
Abstract
To date, only three types of full-length mariner elements have been described in ants, each one in a different genus of the Myrmicinae subfamily: Sinvmar was isolated from various Solenopsis species, Myrmar from Myrmica ruginodis, and Mboumar from Messor bouvieri. In this study, we report the coexistence of three mariner elements (Tnigmar-Si, Tnigmar-Mr, and Tnigmar-Mb) in the genome of a single species, Tapinoma nigerrimum (subfamily Dolichoderinae). Molecular evolutionary analyses of the nucleotide sequence data revealed a general agreement between the evolutionary history of most the elements and the ant species that harbour them, and suggest that they are at the vertical inactivation stage of the so-called Mariner Life Cycle. In contrast, significantly reduced levels of synonymous divergence between Mboumar and Tnigmar-Mb and between Myrmar and Botmar (a mariner element isolated from Bombus terrestris), relative to those observed between their hosts, suggest that these elements arrived to the species that host them by horizontal transfer, long after the species’ split. The horizontal transfer events for the two pairs of elements could be roughly dated within the last 2 million years and about 14 million years, respectively. As would be expected under this scenario, the coding sequences of the youngest elements, Tnigmar-Mb and Mboumar, are intact and, thus, potentially functional. Each mariner element has a different chromosomal distribution pattern according to their stage within the Mariner Life Cycle. Finally, a new defective transposable element (Azteca) has also been found inserted into the Tnigmar-Mr sequences showing that the ant genomes have been invaded by at least four different types of mariner elements.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Transposable elements (TEs) are repeated DNA segments able to change from one locus to another within the genome of their hosts. They fall into two main categories, class I and class II, according to their structure and transposition mechanism involving RNA or DNA molecules as intermediates for their mobility (Wicker et al. 2007).
Mariner elements are class II TEs. Mos1, isolated from Drosophila mauritiana, was the first mariner element discovered (Jacobson et al. 1986). Elements with similar motifs or mariner-like elements (MLEs) belong to a large and varied superfamily named IS630-Tc1-mariner (ITm), which is widespread and abundant in eukaryotic genomes (Bouuaert and Chalmers 2010; Zhou et al. 2011; Diao et al. 2011, among others). The ITm superfamily is further divided into several families based on the characteristics of their catalytic triad, D(Asp)DE(Glu) or DDD (Shao and Tu 2001; Diao et al. 2011). The catalytic motif of the mariner family is DD(34)D (three aspartic acid residues in conserved positions, the last two separated by 34 residues)(Robertson 1996; Lohe et al. 1997). MLEs are about 1.3 kb in length and have one intronless open reading frame (ORF) which encodes a transposase. They are flanked by inverted terminal repeats (ITRs), usually about 28–30 bp long, although they can be considerably larger (Leroy et al. 2003) and generally show the 5′-YYAGRT consensus at their outer ends (Langin et al. 1995). The conservation of two regions at positions 3–8 and 14–18 of the ITRs with sequence logos AGGTBK and WARRK, respectively, has also been reported (Lampe et al. 2001). MLE transposases have two domains, the N-terminal ITR-binding domain containing a helix–turn–helix (HTH) motif, and a C-terminal catalytic domain with the conserved catalytic triad. The conservation of the bipartite nuclear location signal (NLS), flanked by phosphorylation target sites of casein kinase II, has also been suggested (Lohe et al. 1997; Plasterk et al. 1999). The amino-acid sequences WVPHEL and YSPDLAP (I/S/T) separated by about 150 amino-acid residues, are also conserved motifs (Robertson and MacLeod 1993; Augé-Gouillou et al. 2005).
Most MLEs described to date, carry mutations that disrupt their ORFs (generate early stop codons and/or frameshifts) as well as degenerated ITRs, which cause a reduction or complete loss of their ability to transpose. The so-called Mariner Life Cycle includes three stages: the invasion of a new host by horizontal transfer (HT), proliferation in the host genome, and vertical inactivation as a result of the accumulation of mutations (Robertson and Asplund 1996; Hartl 2001).
Three criteria have been used to infer the existence of HT processes: (1) high sequence similarity between the TEs from phylogenetically distant hosts, (2) incongruence between the phylogenies of host and TEs, and (3) discontinuous distribution (patchy distribution) across a group of taxa (reviewed by Loreto et al. 2008). At present, the comparison between the nucleotide divergences at synonymous sites (K S) observed in TEs and in the nuclear genes of the hosts is considered the best method to infer HT processes. The rationale is that the time to the most recent common ancestor of horizontally transferred transposons might be significantly shorter than that between the species that host them, so that they have had less time to accumulate mutations than the nuclear genes of their hosts (Sánchez-Gracia et al. 2005; Bartolomé et al. 2009; reviewed by Schaack et al. 2010).
Hitherto, only three full-length mariner elements have been described in ants, each one in a different genus. They belong to the mauritiana subfamily, included in the mariner family (Lampe et al. 2003; Silva et al. 2005; Rouault et al. 2009). Sinvmar has been isolated from Solenopsis invicta and from other species from the Solenopsis genus (Krieger and Ross 2003), Myrmar from Myrmica ruginodis (Bigot et al. 1994; Rouleux-Bonnin et al. 2005), and Mboumar that has been isolated from Messor bouvieri (Palomeque et al. 2006). Previously, partial mariner elements had been isolated in the Tapinoma sessile and Crematogaster cerasi genomes (Robertson and MacLeod 1993). Among all ant MLEs, only Mboumar was found to codify a full-length active transposase (Palomeque et al. 2006; Muñoz-López et al. 2008). Until now, only three active transposons had been found: Mos1 (Medhora et al. 1991; Hartl 2001), Famar1 from the European earwig, Forfícula auricularia (Barry et al. 2004) and Mboumar-9 (Muñoz-López et al. 2008). In ants, the three types of mariner elements described have been studied only in the ant genera indicated, except the Myrmar element, which has also been found in the bumblebee Bombus terrestris (Hymenoptera, Apidae), a species also included in the order Hymenoptera but phylogenetically far from the ants (Rouleux-Bonnin et al. 2005).
The genetic system of the host species is a significant factor on the dynamics of the MLEs and other TEs (reviewed by Hua-Van et al. 2011). The ants (Hymenoptera, Formicidae) are haplodiploid with haploid males. In haploid males TEs with recessive deleterious effects will be unmasked and they will be exposed to selection. For this reason, they would be expected to be readily eliminated from the haplodiploid genomes (Hurst and Werren 2001), so that the incidence of TEs should be relatively low in these genomes. Bigot et al. (1994) found MLEs on different Hymenopteran species of wasps, bees and on the ant Myrmica ruginodis, as previously mentioned. The authors suggested that this wide distribution of MLEs could be explained by the lack of deleterious effects of these elements on the viability of insects due to their preferential insertion in a specific and conserved sequences where its insertion may be selectively neutral (Bigot et al. 1994; Rouleux-Bonnin et al. 2005). However, other authors have suggested that the conservation of these sequences may simply be the result of the ancient insertion of a mariner element before the divergence of the host species (Haine et al. 2007). Despite the fact that ant males do not undergo meiotic recombination, a high recombination rate has been found in the ants Acromyrmex echinatior and Pogonomyrmex rugosus and in several species of honeybees and wasps (Meznar et al. 2010; Niehuis et al. 2010; Sirviö et al. 2011). Different hypotheses have been proposed to explain this high recombination rate; generally it has been considered as evolutionarily selected among Hymenopteran insects, although the role of selection has not been demonstrated (Sirviö et al. 2006; Niehuis et al. 2010; Sirviö et al. 2011). Overall, the existence of a low or high genetic variability in ants is still a matter of debate (Viginier et al. 2004; Wilfert et al. 2007; Wysocka et al. 2011). Few studies have been made so far on the variability and incidence of MLEs in ant genomes.
This study aims to determine whether on the genome of the ant Tapinoma nigerrimum, coexist the three different MLEs described in other ant species, and to provide an estimate of their genetic diversity, chromosome location and life cycle stage. We conducted this study in T. nigerrimum because the genus Tapinoma belongs to the Dolichoderinae subfamily (Hymenoptera, Formicidae) whereas the genera Myrmica, Solenopsis and Messor belong to the Myrmicinae subfamily (Hymenoptera, Formicidae). The divergence between both subfamilies seems to have occurred about 110 million years ago (Moreau et al. 2006). If any of the three full-length mariner element described in ants were present on Tapinoma, the comparison of the phylogenies of mariners and their host species might provide interesting data on the life cycle of the these mariner elements. To amplify the mariner sequences, primers complementary to conserved regions of the transposons were used. A given species may contain several types of mariner in its genome if it has suffered multiple invasion events. Consequently, the lack of congruence between phylogenies may be due to the comparison of non-orthologous sequences. For this reason and in agreement with other authors (Krieger and Ross 2003) we have used specific primers enabling the amplification of a single type of element. The chromosomal localization of TEs is another important feature. TEs are not distributed at random throughout the chromosomes. They are frequently located in constitutive heterochromatin, especially centromeres and telomeres, where their potential deleterious effect is limited (Charlesworth et al. 1994; Bartolomé et al. 2002; Fontanillas et al. 2007). It has also been suggested that TEs could be involved in centromere and telomere function (reviewed by Hua-Van et al. 2011). Detailed cytogenetic studies are very limited in ants (Lorite and Palomeque 2010) being T. nigerrimum one of the few exceptions, a fact that allows the present study.
Material and methods
Material, DNA extraction, PCR amplification and cloning
Tapinoma nigerrimum and Messor bouvieri adult workers were sampled in the province of Jaén (Spain). Solenopsis invicta was collected in the Louisiana University Campus (Louisiana, USA) and Myrmica ruginodis in Tours (France). Genomic DNA was extracted from pools of 10–15 ants from each sample, following Heinze et al. (1994).
Specific primers were designed in order to amplify the different mariner elements. For Myrmar-like insertions a unique primer was designed using the ITR sequence of the published Myrmar mariners (Bigot et al. 1994; Rouleux-Bonnin et al. 2005) (Mrug-MAR 5′-CCAGGTCTGTAAATATGAAACCGGAAT). Only one primer was necessary since the 5′-ITRs and the 3′-ITRs of these elements are highly conserved. Also, only a unique primer was necessary for amplification of the Mboumar-like mariners. This primer (ITR-MAR 5′-CCAGGTGTGTCGGTAATTCCTTTCCGG) was based on the ITR sequences of Mboumar mariner (Palomeque et al. 2006). On the contrary, since 5′-ITRs and 3′-ITRs of Sinvmar are known to be different (Krieger and Ross 2003), two primers were used for Sinvmar-like mariner amplification (Sinv-mar-1 5′-TTAGGTGTTAAACTTAATTCCTGCCGCT and Sinv-mar-2 5′-AATTGAAGGTAACTTAATTCCTGCCGTT).
PCR amplifications were initially denatured at 92 °C for 2 min and performed using the following cycling profile: 30 cycles at 92 °C (30 s), 50 °C (30 s), 72 °C (2 min), with a final elongation step of 72 °C for 5 min. Reactions were set up in a 50-μl mixture containing 100 ng of genomic DNA, 0.5 mM dNTPs, 50 pmol of the primer and 1 U of Taq polymerase. The amplified fragments were analyzed by electrophoresis in agarose gels, eluted from agarose gel and cloned into the pGEM-T Easy vector (Promega). For all PCRs, only the most intense and clear fragments were studied. Recombinant plasmids were sequenced on both strands by the dideoxy sequencing method.
Three nuclear gene fragments were amplified in the ant species; wingless (wnt-1), abdominal-A (abdA) and long-wavelength rhodopsin (lw Rh). The wnt-1 fragment was amplified using the primers Wg578F (5′-TGCACNGTGAARACYTGCTGGATGCG) and Wg1032R (5′-ACYTCGCAGCACCARTGGAA) (Abouheif and Wray 2002; Ward and Downie 2005). For the abdA gene the primers ant-M (5′-CGGCACCGGCGATATGAGTACGAAATTC) and ant-J (5′-GGGTTGTTGGCAGGATGTCAAAGGATG) (De Menten et al. 2003) were used. The lw Rh gene fragment was amplified using the primers LR143F (5′-GACAAAGTKCCACCRGARATGCT) and LR639ER (5′-YTTACCGRTTCCATCCRAACA) (Ward and Downie 2005). PCR reactions were initially denatured at 94 °C for 2 min and then subjected to 35 cycles at 94 °C (60 s), 45–56 °C (60 s), 72 °C (2 min), with a final elongation step of 72 °C for 5 min. Reactions were set up in a 50-μl mixture containing 100 ng of genomic DNA, 0.5 mM dNTPs, 50 pmol of the primer and 1 U of Taq polymerase. The amplified fragments were analyzed by electrophoresis in agarose gels. Eluted fragments were directly sequenced on both strands using the same primers used for PCR amplification.
Sequence analyses
Multiple-sequence alignments were initially performed using CLUSTALW (Larkin et al. 2007) and MUSCLE (Edgar 2004), and corrected by hand in order to maintain the open reading frame of the coding sequences. Sequence comparisons, ORF search, and other sequence analyses were performed using the available online programs from NCBI (http://www.ncbi.nlm.nih.gov/guide/). The NSP@Network Protein Sequence Analysis program was used for the prediction of helix–turn–helix motifs (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_hth.html). The putative TATA box and polyadenylation signals were determined using the programs HCtata (Hamming Clustering Method for TATA Signal Prediction in Eukaryotic Genes) and HC polya (Hamming Clustering Poly-A Prediction in Eukaryotic Genes). The phosphorylation target sites at casein kinase II were determined using the Motif Finder program (http://motif.genome.jp/). Searches of repetitive element-related sequences were performed using RepeatMasker Web Server (http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker).
Molecular evolutionary analyses
The level of genetic diversity among groups of sequences was estimated by means of π (Nei 1987), which measures the average number of nucleotide differences per site between pairs of sequences, using the Jukes and Cantor correction. Standard errors were estimated by bootstrap (1,000 replications).
Evolutionary distances between groups of sequences were measured as the average number of pairwise nucleotide differences per site. Estimates of nucleotide divergence at all sites were obtained using the Maximum Composite Likelihood method (Tamura et al. 2004), and at synonymous sites (K S) using the Kumar model (Nei and Kumar 2000). Standard errors of these estimates were calculated by bootstrap (1,000 replicates). These calculations were made with the aid of MEGA 5.0 (Tamura et al. 2011).
Phylogenetic relationships among the mariner sequences we explored using neighbor-joining (NJ) and maximum likelihood (ML). The nucleotide substitution models were evaluated using MEGA 5.0, the models with the lowest BIC scores (Bayesian Information Criterion) were considered better to describe the substitution pattern (Tamura et al. 2011). Using this criterion the best model chosen was Tamura 3-parameter assuming a fraction of sites evolutionarily invariable (T92+I). NJ and ML trees were constructed using MEGA 5.0 (Tamura et al. 2011). Bootstrap values for each branch were assessed from 1000 replicates in both cases. The mariner Ammar1 (Genbank accession no. AY154751) from Apis mellifera was used as an out-group in the phylogenetic analyses. This mariner belongs to the mellifera subfamily (Lampe et al. 2003).
In order to determinate the phylogenetic relationships of the mariners described in this report with other mariner elements, phylogenetic analyses were also performed using transposase amino acid sequences from several representative mariner subfamilies (Lampe et al. 2003; Krieger and Ross 2003; Bui et al. 2008) (Table S1). In the phylogenetic analyses, the Fusarium oxysporum impala transposase (GenBank accession no. AAB33090) was used as an out-group as Krieger and Ross (2003). For Mboumar and Tnigmar-Mb mariner their putative transposase sequences were used. For the Solenopsis species mariner, Tnigmar-Si, Myrmar and Tnigmar-Mr the transposase of the more likely ancestral active mariner was built using the consensus sequences. The best amino acid substitution model was evaluated using MEGA 5.0 (Tamura et al. 2011). Results showed that the best-fit amino acid substitution model was the WAG model with a proportion of invariant sites (WAG+I). ML trees were built using MEGA 5.0 and support values were determined by bootstrap analyses with 1,000 replicates.
In situ hybridization procedures
Chromosome spreads were obtained from adult male gonads. In situ hybridization was carried out as described previously (Lorite et al. 2002; Palomeque et al. 2005). The mariner probes (Tnigmar-Si-16, Tnigmar-Mr-3, and Tnigmar-Mb-1) were labeled with biotin-16-dUTP using a biotin nick translation kit (Roche). Fluorescence immunological detection was performed using the avidin–FICT/anti–avidin–biotin system with three rounds of amplification. The preparations were counterstained with propidium iodide and DAPI. FISH was carried out in high-stringency conditions to avoid co-hybridization among different mariner types (Palomeque et al. 2005). The temperature used for hybridization was 37 °C and post-hybridization washes were performed at 42 °C in 50 % formamide. These stringency conditions allow hybridization between DNA–DNA duplex sharing approximately 80–85 % sequence homology (McClean 1998).
Results
Isolation and sequence analyses of mariner elements
Three independent PCR amplification assays were performed using T. nigerrimum DNA as a template and specific primers for the ITR sequences of Sinvmar (Krieger and Ross 2003), Myrmar (Bigot et al. 1994; Rouleux-Bonnin et al. 2005) and Mboumar (Palomeque et al. 2006) mariner TEs. A thin band of about 1.3 kb, three clear amplification bands with different sizes (about 2,200, 900, and 400 bp) and an intense band of about 1.3 kb, were obtained with each primer set, respectively (Fig. S1).
BLAST analyses from the cloned PCR products revealed that the sequences were highly homologous to the elements used to design the primers. Following the nomenclature proposed by Robertson and Asplund (1996) with our own modification, the mariner elements were named Tnigmar-Si, Tnigmar-Mr, and Tnigmar-Mb (T. nigerrimum mariner similar to the S. invicta mariner, M. ruginodis mariner and M. bouvieri mariner, respectively). In addition, a new defective MLE sequence was found in Tapinoma nigerrimum. It was inserted into Tnigmar-Mr. BLAST analyses showed that it displays high homology with a sequence present in the Azteca instabilis ant genome and we named it Tnigmar-Az. The features of this MLE will be exposed later.
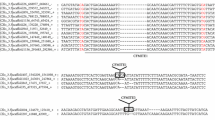
Six Tnigmar-Si sequences were obtained (Tnigmar-Si-1, -3, -5, -16, -17 and -20; Fig. S2, GenBank accession no. HE577153 to HE577158). They are very similar, with just six segregating variants (π = 0.001 ± 0.0006). The ORF of all sequences are interrupted by stop codons and frameshift mutations which are likely to render them inactive. Tnigmar-Si displays little divergence form other mariner elements described in Solenopsis invicta (Sinvmar) as well as in other species form the same genus, such as S. macdonaghi (Smacmar), S. richteri (Sricmar), and S. saevissima (Ssaemar) (Krieger and Ross 2003), with an average synonymous divergence of the order of 0.23 (Table 1). The consensus sequence of Tnigmar-Si is shown in Fig. 1. Conceptual translation and their phylogenetic analyses showed that these elements belong to the mauritiana subfamily of the mariner elements (Fig. 3 and Fig. S3). From here on, they will be referred to as the Solenopsis mariner group.

Alignment of the consensus sequences of the three full length different types of isolated mariner elements: Tnigmar-Si, Tnigmar-Mr, and Tnigmar-Mb. The corresponding ITRs are shaded
In a second set of experiments using the primers specific for Myrmar, we obtained sequences from ten different clones. Six of them were 2,174–2,175 bp long (Tnigmar-Mr-1, -2, -4, -5, -6, and -7. GenBank accession no. HE577159 to HE577164) and were interrupted by the insertion of a 901- to 902-bp fragment of another MLE at position 967 of the alignment (Fig. 2), which we have named Tnigmar-Az (see above). The six Tnigmar-Mr sequences display little sequence diversity, with just seven singleton variants in the remaining 1,279 nucleotides (π = 0.003 ± 0.002). The phylogenetic analysis of Tnigmar-Mr revealed that they were closely related to Myrmar, and also to Botmar of Bombus terrestris (Rouleux-Bonnin et al. 2005) and as shown in Fig. 3. The consensus of Tnigmar-Mr sequences is shown in Fig. 1. The putative ORFs of the Tnigmar-Mr sequences were interrupted by several mutations causing frameshifts and early stop codons. Conceptual translation of Myrmar, Botmar and Tnigmar-Mr elements and their phylogenetic study suggest that they also belong to the mauritiana subfamily of the mariner element and from here on they will be referred to as the Myrmica mariner group (Fig. 3 and Fig. S3). The other four clones (Tnigmar-Mr-3, -8, -9, and -11) corresponded to internally deleted forms of Tnigmar-Mr sequences (Fig. 2 and Fig. S4) (GenBank accession no. HE5771657, HE577165, HE577166, and HE577168).

Schematic representation of the clones obtained using as a primer the ITRs of the mariner transposable element Myrmar isolated from the ant Myrmica ruginodis (Bigot et al. 1994; Rouleux-Bonnin et al. 2005), showing the Azteca element (Tnigmar-Az) inserted into a Tnigmar-Mr mariner (a and b). The duplication of TA dinucleotide and the nucleotide insertion site are also represented. The nucleotide positions of the internal deletions shown by clones of 900–901 bp (c) and 392 bp (d) are also indicated. Only clones marked with an asterisk have been obtained by PCR amplification techniques

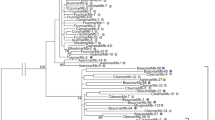
Maximum-likelihood tree depicting the phylogenetic relationships of the mariner elements. The sequences of four elements were retrieved from GenBank: Ssaemar1.1 (Solenopsis saevissima), AF518177; Sricmar1.2 (Solenopsis richteri), AF518176; Smacmar1 (Solenopsis macdonaghi), AF518174 and Ammar1 (AY155490). Ammar1 a mariner element from Apis mellifera was used as outgroup. When several sequences for the same mariner were available consensus sequences were used: Botmar (Bombus terrestris, AJ312716, AJ312717, AJ312720, AJ312722); Sinvmar (Solenopsis invicta, AF518169 to AF518173), and Myrmar (Myrmica ruginodis, AY652423 to AY652426). Branches with bootstrap support values greater than 80 % are indicated. The scale bar represents the number of substitutions per site units. Neighbor-joining (NJ) methods produced similar topologies
Four sequences, Tnigmar-Mb-1, -2, -3 and -5, were obtained with the primers specific for Mbourmar (GenBank accession no. HE577149 to HE577152). Again, the sequences displayed reduced genetic diversity (π = 0.004 ± 0.0011). The Tnigmar-Mb consensus sequence (Fig. 1) included a full-length ORF. This element is nearly identical (K S =0; Table 1) to Mboumar previously described in M. bouvieri (Palomeque et al. 2006). A phylogenetic analysis suggests that these two elements belong to the mauritiana subfamily of the mariner elements, and from here on they will be named the Messor mariner group (Fig. 3 and Fig. S3).
Figure 4 shows the alignment between Mboumar-9 and the sequences isolated from T. nigerrimum. These sequences showed a high nucleotide identity. The ITRs of Tnigmar-Mb elements showed conservation of the consensus 5′-YYAGRT motif (Langin et al. 1995) and the other nucleotide positions considered important in the transposition processes (Lampe et al. 2001). The two ITRs from Tnigmar-Mb were perfect inverted repeat sequences as a result of the amplification technique used, showing two nucleotide differences at 3´-ITR in relation to Mboumar-9 and only one in relation to other Mboumar elements. Tnigmar-Mb also had a putative TATA box (position 57), except on Tnigmar-Mb-5, and a polyadenylation signal (position 1206) like Mboumar-9. A conceptual translation and search for ORF showed that Tnigmar-Mb-5 has a stop codon (Fig. 4). However, Tnigmar-Mb-1, -2, and -3 contained a full-length open reading frame which codified a putative protein with a very strong similarity to the active Mboumar transposase (Muñoz-López et al. 2008). The putative transposase codified by Tnigmar-Mb contains the conserved DD(34)D motif and all the characteristic motifs of an active transposase (Fig. 4.) All together, the data suggest that Tnigmar-Mb is an active TE.

Alignment of the Mboumar-9 sequence (Palomeque et al. 2006) and all Tnigmar-Mb sequences. The ITRs are shaded. The putative proteins codified by Mboumar-9 and Tnigmar-Mb sequences are also shown as well as the TATA box, the bipartite nuclear location signal (NLS), the conserved D,D(34)D motif and other important features. An asterisk indicates the stop codon in the ORF on Tnigmar-Mb-5 sequence
Phylogenetic analyses
The three full-length mariner elements isolated from Tapinoma nigerrimum belong to the mauritiana subfamily although they were clearly different as revealed by the large levels of synonymous nucleotide divergence between them. K S between Tnigmar-Mr and Tnigmar-Mb is 0.76 ± 0.100, and between them and Tnigmar-Si slightly exceeds 1.00 in both cases (Table 1).
The evolutionary relationships among the MLEs found in T. nigerrimum and those from other ants and the bumblebee are depicted in Fig. 3. The phylogenetic tree shows three highly supported clades that correspond to the Solenopsis, Messor and Myrmica mariner groups previously described. Tnigmar-Si and Tnigmar-Mr are well differentiated from the other members of their respective groups, which is consistent with the expected phylogenies of their hosts and supports the hypothesis that these elements have been vertically transmitted (Fig. 3). The phylogenetic analyses using all sequences of each type of mariner also showed the same results (Fig. S5). Contrastingly, Myrmar is more closely related to Botmar that to Tnigmar-Mr which seems at odds with the host’s phylogeny, since Botmar was isolated from Bombus terrestris, a bumblebee from a different Hymenoptera family. Furthermore, Mboumar and Tnigmar-Mb clustered together in a single monophyletic group and they are nearly identical (see above). Such high similarity is unexpected as their host species belong to different ant subfamilies (Myrmicinae and Dolichoderinae, respectively), whose split has been dated about 110 mya (Moreau et al. 2006).
Estimates of K S between the mariner element and same data from nuclear genes are shown in Tables 1 and 2, respectively. We obtained the nucleotide sequences of three single-copy nuclear genes, abdominal-A (abdA), long wavelength rhodopsine (lw Rh) and wingless (wnt-1) from T. nigerrimum, and of lw Rh and wnt-1 from M. bouvieri and M. ruginodis (GenBank accession no. HE963096 to HE963102). We also retrieved from GenBank sequences of these same loci from S. invicta, B. terrestris and other species of all genera (Table S2). Although we were unable to obtain the abdA sequence from M. bouvieri samples after repeated attempts, this locus has been sequenced in other Messor species (M. julianus, M. denticornis and M. andrei; Brady et al. 2006; Table S2). The synonymous differentiation between T. nigerrimun and M. bouvieri at lw Rh and wnt-1 were 0.28 ± 0.083 and 0.40 ± 0.080, respectively. Mean K S at abdA locus between different Messor species (Table S2) and T. nigerrimun was of the order of 0.42, which was not significantly different from that observed at the other two loci between T. nigerrimum and M. bouvieri. These values are in good agreement with our estimate of average K S = 0.36 ± 0.010 across the three loci between various species of these two genera (Table 2).
Chromosomal location of mariner elements
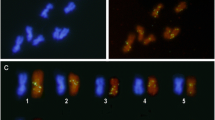
The chromosome number of T. nigerrimum is n = 9. All chromosomes showed heterochromatin on pericentromeric regions according to standard C-banding technique results (Palomeque et al. 1988). The results showed that each type of mariner element has a different hybridization pattern (Fig. 5). Only one or two positive hybridization signals in some chromosomes were found when Tnigmar-Si sequence was used as probe. In addition, the positive signals were not located in the pericentromeric regions (Fig. 5a). On the contrary, Tnigmar-Mr and specially Tnigmar-Mb were more widespread in the genome of T. nigerrimum and numerous hybridization signals were visible in all chromosomes (Fig. 5b, and c). These signals appeared both in pericentromeric regions as in the euchromatic chromosome arms. In this species the rDNA genes are located in the proximal region of short arm of chromosome 6 (Lorite et al. 1997). This region presented hybridization signals when Tnigmar-Mr or Tnigmar-Mb was used as a probe (Fig. 5c and b). However, the distribution patterns of both types of mariner were not the same. First and overall, positive hybridization signals were more numerous when Tnigmar-Mb was used as a probe. In addition there were regions with numerous copies of the Tnigmar-Mb but few or none of the Tnigmar-Mr (e.g., the long arm of chromosome 2), with the opposite pattern (e.g., the long arm of chromosome 4) or with a similar hybridization patterns (e.g., chromosome 5) (Fig. 5b3 and c3).

Chromosomal location of the three types of mariner elements. Metaphase plates of T. nigerrimum with chromosomes stained with DAPI fluorochrome (a, d, and g) and in situ hybridization with Tnigmar-Si (b), Tnigmar-Mr (e), and Tnigmar-Mb (h) as probes. Karyotypes showing the different hybridization pattern obtained with Tnigmar-Si (c), Tnigmar-Mr (f), and Tnigmar-Mb (i). Bar = 5 μm
Analysis of new transposable elements-related sequences
Inserted into Tnigmar-Mr sequences, we found a sequence related to the TE, as mentioned above. Tnigmar-Az was inserted into a TA dinucleotide, as usual for mariner elements (Fig. 2 and Fig. S4). All sequences were very similar, with 901–902 bp in length and with perfect ITRs of 29–30 bp (Fig. S6). They could codify a polypeptide (208 aa) according to the results obtained using the ORF Finder program from NCBI. DELTA-BLAST (Boratyn et al. 2012) analysis showed that the conceptual translated nucleotide presented a possible motif HTH and part of the catalytic motif of the MLEs transposase. The comparison between the 208-aa polypeptide and the transposases of other MLEs suggested that the Tnigmar-Az sequences might have been generated by an internal deletion from a full length mariner which would include part of the catalytic motif, consequently the polypeptide would not active (Fig. S7). On the insertion into Tnigmar-Mr could be involved an active Tnigmar-Az in which the internal deletion took place quickly or a deleted sequence and a trans-acting transposase generated by a functional transposon. The phylogenetic analyses indicated that Tnigmar-Az was near to the marmoratus mariner subfamily (Fig. S3), defined by Bui et al. (2008).
This element was also present in the Azteca instabilis ant genome and we named it Ainsmar-Az. Ainsmar-Az was found inserted into the TA dinucleotide (position 507) from the carbomoylphosphate synthase (CAD) gene (Ward et al. 2010; GenBank accession no. FJ939902). It was inserted into the first intron of the gene sequence according to the study of corresponding protein (GenBank accession no. ADD13205.1) (Fig. S8). A similar sequence was found in a BAC clone from the butterfly Heliconius melpomene (Lepidoptera, Nymphalidae) (GenBank accession no. CU856076) (Fig. S9). It was also inserted into a TA dinucleotide (position 15.872). In addition, the Ainsmar-Az sequence was present only in the gene of A. instabilis, but not in the same gene from related species (Ward et al. 2010, GenBank accession no. FJ939903 and FJ939904). This fact suggests that at some point in time it had to be active. Similar results have been reported in the study of Bmmar2, a type of mariner element isolated from the silkworm Bombyx mori (Lepidoptera, Bombycidae) and in other related species (Kumaresan and Mathavan 2004), with incomplete versions of the transposon inserted in non-coding regions of some genes.
Discussion
In this study, we report the coexistence of three mariner elements (Tnigmar-Si, Tnigmar-Mr, and Tnigmar-Mb) in the genome of a single species, Tapinoma nigerrimum (subfamily Dolichoderinae). They belong to the mauritiana subfamily of MLEs and are closely related to the three different types of mariner TEs that have been described in three ant genera of the Myrmicinae subfamily: Solenopsis, Myrmica, and Messor.
Tnigmar-Si sequences showed high similarity and they were predictably inactive. Similar results have been reported by Krieger and Ross (2003) from other mariner elements described in Solenopsis invicta (Sinvmar) and in other species of the same genus (Krieger and Ross 2003). The high similarity between Tnigmar-Si sequences could be explained by biological processes such as their recent non-autonomous mobilization by transposases coded by other active transposons, or a high rate of gene conversion among insertions. However, the facts that all the substitutions are found just in a single sequence of the samples (singletons) and that a similar scenario was obtained for most other elements in this, as well as other studies (Krieger and Ross 2003; Bigot et al. 1994; Rouleux-Bonnin et al. 2005; Palomeque et al. 2006), mean that we cannot discard the possibility that these singletons substitutions correspond to nucleotide miss-incorporations during the PCR amplification procedure.
The Tnigmar-Mr elements were closely related to Myrmar of M. ruginodis, and also to Botmar of Bombus terrestris (Rouleux-Bonnin et al. 2005). The Tnigmar-Mr sequences also were predictably inactive sequences. A similar situation occurred with the mariner sequences from M. ruginodis and B. terrestris (Bigot et al. 1994; Rouleux-Bonnin et al. 2005). Elements corresponding to internally deleted forms of Tnigmar-Mr were also found. Deletion derivatives have also been isolated in M. ruginodis and B. terrestris by Bigot et al. (1994) and Rouleux-Bonnin et al. (2005) as well as other organisms (Rezende-Teixeira et al. 2010, among others). The transposition mechanism of “cut-and-paste” of these elements requires the repair of the gaps left in the excision process; incorrect repairs may give rise to the observed deletions, as suggested by other authors (Brunet et al. 2002).
A new defective MLE sequence Tnigmar-Az was inserted into Tnigmar-Mr. It showed the structure of a mariner element with an internal deletion. Tnigmar-Az was inserted always in the same position into Tnigmar-Mr sequences suggesting a single transposition event, followed by the subsequent transposition of the surrounding element. A similar sequence was found inserted into an intron of a gene from Azteca instabilis ant. It is the first time in Hymenoptera that a mariner has been described inserted into an intron of a gene. This situation is very common in mammalian genomes (Sironi et al. 2006) and probably also in non-mammalian vertebrates and invertebrates genomes (Sela et al. 2010).
Evidence for horizontal transfer events of MLEs
The patterns of genetic divergence and the phylogenetic relationships of Tnigmar-Mb and Mbourmar, as well as those between Myrmar, Botmar and Tnigmar-Mr do not seem to be consistent with the expectations assuming vertical transmission. In principle, the discrepancies between the phylogenetic relationships of the elements and those of their hosts could have two explanations. One is that the mariner elements described in this study splitted from a common ancestor, before the divergence of the ants and the bumble-bees, and have been transmitted vertically ever since; only one of them, more closely related to Myrmar, has been found in B. terrestris (Botmar). However, the identity between Tnigmar-Mb and Mboumar could not be explained under this scenario. Alternatively, the closer relationships observed between Botmar and Myrmar and between Tnigmar-Mb and Mboumar could be explained by recent HT events of these elements to their current hosts. One way to address this problem is to compare the phylogenies of the elements and the species that harbor them. As a proxy for the patterns of neutral divergence among the host species we estimated K S and compared them with analogous estimates from the elements. With this purpose we estimated K S of three single-copy nuclear genes between the host’s species and the analogous estimates from the mariner elements. We have estimated an average K S = 0.36 ± 0.010 (Table 2) across the three loci between various species of Tapinoma and Messor genera. Under the assumption that coding sequences of TEs and host genes experience a similar mutation rate (this is discussed at length in Bartolomé et al. 2009), the fact that the elements isolated from T. nigerrimum and M. bouvieri are nearly identical, suggests that they have not been inherited by vertical transmission, and that recent HT processes were involved in the evolution of these mariners. The observed K S between Botmar and Myrmar (0.04 ± 0.010 from Table 1) was nearly 20 times smaller than that found between the two host species they were described in (mean K S = 1.05 ± 0.099 from Table 2). Again, the most parsimonious explanation is that a common ancestor of Botmar and Myrmar entered the genomes of one (or both) of these species long after their split. However, the greater divergence between the two elements, together with the fact that none of the sequenced insertions contained an intact coding sequence suggests that this HT event took place a long time ago.
If we consider that the divergence time for the Myrmicinae and Dolichoderinae subfamilies is of around 110 mya (Moreau et al. 2006), and that the mean K S for abdA, lw Rh and wnt-1 between several species representative of the genera Messor, Solenopsis and Myrmica from Myrmicinae, and four species of Tapinoma (Dolichoderinae) is 0.33 ± 0.015 (from Table 2), the mean divergence rate would be of 0.0014 ± 6.5 × 10−5 substitutions per nucleotide per mya (the rate of neutral divergence equals the observed divergence divided by the time to the most recent common ancestor of the species compared multiplied by two; Li 1997). Thus, the HT of the element could be roughly dated around 14.3 ± 3.57 mya. Contrastingly, the neutral divergence rate estimated for other model insect species such as Drosophila is significantly larger (0.011; Tamura et al. 2004). Using this estimate, the HT event would be dated about 1.8 ± 0.455 mya. Given that molecular clock rates are known to vary across organisms and over time (Li 1997; Nei and Kumar 2000), we are inclined to trust the dating from the molecular rate obtained from the ant data, although it was estimated using fewer nuclear loci.
On the other hand, the full identity of the consensus coding sequences of Tnigmar-Mb and Mboumar suggests that the time since the HT event approximates zero. If we assume that neutral mutations follow a Poisson distribution, the probability of observing exactly zero changes is e −m, where m is the expected number of changes. If we allow for the probability of zero to be 0.05, m = 2.996. The upper limit for the time since the HT event can then be estimated as (2.996/512)/(2 × 0.0014) = 2 million years, where 512 is the total number of synonymous sites analyzed. However, caution is recommended when interpreting these results. It should also be noted that the HT hypothesis is further supported by the fact that the consensus sequence of the four Tnigmar-Mb elements have a full length intact ORF, which means that they are or have recently been potentially active, as demonstrated in vitro for Mboumar (Muñoz-López et al. 2008). It is likely that TE activity is needed for HT, as extrachromosomal copies of the elements produced during transposition are expected to be more easily transported to new hosts, and new copies need to be integrated into their genomes.
Chromosome distribution and Mariner Life Cycle
The chromosome localization of Tnigmar-Si, Tnigmar-Mr, and Tnigmar-Mb mariner TEs showed that each type of mariner element has a different hybridization pattern, which could reflect that these elements are at different stages of the Mariner Life Cycle in the host genomes. When a young and active transposon invades a new host by horizontal transmission, it has to multiply within the genome and colonize the germ line to expand within the species and population. At the same time mutations may be accumulated giving rise to inactive or partially inactive copies (reviewed by Miskey et al. 2005). At this stage the transposon copies should be numerous and scattered all over the genome. Consequently, widespread hybridization signals should be detected on the host’s chromosomes. The hybridization patterns obtained in the study of Tnigmar-Mb element suggest that it could be at this stage of the mariner cycle. Mutation accumulation causes the vertical inactivation of the elements, which eventually determines their stochastic loss from the genomes. Thus, the transposon to survive could be horizontally transferred to new host and the cycle would begin again (reviewed by Miskey et al. 2005). MLEs, especially in an inactive state, may persist in a genome through evolutionary time including speciation processes (Hartl et al. 1997). Although we cannot discard the possibility that other intact copies may still exist undetected in the ants’ genomes, the fact that all Tnigmar-Si and Tnigmar-Mr copies were defective due to accumulation of null mutations suggest that these two families could be in the decaying phase of their cycle, with comparatively reduced copy numbers, approaching their stochastic loss from their host genomes.
References
Abouheif E, Wray GA (2002) Evolution of the gene network underlying wing polyphenism in ants. Science 297:249–252
Augé-Gouillou C, Brillet B, Germon S, Hamelin MH, Bigot Y (2005) Mariner Mos 1transposase dimerizes prior to ITR binding. J Mol Biol 351:117–130
Barry EG, Witherspoon DJ, Lampe DJ (2004) A bacterial genetic screen identifies functional coding sequences of the insect mariner transposable element Famar1 amplified from the genome of the earwig, Forficula auricularia. Genetics 166:823–833
Bartolomé C, Maside X, Charlesworth B (2002) On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol Biol Evol 19:926–937
Bartolomé C, Bello X, Maside X (2009) Widespread evidence for horizontal transfer of transposable elements across Drosophila genomes. Genome Biol 10:R22
Bigot Y, Hamelin MH, Capy P, Periquet G (1994) Mariner-like elements in hymenopteran species, insertion site and distribution. Proc Natl Acad Sci USA 91:3408–3412
Boratyn GM, Schaffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL (2012) Domain enhanced lookup time accelerated BLAST. Biol Direct 17:12
Brady SG, Schultz TR, Fisher BL, Ward PS (2006) Evaluating alternative hypotheses for the early evolution and diversification of ants. Proc Natl Acad Sci USA 103:18172–18177
Brunet F, Giraud T, Godin F, Capy P (2002) Do deletions of Mos1-like elements occur randomly in the Drosophilidae family? J Mol Evol 54:227–234
Bouuaert C, Chalmers R (2010) Transposition of the human Hsmar1 transposon: rate-limiting steps and the importance of the flanking TA dinucleotide in second strand cleavage. Nucleic Acids Res 38:190–202
Bui QT, Casse N, Leignel V, Nicolas V, Chénais B (2008) Widespread occurence of mariner transposons in coastal crabs. Mol Phylogenet Evol 47:1181–1189
Charlesworth B, Jarne P, Assimacopoulos S (1994) The distribution of transposable elements within and between chromosomes in a population of Drosophila melanogaster. Drosophila melanogaster: III. Genet Res 64:183–197
De Menten L, Niculita H, Gilbert M, Delneste D, Aron S (2003) Fluorescence in situ hybridization: a new method for determining primary sex ratio in ants. Mol Ecol 12:1637–1648
Diao Y, Qi Y, Ma Y, Xia A, Sharakhov I, Chen X, Biedler J, Ling E, Tu ZJ (2011) Next-generation sequencing reveals recent horizontal transfer of a DNA transposon between divergent mosquitoes. PLoS One 6:e16743
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Fontanillas P, Hartl DL, Reuter M (2007) Genome organization and gene expression shape the transposable element distribution in the Drosophila melanogaster euchromatin. PLoS Genet 3:e210
Haine ER, Kabat P, Cook JM (2007) Diverse mariner-like elements in fig wasps. Insect Mol Biol 16:743–752
Hartl DL (2001) Discovery of the transposable element mariner. Genetics 157:471–476
Hartl DL, Lohe AR, Lozovskaya ER (1997) Modern thoughts on an ancyent marinere: function, evolution, regulation. Annu Rev Genet 31:337–358
Heinze J, Gadau J, Hölldobler B, Nanda I, Schmid M, Scheller K (1994) Genetic variability in the ant Camponotus floridanus detected by multilocus fingerprinting. Naturwissenschaften 81:34–36
Hua-Van A, Le Rouzic A, Boutin TS, Filée J, Capy P (2011) The struggle for life of the genome's selfish architects. Biol Direct 6:19
Hurst GDD, Werren JH (2001) The role of selfish genetic elements in eukaryotic evolution. Nat Rev Genet 2:597–606
Jacobson JW, Medhora MM, Hartl DL (1986) Molecular structure of a somatically unstable transposable element in Drosophila. Proc Natl Acad Sci USA 83:8684–8688
Kumaresan GS, Mathavan S (2004) Molecular diversity and phylogenetic analysis of mariner-like transposons in the genome of the silkworm Bombyx mori. Insect Mol Biol 13:259–271
Krieger MJB, Ross KG (2003) Molecular evolutionary analyses of mariners and other transposable elements in fire ants (Hymenoptera, Formicidae). Insect Mol Biol 12:155–165
Langin T, Capy P, Daboussi MJ (1995) The transposable element Impala, a fungal member of the Tc1-mariner superfamily. Mol Gen Genet 246:19–28
Lampe DJ, Walden KKO, Robertson HM (2001) Loss of transposase-DNA interaction may underlie the divergence of mariner family transposable elements and the ability of more than one mariner to occupy the same genome. Mol Biol Evol 18:954–961
Lampe DJ, Witherspoon DJ, Soto-Adames FN, Robertson HM (2003) Recent horizontal transfer of mellifera subfamily mariner transposons into insect lineages representing four different orders shows that selection acts only during horizontal transfer. Mol Biol Evol 20:554–562
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Leroy H, Castagnone-Sereno P, Renault S, Augé-Gouillou C, Bigot Y, Abad P (2003) Characterization of Mcmar1, a mariner-like element with large inverted terminal repeats (ITRs) from the phytoparasitic nematode Meloidogyne chitwoodi. Gene 304:35–41
Li WS (1997) Molecular evolution. Chapters 7 and 8. Sinauer Associates, Inc, Sunderland
Lohe AR, De Aguiar D, Hartl DL (1997) Mutations in the mariner transposase, the “D, D(35)E” consensus sequence is nonfunctional. Proc Natl Acad Sci USA 94:1293–1297
Loreto EL, Carareto CM, Capy P (2008) Revisiting horizontal transfer of transposable elements in Drosophila. Heredity 100:545–554
Lorite P, Aránega AE, Luque F, Palomeque T (1997) Analysis of the nucleolar organizing regions in the ant Tapinoma nigerrimum (Hymenoptera, Formicidae). Heredity 78:578–582
Lorite P, Carrillo JA, Garneria I, Petitpierre E, Palomeque T (2002) Satellite DNA in the elm leaf beetle Xanthogaleruca luteola (Coleoptera, Chrysomelidae): characterization, interpopulation analysis and chromosome location. Cytogenet Genome Res 98:302–307
Lorite P, Palomeque T (2010) Karyotype evolution in ants (Hymenoptera: Formicidae) with a review of the known ant chromosome numbers. Myrmecol News 13:89–102
McClean P (1998). DNA — basics of structure and analysis. Nucleic acid hybridizations. Retrieved from http://www.ndsu.nodak.edu/instruct/mcclean/plsc731/dna/dna6.htm 12/7/2012
Miskey C, Izsvák Z, Kawakami K, Ivics Z (2005) DNA transposons in vertebrate functional genomics. Cell Mol Life Sci 62:629–641
Meznar ER, Gadau J, Koeniger N, Rueppell OJ (2010) Comparative linkage mapping suggests a high recombination rate in all honeybees. J Hered 101(Suppl 1):S118–S126
Medhora M, Maruyama K, Hartl DL (1991) Molecular and functional analysis of the mariner mutator element Mos1 in Drosophila. Genetics 128:311–318
Moreau CS, Bell CD, Vila R, Archibald SB, Pierce NE (2006) Phylogeny of the ants: diversification in the age of Angiosperms. Science 312:101–104
Muñoz-López M, Siddique A, Bischerour J, Lorite P, Chalmers R, Palomeque T (2008) Transposition of Mboumar-9: identification of a new naturally-active mariner-family transposón. J Mol Biol 382:567–572
Nei M (1987) Molecular evolutionary genetics. Columbia Univ, Press, New York
Nei M, Kumar S (2000) Molecular evolutionary and phylogenetics. Chapter 10. Oxford University Press, New York
Niehuis O, Gibson JD, Rosenberg MS, Pannebakker BA, Koevoets T, Judson AK, Desjardins CA, Kennedy K, Duggan D, Beukeboom LW, van de Zande L, Shuker DM, Werren JH, Gadau J (2010) Recombination and its impact on the genome of the haplodiploid parasitoid wasp Nasonia. PLoS One 5:e8597
Palomeque T, Chica E, Cano MA, Díaz de la Guardia R (1988) Karyotypes, C-banding, and chromosomal location of active nucleolar organizing regions in Tapinoma (Hymenoptera, Formicidae). Genome 30:277–280
Palomeque T, Muñoz-López M, Carrillo JA, Lorite P (2005) Characterization and evolutionary dynamics of a complex family of satellite DNA in the leaf beetle Chrysolina carnifex (Coleoptera, Chrysomelidae). Chromosome Res 13:795–807
Palomeque T, Carrillo JA, Muñoz-López M, Lorite P (2006) Detection of a mariner-like element and a miniature inverted-repeat transposable element (MITE) associated with the heterochromatin from ants of the genus Messor and their possible involvement for satellite DNA evolution. Gene 371:194–205
Plasterk RH, Izsvák Z, Ivics Z (1999) Resident aliens, the Tc1/mariner superfamily of transposable elements. Trends Genet 15:326–332
Rezende-Teixeira P, Lauand C, Siviero F, Machado-Santelli GM (2010) Normal and defective mariner-like elements in Rhynchosciara species (Sciaridae, Diptera). Genet Mol Res 9:849–857
Robertson HM (1996) Members of the pogo superfamily of DNA-mediated transposons in the human genome. Mol Gen Genet 252:761–766
Robertson HM, MacLeod EG (1993) Five major subfamilies of mariner transposable elements in insects, including the Mediterranean fruit fly, and related arthropods. Insect Mol Biol 2:125–139
Robertson HM, Asplund KL (1996) Bmmar1, a basal lineage of the mariner family of transposable elements in the silkworm moth, Bombyx mori. Insect Biochem Mol Biol 26:945–954
Rouleux-Bonnin F, Petit A, Demattei MV, Bigot Y (2005) Evolution of full-length and deleted forms of the mariner-like element, Botmar1, in the genome of the bumble bee, Bombus terrestris (Hymenoptera: Apidae). J Mol Evol 60:736–747
Rouault JD, Casse N, Chénais B, Hua-Van A, Filée J, Capy P (2009) Automatic classification within families of transposable elements: application to the mariner family. Gene 448:227–232
Sánchez-Gracia A, Maside X, Charlesworth B (2005) High rate of horizontal transfer of transposable elements in Drosophila. Trends Genet 21:200–203
Schaack S, Gilbert C, Feschotte C (2010) Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol 25:537–546
Sela N, Kim E, Ast G (2010) The role of transposable elements in the evolution of non-mammalian vertebrates and invertebrates. Genome Biol 11:R59
Shao H, Tu Z (2001) Expanding the diversity of the IS630-Tc1-mariner superfamily: discovery of a unique DD37E transposon and reclassification of the DD37D and DD39D transposons. Genetics 159:1103–1115
Silva JC, Bastida F, Bidwell SL, Johnson PJ, Carlton JM (2005) A potentially functional mariner transposable element in the protist Trichomonas vaginalis. Mol Biol Evol 22:126–134
Sironi M, Menozzi G, Comi GP, Cereda M, Cagliani R, Bresolin N, Pozzoli U (2006) Gene function and expression level influence the insertion/fixation dynamics of distinct transposon families in mammalian introns. Genome Biol 7:R120
Sirviö A, Gadau J, Rueppell O, Lamatsch D, Boomsma JJ, Pamilo P, Page RE Jr (2006) High recombination frequency creates genotypic diversity in colonies of the leaf-cutting ant Acromyrmex echinatior. J Evol Biol 19:1475–1485
Sirviö A, Johnston JS, Wenseleers T, Pamilo P (2011) A high recombination rate in eusocial Hymenoptera: evidence from the common wasp Vespula vulgaris. BMC Genet 12:95
Tamura K, Subramanian S, Kumar S (2004) Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol Biol Evol 21:36–44
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony methods. Mol Biol Evol 28:2731–2739
Viginier B, Peeters C, Brazier L, Doums C (2004) Very low genetic variability in the Indian queenless ant Diacamma indicum. Mol Ecol 13:2095–2100
Ward PS, Downie DA (2005) The ant subfamily Pseudomyrmecinae (Hymenoptera: Formicidae): phylogeny and evolution of big-eyed arboreal ants. Syst Entomol 30:310–335
Ward PS, Brady SG, Fisher BL, Schultz TR (2010) Phylogeny and biogeography of dolichoderine ants: effects of data partitioning and relict taxa on historical inference. Syst Biol 59:342–362
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Wilfert L, Gadau J, Schmid-Hempel P (2007) Variation in genomic recombination rates among animal taxa and the case of social insects. Heredity 98:189–197
Wysocka A, Krzysztofiak L, Krzysztofiak A, Zołnierkiewicz O, Ojdowska E, Sell J (2011) Low genetic diversity in Polish populations of sibling ant species: Lasius niger (L.) and Lasius platythorax Seifert (Hymenoptera, Formicidae). Insec Soc 58:191–195
Zhou MB, Zhong H, Tang DQ (2011) Isolation and characterization of seventy-nine full-length mariner-like transposase genes in the Bambusoideae subfamily. J Plant Res 124:607–617
Acknowledgements
We thank Dr. Dale K. Pollet (Louisiana State University Agricultural Center, Baton Rouge, LA, USA) for providing us samples of Solenopsis invicta. We are also grateful to Alain Lenoir (Université François Rabelais, Tours, France) for providing us samples of Myrmica ruginodis. This work was supported by the Spanish Junta de Andalucía (through the programs “Ayudas a Grupos de Investigación”, Group BIO220 and “Incentivos a proyectos de investigación de excelencia”, project CVI-6807, co-funded by the European Regional Development Fund), by the Spanish Ministerio de Educación e Innovación (through project CGL2011-23841, co-funded by the European Regional Development Fund) and by the University of Jaén (through the program “Programa propio de ayudas para la realización de proyectos de investigación”, UJA-08-16-03).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Sven Thatje
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 6186 kb)
Rights and permissions
About this article
Cite this article
Lorite, P., Maside, X., Sanllorente, O. et al. The ant genomes have been invaded by several types of mariner transposable elements. Naturwissenschaften 99, 1007–1020 (2012). https://doi.org/10.1007/s00114-012-0982-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00114-012-0982-5