, two RAPD markers (NAUrp011_754 and NAUrp751_764) distributed at one side of qRCd9 with a distance of 4.8 and 7.0 cM, respectively, and the RAPD marker NAUrp011_754 located 4.8 cM to qRCd9 was the closest one. On the other side of qRCd9, the SRAP marker EM5me6_286 was most closely linked with a distance of 5.6 cM and the SSR marker BRMS058_620 was located at a distance of 7.6 cM to qRCd9.
Two putative QTLs associated with shoot Cd accumulation (SCd) were detected on LGs 1 (qSCd1) and 3 (qSCd3), respectively. A major QTL qSCd3 with an LOD value of 7.64 was identified on LG3 flanked by EM16ga18_383 and Na10A08_455, which explained 29.53 % of the PV (Table 4; Fig. 4). The other QTL (qSCd1) flanked by Ol10F09_445 and PM17ga27_569 was located on LG1 and explained 17.08 % of the PV with an LOD value of 4.37. The QTL qSCd1 showed dominance and increased SCd by the presence of the Nau-Yh allele, whereas qSCd3 showed overdominance and increased SCd by the presence of the Nau-Dysx allele.

A major QTL for shoot Cd accumulation mapped on LG 3 in the F2 population derived from the cross Nau-Dysx (high Cd) × Nau-Yh (low Cd) was identified using composite interval mapping. The LOD values from each centiMorgan of the linkage groups (LGs) were plotted against the LGs
In addition, one SRAP marker (EM16ga18_383) and two RAPD markers (NAUrp1106_792 and NAUrp829_696) distributed at one side of qSCd3 with a distance of 3.2, 7.5, and 9.8 cM, respectively, and the SRAP marker EM16ga18_383 was the closest one with a distance of 3.2 cM to qSCd3. On the other side of qSCd3, the EST-SSR marker Na10A08_455 was the closest one with a distance of 7.4 cM and the SRAP marker PM2em11_312 was located at a distance of 9.6 cM to qSCd3.
Discussion
Cadmium is a vital toxicant that presents a potential risk to human health, as Cd consumption may cause many serious sicknesses including ‘Itai-itai’ disease. Radish is an important fleshy root vegetable worldwide. To minimize human intake of Cd, there is an urgent need to understand the genetic molecular mechanisms of Cd accumulation to devise a breeding program for developing low-Cd content radish cultivars.
Constructing a high-density linkage map is a vital prerequisite for genetically dissecting the location and organization of genes/QTLs associated with complex agronomic traits. Genetic linkage maps have been constructed for many economically important vegetable crop species including a few Brassicaceae vegetables, such as Brassica napus (Suwabe et al. 2006; Smooker et al. 2011), B. oleracea (Gao et al. 2007), and B. rapa (Choi et al. 2007). However, limited information on molecular genetics of important complicated traits in radish is available (Kamei et al. 2010). Only a few genetic linkage maps of radish have been reported (Bett and Lydiate 2003; Tsuro et al. 2005; Budahn et al. 2009; Kamei et al. 2010; Shirasawa et al. 2011; Li et al. 2011). Recently, an intraspecific radish linkage map was constructed using an F2 population and RAPD, dpRAPD, AFLP, and SSR marker systems (Budahn et al. 2009). That map consists of 545 markers, encompassing 1,517.1 cM and distributed on nine linkage groups with an average distance of 2.8 cM between makers. Moreover, Shirasawa et al. (2011) reported a radish map that consisted of 630 SSR loci and 213 RAPD, RFLP, and trait marker loci, covering 1,129.2 cM and distributed on nine linkage groups with an average distance of 1.3 cM between makers. Li et al. (2011) constructed another radish map that consisted of 746 SNP markers, covering 806.7 cM and distributed on nine linkage groups with an average distance of 1.1 cM between makers. In this study, a comprehensive genetic linkage map was constructed with 532 loci using an F2 population. The map had a total coverage of 1,678.2 cM with an average distance of 3.4 cM between adjacent markers and distributed on nine linkage groups. To our knowledge, this map comprises the longest genetic distance among all published linkage maps of radish species (Shirasawa et al. 2011; Li et al. 2011). The loci of the present map were evenly distributed on the entire genome rather than assembling in clusters. However, the coverage of markers varied per linkage group, with LG1, LG6, and LG7 having the best coverage overall and LG8 having the least coverage (Table 3; Fig. 2). Our result was very similar to the one observed in the previous intraspecific maps (Budahn et al. 2009), which might be due to the nonrandom distribution of polymorphic loci and lack of marker polymorphism between mapping parents on some linkage groups (Budahn et al. 2009; Tsuro et al. 2005; Shirasawa et al. 2011).
Parental lines that show wide phenotypic variations in the target agronomic traits are necessary for QTL analysis, because QTL detection is based on natural allelic differences between parental lines (Ashikari et al. 2005; Ishikawa et al. 2010). The two radish lines used in this study have striking contrasting characteristics on Cd accumulation in roots and shoots, in which Nau-Yh is a low-Cd-accumulating cultivar and Nau-Dysx is a high-Cd-accumulating cultivar. To our knowledge, this is the first report on QTL mapping for Cd accumulation in roots and shoots of radish. A major QTL (qRCd9) for root Cd accumulation was detected with a high LOD value of 23.64, which showed overdominance and explained a large proportion of the PV (R 2 = 48.64 %) in the F2 population (Table 4). In addition, the positive additive value of qRCd9 indicates that the major gene increasing root Cd accumulation is located on qRCd9 of the Nau-Dysx allele. Therefore, it can be concluded that the gene located on the qRCd9 region between NAUrp011_754 and EM5me6_286 on LG9 is responsible for controlling root Cd accumulation. Moreover, three minor QTLs (qRCd1, qRCd4, and qRCd6) for root Cd accumulation were also detected on LGs 1, 4, and 6 and these three QTLs could explain 26.42, 9.86, and 14.38 % of the phenotypic variance, respectively. Two of these QTLs (qRCd1 and qRCd4) showed overdominance, whereas the other QTL qRCd6 showed dominance (Table 4).
In recent years, a few studies have reported that some important agronomic traits including RL, SH, RDW, SDW, and TDW influence the root/shoot Cd accumulation at different levels, indicating that the identification of target QTLs for these agronomic traits may provide valuable approach for understanding the genetic molecular mechanisms of Cd accumulation in plants (Ishikawa et al. 2010; Xue et al. 2009). In the present study, both qRCd1 and qRL1 were located on LG1 for RCd and RL, respectively. However, qRCd1 flanked by Ni2E05_525 and PM17em10_456, and qRL1 flanked by Na10F06_550 and Fc9odd54_388, indicating that these two QTLs were not located on the same chromosomal region of LG1. Moreover, qRCd1 increased RCd by the presence of Nau-Dysx allele, while qRL1 increased RL by the presence of Nau-Yh allele, indicating that the addictive effects of the two QTLs come from different parent’s alleles. Similar results were also observed between qRCd6 and qRDW6 on LG6 and qRCd9, and qRDW9 on LG9. Moreover, the QTL qRL1 showed dominance, whereas the other two QTLs (qRDW6 and qRDW9) showed overdominance. Furthermore, these three QTLs (qRL1, qRDW6 and qRDW9) were involved in agronomic traits that may influence the RCd. These results indicated that the RCd had significant positive correlations with RL and RDW, respectively (Table 2). Additionally, SCd had significant positive correlations with SH and SDW, respectively (Table 2). However, the QTLs associated with SCd, SH, and SDW were not successfully mapped on the same linkage groups (Table 4; Fig. 2).
Previous studies have detected several QTLs that associated with Cd accumulation and translocation in other species (Ishikawa et al. 2005, 2010; Wiebe et al. 2010). In rice, six putative QTLs controlling Cd accumulation have been detected on chromosomes 3, 6, and 8 (Ishikawa et al. 2005, 2010) and chromosomes 2, 5, and 11 (Ueno et al. 2009), respectively. In wheat, a major QTL (Cdu1) associated with seed Cd accumulation has been identified and mapped to 5BL (Clarke et al. 1997; Knox et al. 2009). Recently, QTLs for the translocation of Cd from source to sink regions were detected in rice (Kashiwagi et al. 2009; Tezuka et al. 2010). A major QTL (qCdT7) controlling Cd translocation from roots to shoots has been identified on rice chromosome 7, and this QTL explained a large proportion of the phenotypic variation (R 2 = 88 %), indicating that low Cd accumulation is a dominant trait (Tezuka et al. 2010). Courbot et al. (2007) reported that the heavy metal transporting ATPase4 (HMA4) was co-localized with a major QTL whose transcript levels were shown to be higher in roots and shoots of Arabidopsis halleri. Nakanishi et al. (2006) found that two rice Fe2+ transporters, OsIRT1 (Iron-Regulated Transporter 1) and OsIRT2, also had the ability to transport Cd in yeast. Takahashi et al. (2011) reported that OsNramp1 iron transporter was involved in high-level Cd accumulation in rice. However, the candidate genes for heavy metal transport located on qRCd9 have not been validated in radish. BLASTn analysis revealed that genomic homology exists between radish ESTs and the four Cd-related genes, but none of them has been successfully cloned in radish to date. With the similar physiological and genetic mechanisms observed, mapping and isolating of these genes in radish should be a vital subject of priority.
Recently, a few molecular markers linked to the target QTLs associated with Cd accumulation have been identified in some crop species, such as wheat (Wiebe et al. 2010), soybean (Jegadeesan et al. 2010), and oat (Tanhuanpää et al. 2007), which have the potential to develop low-Cd content cultivars in breeding programs. In this study, four PCR-based markers (two RAPD, one SRAP and one BRMSSR marker) were successfully mapped within 10.0 cM region to the major QTL qRCd9, and the RAPD marker NAUrp011_754 was the closest one with a linkage distance of 4.8 cM to qRCd9. These molecular markers tightly linked to the major QTL for Cd accumulation in radish could be further applied in MAS for developing low-Cd content radish cultivars and identifying gene(s) responsible for low Cd accumulation, which would be more efficient and cost-effective comparing with the traditional breeding process. Moreover, the result of this study will also provide valuable information for better understanding the genetic molecular mechanisms for Cd accumulation in radish.
References
Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M (2005) Cytokinin oxidase regulates rice grain production. Science 309:741–745
Bassam BJ, Anollés GC, Gresshoff PM (1991) Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal Biochem 196:80–83
Besson-Bard A, Gravot A, Richaud P, Auroy P, Duc C, Gaymard F, Taconnat L, Renou JP, Pugin A, Wendehenne D (2009) Nitric oxide contributes to cadmium toxicity in Arabidopsis by promoting cadmium accumulation in roots and by up-regulating genes related to iron uptake. Plant Physiol 149:1302–1315
Bett KE, Lydiate DJ (2003) Genetic analysis and genome mapping in Raphanus. Genome 46:423–430
Budahn H, Peterka H, Mousa MA, Ding Y, Zhang S, Li J (2009) Molecular mapping in oil radish (Raphanus sativus L.) and QTL analysis of resistance against beet cyst nematode (Heterodera schachtii). Theor Appl Genet 118:775–782
Choi SR, Teakle GR, Plaha P, Kim JH, Allender CJ, Beynon E, Piao ZY, Soengas P, Han TH, King GJ, Baker GC, Hand P, Lydiate DJ, Batley J, Edwards D, Koo DH, Bang JW, Park B-S, Lim YP (2007) The reference genetic linkage map for the multinational Brassica rapa genome sequencing project. Theor Appl Genet 115:777–792
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Clarke JM, Leisle D, Kopytko GL (1997) Inheritance of cadmium concentration in five durum wheat crosses. Crop Sci 37:1722–1725
Courbot M, Willems G, Motte P, Arvidsson S, Roosens N, Saumitou-Laprade P, Verbruggen N (2007) A major quantitative trait locus for cadmium tolerance in Arabidopsis halleri colocalizes with HMA4, a gene encoding a heavy metal ATPase. Plant Physiol 144:1052–1065
Deckert J (2005) Cadmium toxicity in plants: is there any analogy to its carcinogenic effect in mammalian cells? Biometals 18:475–481
Deniau AX, Pieper B, Ten Bookum WM, Lindhout P, Aarts MGM, Schat H (2006) QTL analysis of cadmium and zinc accumulation in the heavy metal hyperaccumulator Thlaspi caerulescens. Theor Appl Genet 113:907–920
Fourmann M, Charlot F, Froger N, Delourme R, Brunel D (2001) Expression, mapping, and genetic variability of Brassica napus disease resistance gene analogues. Genome 44:1083–1099
Gao MQ, Li GY, Yang B, Qiu D, Farnham M, Quiros CF (2007) High-density Brassica oleracea linkage map: identification of useful new linkages. Theor Appl Genet 115:277–287
Grant CA, Buckley WT, Bailey LD, Selles F (1998) Cadmium accumulation in crops. Can J Plant Sci 78:1–17
Grant CA, Clarke JM, Duguid S, Chaney RL (2008) Selection and breeding of plant cultivars to minimize cadmium accumulation. Sci Total Environ 390:301–310
Ishikawa S, Ae N, Yano M (2005) Chromosomal regions with quantitative trait loci controlling cadmium concentration in brown rice (Oryza sativa). New Phytol 168:345–350
Ishikawa S, Abe T, Kuramata M, Yamaguchi M, Ando T, Yamamoto T, Yano M (2010) A major quantitative trait locus for increasing cadmium-specific concentration in rice grain is located on the short arm of chromosome 7. J Exp Bot 61:923–934
Jegadeesan S, Yu KF, Poysa V, Gawalko E, Morrison MJ, Shi C, Cober E (2010) Mapping and validation of simple sequence repeat markers linked to a major gene controlling seed cadmium accumulation in soybean [Glycine max (L.) Merr]. Theor Appl Genet 121:283–294
Kamei A, Tsuro M, Kubo N, Hayashi T, Wang N, Fujimura T, Hirai M (2010) QTL mapping of clubroot resistance in radish (Raphanus sativus L.). Theor Appl Genet 120:1021–1027
Kashiwagi T, Shindoh K, Hirotsu N, Ishimaru K (2009) Evidence for separate pathways in determining cadmium accumulation in grain and aerial plant parts in rice. BMC Plant Biol 9:8. doi:10.1186/1471-2229-9-8
Knox RE, Pozniak CJ, Clarke FR, Clarke JM, Houshmand S, Singh AK (2009) Chromosomal location of the cadmium uptake gene (Cdu1) in durum wheat. Genome 52:741–747
Kosambi DD (1944) The estimation of map distance from recombination values. Ann Eugen 12:172–175
Li GY, Quiros CF (2001) Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theor Appl Genet 103:455–461
Li F, Hasegawa Y, Saito M, Shirasawa S, Fukushima A, Ito T, Fujii H, Kishitani S, Kitashiba H, Nishio T (2011) Extensive chromosome homoeology among Brassiceae species were revealed by comparative genetic mapping with high-density EST-based SNP markers in radish (Raphanus sativus L.). DNA Res 18:401–411
Liu L, Guo W, Zhu X, Zhang T (2003) Inheritance and fine mapping of fertility-restoration for cytoplasmic male sterility in Gossypium hirsutum L. Theor Appl Genet 106:461–469
Liu L, Zhu X, Gong Y, Song X, Wang Y, Zhao L, Wang L (2007) Genetic diversity analysis of radish germplasm with RAPD, AFLP and SRAP markers. Acta Hortic 760:125–130
Liu L, Yang J, Hou X, Gong Y, Zhu X, Wang F, Zhang C, Chu R (2008) Genotypic differences of Cd uptake and distribution in radish (Raphanus sativus L.). Bioinformatics and Biomedical Engineering (ICBBE) 4696–4699. doi:10.1109/ICBBE.2008.332
McGrath SP, Zhao FJ, Lombi E (2001) Plant and rhizosphere processes involved in phytoremediation of metal-contaminated soils. Plant Soil 232:207–214
Nakanishi H, Ogawa I, Ishimaru Y, Mori S, Nishizawa NK (2006) Iron deficiency enhances cadmium uptake and translocation mediated by the Fe2+ transporters OsIRT1 and OsIRT2 in rice. Soil Sci Plant Nutr 52:464–469
Saal B, Struss D (2005) RGA- and RAPD-derived SCAR markers for a Brassica B-genome introgression conferring resistance to blackleg in oilseed rape. Theor Appl Genet 111:281–290
Shirasawa K, Oyama M, Hirakawa H, Sato S, Tabata S, Fujioka T, Kimizuka-Takagi C, Sasamoto S, Watanabe A, Kato M, Kishida Y, Kohara M, Takahashi C, Tsuruoka H, Wada T, Sakai T, Isobe S (2011) An EST-SSR linkage map of Raphanus sativus and comparative genomics of the Brassicaceae. DNA Res 18:221–232
Smooker AM, Wells R, Morgan C, Beaudoin F, Cho K, Fraser F, Bancroft I (2011) The identification and mapping of candidate genes and QTL involved in the fatty acid desaturation pathway in Brassica napus. Theor Appl Genet 122:1075–1090
Suwabe K, Iketani H, Nunome T, Kage T, Hirai M (2002) Isolation and characterization of microsatellites in Brassica rapa L. Theor Appl Genet 104:1092–1098
Suwabe K, Tsukazaki H, Iketani H, Hatakeyama K, Kondo M, Fujimura M, Nunome T, Fukuoka H, Hirai M, Matsumoto S (2006) Simple sequence repeat-based comparative genomics between Brassica rapa and Arabidopsis thaliana: the genetic origin of clubroot resistance. Genet 173:309–319
Takahashi R, Ishimaru Y, Senoura T, Shimo H, Ishikawa S, Arao T, Nakanishi H, Nishizawa NK (2011) The OsNRAMP1 iron transporter is involved in Cd accumulation in rice. J Exp Bot 62:4843–4850
Tanhuanpää P, Kalendar R, Schulman AH, Kiviharju E (2007) A major gene for grain cadmium accumulation in oat (Avena sativa L.). Genome 50:588–594
Tezuka K, Miyadate H, Katou K, Kodama I, Matsumoto S, Kawamoto T, Masaki S, Satoh H, Yamagucji M, Sakurai K, Takahashi H, Satoh-Nagasawa N, Watanabe A, Fujimura T, Akagi H (2010) A single recessive gene controls cadmium translocation in the cadmium hyperaccumulating rice cultivar Cho-Ko-Kohu. Theor Appl Genet 120:1175–1182
Tsuro M, Suwabe K, Kubo N, Matsumoto S, Hirai M (2005) Construction of a linkage map of radish (Raphanus sativus L.), based on AFLP and Brassica-SSR markers. Breed Sci 55:107–111
Tsuro M, Suwabe K, Kubo N, Matsumoto S, Hirai M (2008) Mapping of QTLs controlling for root shape and red pigmentation in radish, Raphanus sativus L. Breed Sci 58:55–61
Ueno D, Kono I, Yokosho K, Ando T, Yano M, Ma JF (2009) A major quantitative trait locus controlling cadmium translocation in rice (Oryza sativa). New Phytol 182:644–653
Van Ooijen JW, Voorrips RE (2001) JoinMap Version 3.0, Software for the calculation of genetic linkage maps. Plant Research International, Wageningen
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTL. J Hered 93:77–78
Wang L, He Q (2005) China radish. Scientific and Technical Documents Publishing House, Beijing
Wang N, Hu J, Ohsawa R, Ohta M, Fujimura T (2007a) Identification and characterization of microsatellite markers derived from expressed sequence tags (ESTs) of radish (Raphanus sativus L.). Mol Ecol Notes 7:503–506
Wang S, Basten CJ, Zeng ZB (2007b) Windows QTL cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC
Wiebe K, Harris NS, Faris JD, Clarke JM, Knox RE, Taylor GJ, Pozniak CJ (2010) Targeted mapping of Cdu1, a major locus regulating grain cadmium concentration in durum wheat (Triticum turgidum L. var durum). Theor Appl Genet 121:1047–1058
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18:6531–6535
Xue D, Chen M, Zhang G (2009) Mapping of QTLs associated with cadmium tolerance and accumulation during seedling stage in rice (Oryza sativa L.). Euphytica 165:587–596
Yamamoto T, Yonemaru J, Yano M (2009) Towards the understanding of complex traits in rice: Substantially or superficially? DNA Res 16:141–154
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgments
This work was in part supported by grants from the Program for NCET, MOE (NCET-10-047), the National Key Technologies R & D Program of China (2009BADB8B03), Key Technology R & D Program of Jiangsu Province (BE2010328) and the Priority Academic Program Development of Jiangsu Higher Education Institutions(PAPD). We thank Dr. J. Hu from USDA-ARS for his critical review and helpful comments during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by I. Rajcan.
Rights and permissions
About this article
Cite this article
Xu, L., Wang, L., Gong, Y. et al. Genetic linkage map construction and QTL mapping of cadmium accumulation in radish (Raphanus sativus L.). Theor Appl Genet 125, 659–670 (2012). https://doi.org/10.1007/s00122-012-1858-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-012-1858-y