
Abstract
The role of inflammatory signaling pathways in synaptic plasticity has long been identified. Yet, it remains unclear how inflammatory cytokines assert their pleiotropic effects on neural plasticity. Moreover, the neuronal targets through which inflammatory cytokines assert their effects on plasticity remain not well-understood. In an attempt to learn more about the plasticity-modulating effects of the pro-inflammatory cytokine tumor necrosis factor (TNF), we used two-pathway long-term potentiation (LTP) experiments at Schaffer collateral-CA1 synapses to test for concentration-dependent effects of TNF on synaptic plasticity. We report that high concentrations of TNF (1 μg/mL) impair the ability of mouse CA1 pyramidal neurons to express synaptic plasticity without affecting baseline synaptic transmission and/or previously established LTP. Interestingly, 100 ng/mL of TNF has no apparent effect on LTP, while low concentrations (1 ng/mL) promote the ability of neurons to express LTP. These dose-dependent metaplastic effects of TNF are modulated by intracellular calcium stores: Pharmacological activation of intracellular calcium stores with ryanodine (10 μM) reverses the negative effects of TNF[high], and the plasticity-promoting effects of TNF[low] are blocked when intracellular calcium stores are depleted with thapsigargin (1 μM). Consistent with this result, TNF does not promote plasticity in synaptopodin-deficient preparations, which show deficits in neuronal calcium store-mediated synaptic plasticity. Thus, we propose that TNF mediates its pleiotropic effects on synaptic plasticity in a concentration-dependent manner through signaling pathways that are modulated by intracellular calcium stores and require the presence of synaptopodin. These results demonstrate that TNF can act as mediator of metaplasticity, which is of considerable relevance in the context of brain diseases associated with increased TNF levels and alterations in synaptic plasticity.
Key messages
• TNF modulates the ability of neurons to express synaptic plasticity.
• High concentrations of TNF impair synaptic plasticity.
• Low concentrations of TNF improve synaptic plasticity.
• TNF does not affect previously established long-term potentiation.
• Plasticity effects of TNF are modulated by intracellular calcium stores.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite a wealth of information on the cellular and molecular mechanisms of neural plasticity, the relevance of synaptic plasticity under pathological conditions remains a matter of debate [8, 10, 21, 36, 39, 42, 50]. A better understanding of molecules and mechanisms that modulate the ability of neurons to express synaptic plasticity in pathological brain states is urgently needed. This knowledge could be used to restore physiological brain function and/or promote recovery after brain injury in clinically relevant settings [23, 39]. Considering that neurological and psychiatric diseases are often accompanied by inflammatory responses [52, 61], we here sought to test for the role of the pro-inflammatory cytokine tumor necrosis factor (TNF) in modulating the ability of neurons to express synaptic plasticity.
TNF has been previously linked to synaptic transmission and plasticity (for details see, [3, 5, 12, 20, 47, 49, 53, 63, 65]). However, conflicting results have been reported, and as a result, it remains unclear whether TNF promotes and/or occludes synaptic plasticity. Also, a role for TNF in both long-term potentiation (LTP) and homeostatic synaptic plasticity has been suggested [5, 26, 30, 35, 59, 60, 71], while the neuronal targets through which TNF mediates its effects on plasticity remain not well-understood. We have recently shown that systemic inflammation is accompanied by a strong increase in brain TNF expression (~ 40-fold increase in TNF mRNA; [62]) and alterations in LTP of Schaffer collateral-CA1 synapses [40, 62]. Conversely, in a model of traumatic brain injury, i.e., in vitro entorhinal cortex lesion [69], a moderate increase in TNF was linked to increased synaptic strength in denervated neurons [6, 7]. Interestingly, in both pathological settings, changes in the expression of the neuronal protein synaptopodin were observed, which reflected TNF-mediated changes in plasticity [62, 70]. Synaptopodin is an actin-modulating protein [14, 43] and essential component of stacked smooth endoplasmic reticulum (sER) structures, such as the spine apparatus organelle [15, 16, 19, 57], and cisternal organelles [4, 29, 34, 45]. Similar to TNF, synaptopodin has been linked to the ability of neurons to express LTP [15, 25, 67] and homeostatic synaptic plasticity [70]. In this context, evidence has been provided that synaptopodin acts through the regulation of neuronal sER calcium stores [24, 27, 28, 66, 68].
Based on these observations, it appeared well-warranted to test (1) whether TNF asserts differential concentration-dependent effects on synaptic plasticity, (2) whether intracellular calcium stores are involved in mediating the effects of TNF on synaptic plasticity, and (3) if the presence of synaptopodin is required for TNF-mediated modulation of synaptic plasticity to occur.
Materials and methods
Ethics statement
All experiments were approved by the Institutional Animal Care and Use Committee of The Chaim Sheba Medical Center (Tel HaShomer, Israel), which adheres to the Israeli law on the use of laboratory animals and NIH rules. Experimental procedures were performed also according to the German animal welfare legislation as approved by the animal welfare officer at Albert-Ludwigs-University Freiburg, Faculty of Medicine. Human material was not employed in this study.
Animals
Four- to five-month-old male C57BL/6 mice and synaptopodin-deficient mice (Synpo−/−; available at Jackson Laboratory; [15]) and their age- and time-matched wild-type littermates were used. Mice were maintained in a 12-h light/dark cycle with food and water available ad libitum. Every effort was made to minimize distress and pain of animals.
Pharmacology
The following drugs were used at the indicated final concentrations: thapsigargin (Alomone Labs, Jerusalem, Israel), ryanodine (Tocris, UK), recombinant mouse TNF-alpha (Genescript, USA).
Electrophysiology in acute brain slices
Extracellular recordings in acute slices prepared from dorsal hippocampus were performed as previously described (e.g., [38]). Following anesthesia with ketamine/xylazine (100 mg/kg and 10 mg/kg, respectively), animals were rapidly decapitated, the brains removed, and 400-μm slices prepared using a vibroslicer. Slices were incubated for 1.5 h in a humidified, carbogenated gas atmosphere (5% CO2 and 95% O2) at 33 ± 1 °C and perfused with artificial cerebrospinal fluid (ACSF) containing (in mM): 124 NaCl, 2 KCl, 26 NaHCO3, 1.24 KH2PO4, 2.5 CaCl2, 2 MgSO4, and 10 glucose (pH 7.4) in a standard interface chamber. Recordings were made with a glass pipette containing 0.75 M NaCl (4 MΩ) placed in stratum radiatum of CA1. Stimulation of Schaffer collaterals was evoked using a pulse stimulator and delivered through bipolar nichrome electrodes. Input-output curves were run on each slice prior to beginning of each experiment. Before applying the tetanic stimulation, baseline values were recorded at a frequency of 0.033 Hz. LTP was induced by high-frequency stimulation (HFS) consisting of 100 pulses, delivered at a frequency of 100 Hz (1 s). Short HFS (sHFS) consisted of 25 pulses, delivered at 100 Hz.
Quantification and statistics
Data were analyzed off-line using Spike 2 software (Cambridge Electronic Design). Excitatory postsynaptic potential (EPSP) slope changes after tetanic stimulation were calculated with respect to baseline. Statistical comparisons between the two groups were performed using a t test (unpaired, two-tailed). p values smaller 0.05 were considered a significant difference between means.
Results
Concentration-dependent effects of TNF on synaptic plasticity
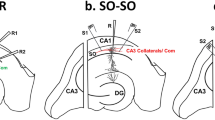
The effects of TNF were studied in a two-pathway experimental setting, in which two stimulating electrodes are used to independently probe and potentiate Schaffer collateral-CA1 synapses, while recording field EPSPs in CA1 stratum radiatum (see schematic in Fig. 1(A)). After acquiring a baseline of evoked EPSPs at both pathways, LTP is induced by delivering an electric stimulus consisting of 100 pulses at twice the test intensity (at a frequency of 100 Hz) to one of the two pathways (Fig. 1(B)). This leads to a robust increase in EPSP slope, leaving synaptic transmission probed at the other pathway unaffected. Eventually, the other pathway is probed/potentiated using the exact same stimulus (see arrows in Fig. 1(B)), leading to comparable levels of LTP at the second pathway without affecting pre-established LTP (Fig. 1(B)). Using this experimental setting, TNF was washed in at distinct concentrations (1 μg/mL, 100 ng/mL, 1 ng/mL, 0.1 ng/mL) after the potentiation of the first pathway. Hence, we were able to systematically test for the effects of TNF on the ability of neurons to express LTP, and for its impact on LTP that is established prior to the exposure to TNF (Fig. 2(A–D)).

Two-pathway long-term potentiation (LTP) experiments at Schaffer collateral-CA1 synapses. (a) Schematic illustration of the experimental setting. Two stimulating electrodes (stim.) are used to independently probe and potentiate Schaffer collateral-CA1 synapses of acute mouse hippocampal slices, while recording (rec.) field excitatory postsynaptic potentials (EPSP) in CA1 stratum radiatum (dentate gyrus, DG). (b) LTP is induced by delivering an electric stimulus consisting of 100 pulses at twice the test intensity at a frequency of 100 Hz (see arrows). Averaged EPSPs are plotted vs. time. Note, representative traces at indicated times (a, b). Details provided in the main text (n = 12 slices; from 4 animals each)

Tumor necrosis factor (TNF) affects hippocampal long-term potentiation (LTP) in a concentration-dependent manner. (a) TNF applied at a concentration of 1 μg/mL reduces the ability of neurons to express LTP, while having no apparent effect on LTP established earlier at the other pathway. (b) At a concentration of 100 ng/mL, TNF does not affect LTP. (c) TNF at 1 ng/mL enhances LTP, while (d) LTP is not affected when slices are exposed to TNF concentrations of 0.1 ng/mL. Averaged EPSPs are plotted vs. time. Representative traces at indicated times (a, b) are shown on top of each section (n = 12 slices; from 4 animals each). Arrows indicate the time of tetanus delivery (100 pulses at twice the test intensity at 100 Hz). (e) Application of a short HFS (sHFS; 25 pulses at twice the test intensity at 100 Hz) in the presence of TNF[high] (1 μg/mL) neither enhances nor depresses the effects of sHFS on LTP. (f) The presence of 1 ng/mL TNF[low] enhances the ability of sHFS to induce LTP. Averaged EPSPs are plotted vs. time. Representative traces at indicated times (a, b) are shown on top of each section (n = 12 slices; from 4 animals each). Arrows indicate the time of (s)HFS delivery
Treating hippocampal slices with a high concentration of TNF resulted in reduced LTP (Fig. 2(A)). Specifically, a tetanic stimulation applied after exposure to 1 μg/mL of TNF (referred to as TNF[high]) resulted in a level of potentiation of 1.32 ± 0.07 compared to the 1.76 ± 0.06 at the first pathway (p < 0.01; n = 12 slices from 4 animals). Notably, TNF[high] did not affect LTP that was established at the first pathway prior to the treatment in the same set of slices (Fig. 2(A)). Hence, toxic effects of TNF[high] do not trivially explain our results. Rather, 1 μg/mL TNF affects the ability of neurons to express new LTP, while leaving pre-established LTP unaltered.
In an independent set of experiments, we tested for the effects of 100 ng/mL TNF and found no significant effect on the ability of neurons to express LTP and/or previously established LTP (1.83 ± 0.07 vs. 1.72 ± 0.07, p = 0.08, n = 12 slices from 4 animals; Fig. 2(B); c.f. Fig. 1(B)). These results suggest that a high concentration of TNF is required to induce alterations in the ability of neurons to express LTP.
Strikingly, the opposite result, i.e., an enhancement of LTP, was observed when TNF was washed in at a concentration of 1 ng/mL (Fig. 2(C)). In this setting, a tetanic stimulation to the second pathway following the application of 1 ng/mL TNF (referred to as TNF[low]) resulted in a potentiation of 2.26 ± 0.08 vs 1.72 ± 0.07 of the control pathway (Fig. 1(C); p < 0.001; n = 12 slices from 4 animals). Similar to what we observed with TNF[high], no effect on established LTP at the other pathway was observed with TNF[low]. The plasticity-enhancing effect of TNF was not statistically different between the two pathways at a concentration of 0.1 ng/mL (1.82 ± 0.07 vs 2.06 ± 0.08, p = 0.06, n = 12 slices from 4 animals; Fig. 2(D)). Taken together, we conclude that TNF asserts differential effects on the ability of neurons to express LTP, without affecting baseline synaptic transmission and/or previously established LTP (at all concentrations tested here): TNF[low] promotes new LTP, while TNF[high] impedes LTP induction.
Low-concentration TNF reduces the threshold for plasticity induction
In another set of experiments, short high-frequency stimulation was delivered (sHFS; 25 pulses at twice the test intensity at a frequency of 100 Hz), which does not induce a strong and robust LTP response per se (Fig. 2(E, F)). In this setting, application of sHFS in presence of TNF[high] neither enhanced nor depressed the effects of sHFS (Fig. 2(E)). In contrast, the presence of TNF[low] enhanced the ability of sHFS to induce LTP, and sHFS resulted in a full-blown LTP response reaching a level of 1.64 ± 0.08 vs. 1.12 ± 0.07 (Fig. 1(F); p < 0.001; n = 12 from 4 animals). Hence, low concentrations of TNF affect the ability of neurons to express plasticity by reducing the threshold for plasticity induction.
Intracellular Ca2+ stores mediate the plasticity-enhancing effects of low-concentration TNF
Previous work has indicated that TNF may act on neural tissue through intracellular calcium stores [11, 18, 48, 62]. In fact, it is well-accepted that intracellular calcium stores modulate the ability of neurons to express plasticity (reviewed in [17, 39, 55]). Specifically, metaplastic changes in the threshold of plasticity induction have been reported to depend on intracellular calcium homeostasis [22, 32, 33, 41, 51].
Indeed, enhancing calcium-release from intracellular stores with 10 μM ryanodine improved the ability of neurons to express synaptic plasticity in our experimental setting (Fig. 3(A)), similar to what we observed with TNF[low] (c.f. Fig. 2(C)). In turn, depletion of intracellular calcium stores with 1 μM thapsigargin—unlike TNF[high] (c.f. Fig. 2(A))—resulted in a slow rundown of pre-established LTP, and no major effect on the ability to induce new LTP was observed (Fig. 3(B)). Thus, activation of calcium stores mimics the effects of TNF[low], while pharmacological calcium store depletion does not reflect TNF[high] effects on synaptic plasticity. We therefore hypothesized that intracellular calcium stores mediate the plasticity-enhancing effects of TNF[low], while other mechanisms may account for the effects of TNF[high].

Intracellular calcium stores mediate the plasticity-enhancing effects of low-centration Tumor necrosis factor (TNF). (a) Activation of calcium stores with 10 μM ryanodine enhances LTP, similar to what is observed in response to 1 ng/mL TNF, while (b) pharmacological depletions of intracellular calcium stores with 1 μM thapsigargin trigger a slowly decaying LTP. Unlike 1 μg/mL TNF, in the presence of 1 μM thapsigargin, the ability to induce LTP at the second pathway is not affected. (c) Application of 1 μM thapsigargin attenuates the plasticity-enhancing effects of 1 ng/mL TNF. (d) When TNF-enhanced LTP is induced at the first pathway, a considerable rundown of LTP is observed in the presence of 1 μM thapsigargin and the ability to express enhanced LTP is completely blocked at the second pathway in the presence of 1 μM thapsigargin and 1 ng/mL TNF. Averaged EPSPs are plotted vs. time. Representative traces at indicated times (a, b) are shown on top of each section (n = 12 slices; from 4 animals each). Arrows indicate the time of high frequency stimulation
In view of these results, we reasoned that the depletion of intracellular calcium stores with 1 μM thapsigargin should occlude the plasticity-enhancing effects of TNF[low]. Indeed, applying 1 ng/mL TNF in the presence of 1 μM thapsigargin attenuated the plasticity-enhancing effects of TNF[low], resulting in an LTP level of 1.74 ± 0.08 (Fig. 3(C)). Moreover, when 1 μM thapsigargin was washed in after the induction of TNF[low]-enhanced LTP (Fig. 3(D)), a substantial rundown of LTP was observed, and the ability to induce TNF[low]-enhanced LTP at the second pathway was blocked (Fig. 3(D)). These experiments also showed that priming LTP induction at the first pathway is not required for TNF[low]-enhanced synaptic plasticity to occur (c.f. Fig. 2(F)). We conclude that functional intracellular calcium stores are involved in mediating the plasticity-enhancing effects of TNF[low].
Low-concentration TNF does not enhance plasticity in synaptopodin-deficient neurons
Pharmacology acts on intracellular calcium stores in all neural compartments (i.e., neurons, astrocytes, oligodendrocytes, and microglia). In order to test for the neuronal targets of TNF and based on our earlier work on the role of synaptopodin-associated neuronal intracellular calcium stores in synaptic plasticity (e.g., [31, 62]), we next assessed the plasticity-enhancing effects of TNF[low] on hippocampal slices prepared from synaptopodin-deficient mice and their age/time-matched wild-type littermates.
Once more, the plasticity-enhancing effect of 1 ng/mL TNF[low] was observed in Synpo+/+ preparations (Fig. 4(A); c.f. Fig. 2(C)). The ability of Synpo−/− neurons to express LTP was reduced, consistent with earlier findings [15, 25], and TNF[low] was not able to further enhance LTP (Fig. 4(B)). Specifically, LTP levels upon treatment with 1 ng/mL TNF were 1.56 ± 0.05 in Synpo−/− vs. 2.07 ± 0.07 in Synpo+/+ (p < 0.001; n = 6 slices from 3 animals each). Hence, TNF[low] requires the presence of synaptopodin to assert its plasticity-promoting effects.

Low-concentration tumor necrosis factor (TNF) does not induce metaplasticity in synaptopodin-deficient neurons. (a) TNF (1 ng/mL) enhances LTP in age- and time-matched wild-type (Synpo+/+), but not (b) in synaptopodin-deficient mice (Synpo−/−). Averaged EPSPs are plotted vs. time. Representative traces at indicated times (a, b) are shown on top of each section (n = 6 slices; from 3 animals each). Arrows indicate the time of stimulation
Pharmacological activation of intracellular calcium stores counters the effects of TNF[high]
Finally, we used wild-type hippocampal slices to test for the effects of thapsigargin (Fig. 5(A)) and ryanodine (Fig. 5(B)) on TNF[high]-mediated alterations in synaptic plasticity. While the negative effects of TNF[high] are detectable in the presence of thapsigargin (Fig. 5(A); 1.33 ± 0.07 vs. 1.56 ± 0.07, p < 0.001; n = 9 slices from 3 animals), enhancing calcium release from intracellular stores counters the negative effects of TNF[high]: Application of 10 μM ryanodine prior to 1 μg/mL TNF[high] resulted in enhanced LTP (Fig. 5; 2.25 ± 0.07 vs. 1.79 ± 0.07, p < 0.001; n = 12 from 4 animals), comparable to what we observed in our ryanodine-only experiments (c.f. Fig. 3(A)).

Pharmacological activation of calcium stores counters the effects of high-concentration tumor necrosis factor (TNF). (a) In the presence of 1 μM thapsigargin and 1 μg/mL TNF a reduction in LTP is observed (n = 9 slices, from 3 animals each; c.f. Fig. 3(C)). (b) In the presence of 10 μM ryanodine and 1 μg/mL TNF, an enhancement of LTP is observed (n = 12 slices, from 4 animals each). Averaged EPSPs are plotted vs. time. Note, representative traces at indicated times (a, b). Arrows indicate the time of tetanus delivery
Discussion
The results of the present study reveal that the pro-inflammatory cytokine TNF can have opposite effects on synaptic plasticity, depending on the concentration. The plasticity-enhancing effect of 1 ng/mL TNF[low] is not observed when calcium stores are pharmacologically depleted. Likewise, in synaptopodin-deficient neurons, which do not form stacked sER morphologies (recently reviewed in [24]), TNF[low] does not promote plasticity. While the mechanisms through which 1 μg/mL TNF[high] affects plasticity warrant further investigation, our experiments indicate that pharmacological activation of calcium stores counters the negative effects of TNF[high]. These results provide new important insight on the role of TNF in synaptic plasticity by identifying TNF as a mediator of metaplasticity [1, 23], which acts, at least in part, through intracellular calcium stores and synaptopodin.
A remarkable finding of our study is the observation that TNF treatment does not affect synaptic transmission and/or pre-established LTP. Rather, TNF modulates the ability of neurons to express plasticity in a concentration-dependent manner. This finding is consistent with previous work in the field of homeostatic synaptic plasticity, which has indicated a permissive role of TNF in synaptic plasticity: TNF modulates the ability of neurons to express homeostatic synaptic plasticity, rather than being instructive, i.e., inducing changes in synaptic strength per se [6, 58]. Yet, robust evidence exists that TNF can change synaptic strength (e.g., [5, 59]). For example, Beattie et al. [5] showed that low concentrations of TNF increase the surface expression of AMPA receptors at excitatory postsynapses in primary hippocampal neurons. It is interesting to speculate though that TNF signaling may have changed the plasticity threshold in these preparations, rather than triggering synaptic accumulation of glutamate receptors per se. Hence, network activity that is not sufficient to induce LTP under control conditions may have triggered the strengthening of glutamatergic neurotransmission in the presence of TNF. Indeed, this suggestion is supported by the results of our experiments, in which we used a mild stimulus to demonstrate that TNF[low] reduces the threshold for plasticity induction (c.f. Fig. 2(E, F)). Although more work is required to clarify the precise role of TNF in synaptic plasticity, the results of the present study provide the first experimental evidence that TNF is a component of a signaling pathway which affects the ability of neurons to express plasticity, e.g., by reducing the threshold of plasticity induction, without modifying baseline synaptic transmission and/or pre-established LTP. Hence, we are confident to conclude that besides its suggested roles in Hebbian and homeostatic plasticity, TNF also acts as a “metaplasticity mediator.”
Apparently, the precise cellular and molecular mechanisms through which TNF asserts its metaplastic effects warrant further investigation. Two canonical receptors mediate the effects of TNF. TNFR1 predominantly binds soluble TNF and is constitutively expressed on most cells of the body (including neurons and astrocytes), while TNFR2 binds with high affinity to membrane-bound TNF and is mainly expressed on cells of the immune system [2, 37, 53]. The rather rapid response to TNF, i.e., within ~ 10 min, in our experimental setting, and the fact that we used soluble recombinant TNF argue for neuronal TNFR1. However, it is difficult to conceive how TNFR1- and/or TNFR2-mediated signaling pathways could mediate the concentration-dependent effects of soluble TNF. Also, at 100 ng/mL, no apparent effect of TNF on synaptic plasticity was detected. Whether TNFR1 and TNFR2 have different affinities to soluble TNF and/or yet unknown non-canonical pathways contribute to the plasticity-modulating effects of TNF remains unknown at this point.
Regardless of these considerations, our experiments suggest a role of intracellular calcium stores in TNF-induced metaplasticity (c.f. [11, 18, 48]). Pharmacological release of calcium from intracellular stores prevented the negative effects of TNF[high] on synaptic plasticity, while the plasticity-promoting effects of TNF[low] were abolished when calcium stores were depleted. However, the results of the present study do not support a simple model in which TNF[low] activates intracellular calcium stores while TNF[high] acts by depleting intracellular stores. Even though the evidence which indicates that calcium stores mediate the plasticity-enhancing effects of TNF[low] seems robust, 1 μM thapsigargin does not mimic the effects of TNF[high]. Also, negative effects of TNF[high] on synaptic plasticity are detectable in the presence of thapsigargin. It is interesting to speculate in this context that TNF may act on a more refined level of sER morphology. Considering that spine ER is a highly dynamic structure which can enter and leave individual spines within minutes ([44, 64]; see also [68]), it is possible that TNF[high] may cause a retraction of spine ER, while dendritic ER tubules may enter spine compartments and changes their confirmation in response to TNF[low]. Such rapid regulation of sER structure could modify spine calcium signaling and homeostasis, which could explain the fast metaplastic adjustment induced by TNF.
Indeed, the work on synaptopodin supports the suggestion that TNF may act through specific changes in sER morphologies. Previous work disclosed that changes in synaptopodin cluster sizes reflect changes in the number of stacked spine ER cisternae ([70]). Moreover, a reduction in synaptopodin cluster sizes was observed in a lipopolysaccharide model of system inflammation, which is accompanied by a strong increase in brain TNF mRNA levels and alterations in the ability of neurons to express LTP [62]. Conversely, increased synaptopodin cluster sizes were reported in a model of in vitro denervation [70], which depends on local TNF release [6, 7]. Consistent with this suggestion, the results of the present study show that TNF[low] requires the presence of synaptopodin to induce its plasticity-enhancing effects. These observations motivate a rigorous study on the cellular and molecular mechanisms through which distinct concentrations of TNF modulate synaptopodin-associated calcium stores and dendritic spine plasticity.
It is interesting to note in this context that recent work has implicated intracellular calcium homeostasis and alterations in sER calcium stores to play a major role in pathological brain states [13, 39, 46, 54]. Indeed, a recent study reported that metaplastic activation of ryanodine receptors restores plasticity in an animal model of Alzheimer’s disease [33]. Considering the proposed role of TNF as a therapeutic target in Alzheimer’s disease (for recent reviews, see [9, 56]), we are confident that a better understanding on the effects of TNF and other inflammatory cytokines on glial and neuronal sER calcium stores may provide an important biological basis for the development of new therapeutic strategies based on metaplasticity induction and neuromodulation.
References
Abraham WC (2008) Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci 9:387
Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3:745–756
Albensi BC, Mattson MP (2000) Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse 35:151–159
Bas Orth C, Schultz C, Muller CM, Frotscher M, Deller T (2007) Loss of the cisternal organelle in the axon initial segment of cortical neurons in synaptopodin-deficient mice. J Comp Neurol 504:441–449
Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC (2002) Control of synaptic strength by glial TNFalpha. Science 295:2282–2285
Becker D, Zahn N, Deller T, Vlachos A (2013) Tumor necrosis factor alpha maintains denervation-induced homeostatic synaptic plasticity of mouse dentate granule cells. Front Cell Neurosci 7:257
Becker D, Deller T, Vlachos A (2015) Tumor necrosis factor (TNF)-receptor 1 and 2 mediate homeostatic synaptic plasticity of denervated mouse dentate granule cells. Sci Rep 5:12726
Bliss TV, Collingridge GL, Kaang BK, Zhuo M (2016) Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat Rev Neurosci 17:485–496
Chang R, Yee KL, Sumbria RK (2017) Tumor necrosis factor alpha inhibition for Alzheimer’s disease. J Cent Nerv Syst Dis 9:1179573517709278
Chung WS, Welsh CA, Barres BA, Stevens B (2015) Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci 18:1539–1545
Cumiskey D, Butler MP, Moynagh PN, O’connor J, J. (2007) Evidence for a role for the group I metabotropic glutamate receptor in the inhibitory effect of tumor necrosis factor-alpha on long-term potentiation. Brain Res 1136:13–19
Cunningham AJ, Murray CA, O’neill LA, Lynch MA, O’connor JJ (1996) Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett 203:17–20
Del Prete D, Checler F, Chami M (2014) Ryanodine receptors: physiological function and deregulation in Alzheimer disease. Mol Neurodegener 9:21
Deller T, Mundel P, Frotscher M (2000) Potential role of synaptopodin in spine motility by coupling actin to the spine apparatus. Hippocampus 10:569–581
Deller T, Korte M, Chabanis S, Drakew A, Schwegler H, Stefani GG, Zuniga A, Schwarz K, Bonhoeffer T, Zeller R, Frotscher M, Mundel P (2003) Synaptopodin-deficient mice lack a spine apparatus and show deficits in synaptic plasticity. Proc Natl Acad Sci U S A 100:10494–10499
Fifkova E, Markham JA, Delay RJ (1983) Calcium in the spine apparatus of dendritic spines in the dentate molecular layer. Brain Res 266:163–168
Finch EA, Tanaka K, Augustine GJ (2012) Calcium as a trigger for cerebellar long-term synaptic depression. Cerebellum 11:706–717
Glazner GW, Camandola S, Geiger JD, Mattson MP (2001) Endoplasmic reticulum D-myo-inositol 1,4,5-trisphosphate-sensitive stores regulate nuclear factor-kappaB binding activity in a calcium-independent manner. J Biol Chem 276:22461–22467
Gray EG (1959) Axo-somatic and axo-dendritic synapses of the cerebral cortex: an electron microscope study. J Anat 93:420–433
He P, Liu Q, Wu J, Shen Y (2012) Genetic deletion of TNF receptor suppresses excitatory synaptic transmission via reducing AMPA receptor synaptic localization in cortical neurons. FASEB J 26:334–345
Hong S, Dissing-Olesen L, Stevens B (2016) New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 36:128–134
Hulme SR, Jones OD, Ireland DR, Abraham WC (2012) Calcium-dependent but action potential-independent BCM-like metaplasticity in the hippocampus. J Neurosci 32:6785–6794
Hulme SR, Jones OD, Abraham WC (2013) Emerging roles of metaplasticity in behaviour and disease. Trends Neurosci 36:353–362
Jedlicka P, Deller T (2017) Understanding the role of synaptopodin and the spine apparatus in Hebbian synaptic plasticity - new perspectives and the need for computational modeling. Neurobiol Learn Mem 138:21–30
Jedlicka P, Schwarzacher SW, Winkels R, Kienzler F, Frotscher M, Bramham CR, Schultz C, Bas Orth C, Deller T (2009) Impairment of in vivo theta-burst long-term potentiation and network excitability in the dentate gyrus of synaptopodin-deficient mice lacking the spine apparatus and the cisternal organelle. Hippocampus 19:130–140
Kaneko M, Stellwagen D, Malenka RC, Stryker MP (2008) Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron 58:673–680
Korkotian E, Segal M (2011) Synaptopodin regulates release of calcium from stores in dendritic spines of cultured hippocampal neurons. J Physiol 589:5987–5995
Korkotian E, Frotscher M, Segal M (2014) Synaptopodin regulates spine plasticity: mediation by calcium stores. J Neurosci 34:11641–11651
Kosaka T (1980) The axon initial segment as a synaptic site: ultrastructure and synaptology of the initial segment of the pyramidal cell in the rat hippocampus (CA3 region). J Neurocytol 9:861–882
Kronschläger MT, Drdla-Schutting R, Gassner M, Honsek SD, Teuchmann HL, Sandkühler J (2016) Gliogenic LTP spreads widely in nociceptive pathways. Science 354:1144–1148
Lenz M, Ben Shimon M, Deller T, Vlachos A, Maggio N (2017) Pilocarpine-induced status epilepticus is associated with changes in the actin-modulating protein synaptopodin and alterations in long-term potentiation in the mouse hippocampus. Neural Plast 2017:2652560
Li Q, Rothkegel M, Xiao ZC, Abraham WC, Korte M, Sajikumar S (2014) Making synapses strong: metaplasticity prolongs associativity of long-term memory by switching synaptic tag mechanisms. Cereb Cortex 24:353–363
Li Q, Navakkode S, Rothkegel M, Soong TW, Sajikumar S, Korte M (2017) Metaplasticity mechanisms restore plasticity and associativity in an animal model of Alzheimer’s disease. Proc Natl Acad Sci U S A 114:5527–5532
Lindsey JD, Ellisman MH (1985) The neuronal endomembrane system. III. The origins of the axoplasmic reticulum and discrete axonal cisternae at the axon hillock. J Neurosci 5:3135–3144
Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, Wei X, Xu T, Xin WJ, Pang RP, Li YY, Qin ZH, Murugan M, Mattson MP, Wu LJ, Liu XG (2017) TNF-α differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J Neurosci 37:871–881
Lu B, Nagappan G, Guan X, Nathan PJ, Wren P (2013) BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci 14:401–416
Macewan DJ (2002) TNF receptor subtype signalling: differences and cellular consequences. Cell Signal 14:477–492
Maggio N, Segal M (2007) Unique regulation of long term potentiation in the rat ventral hippocampus. Hippocampus 17:10–25
Maggio N, Vlachos A (2014) Synaptic plasticity at the interface of health and disease: new insights on the role of endoplasmic reticulum intracellular calcium stores. Neuroscience 281:135–146
Maggio N, Shavit-Stein E, Dori A, Blatt I, Chapman J (2013) Prolonged systemic inflammation persistently modifies synaptic plasticity in the hippocampus: modulation by the stress hormones. Front Mol Neurosci 6:46
Maggio N, Itsekson Z, Ikenberg B, Strehl A, Vlachos A, Blatt I, Tanne D, Chapman J (2014) The anticoagulant activated protein C (aPC) promotes metaplasticity in the hippocampus through an EPCR-PAR1-S1P1 receptors dependent mechanism. Hippocampus 24:1030–1038
Monday HR, Castillo PE (2017) Closing the gap: long-term presynaptic plasticity in brain function and disease. Curr Opin Neurobiol 45:106–112
Mundel P, Heid HW, Mundel TM, Kruger M, Reiser J, Kriz W (1997) Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol 139:193–204
Ng AN, Toresson H (2011) Endoplasmic reticulum dynamics in hippocampal dendritic spines induced by agonists of type I metabotropic glutamate but not by muscarinic acetylcholine receptors. Synapse 65:351–355
Palay SL, Sotelo C, Peters A, Orkand PM (1968) The axon hillock and the initial segment. J Cell Biol 38:193–201
Pchitskaya E, Popugaeva E, Bezprozvanny I (2018) Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70:87–94
Pickering M, Cumiskey D, O’connor JJ (2005) Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol 90:663–670
Pollock J, Mcfarlane SM, Connell MC, Zehavi U, Vandenabeele P, Macewan DJ, Scott RH (2002) TNF-alpha receptors simultaneously activate Ca2+ mobilisation and stress kinases in cultured sensory neurones. Neuropharmacology 42:93–106
Pribiag H, Stellwagen D (2014) Neuroimmune regulation of homeostatic synaptic plasticity. Neuropharmacology 78:13–22
Roeper J (2018) Closing gaps in brain disease-from overlapping genetic architecture to common motifs of synapse dysfunction. Curr Opin Neurobiol 48:45–51
Sajikumar S, Li Q, Abraham WC, Xiao ZC (2009) Priming of short-term potentiation and synaptic tagging/capture mechanisms by ryanodine receptor activation in rat hippocampal CA1. Learn Mem 16:178–186
Salter MW, Stevens B (2017) Microglia emerge as central players in brain disease. Nat Med 23:1018–1027
Santello M, Volterra A (2012) TNFalpha in synaptic function: switching gears. Trends Neurosci 35:638–647
Santos LE, Ferreira ST (2017) Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 136:350–360
Segal M, Korkotian E (2016) Roles of calcium stores and store-operated channels in plasticity of dendritic spines. Neuroscientist 22:477–485
Shamim D, Laskowski M (2017) Inhibition of inflammation mediated through the tumor necrosis factor alpha biochemical pathway can lead to favorable outcomes in Alzheimer disease. J Cent Nerv Syst Dis 9:1179573517722512
Spacek J (1985) Three-dimensional analysis of dendritic spines. II. Spine apparatus and other cytoplasmic components. Anat Embryol (Berl) 171:235–243
Steinmetz CC, Turrigiano GG (2010) Tumor necrosis factor-alpha signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci 30:14685–14690
Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial TNF-alpha. Nature 440:1054–1059
Stellwagen D, Beattie EC, Seo JY, Malenka RC (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 25:3219–3228
Stephenson J, Nutma E, Van Der Valk P, Amor S (2018) Inflammation in CNS neurodegenerative diseases. Immunology 154:204–219
Strehl A, Lenz M, Itsekson-Hayosh Z, Becker D, Chapman J, Deller T, Maggio N, Vlachos A (2014) Systemic inflammation is associated with a reduction in synaptopodin expression in the mouse hippocampus. Exp Neurol 261:230–235
Tancredi V, D’arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F (1992) Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett 146:176–178
Toresson H, Grant SG (2005) Dynamic distribution of endoplasmic reticulum in hippocampal neuron dendritic spines. Eur J Neurosci 22:1793–1798
Turrigiano GG (2006) More than a sidekick: glia and homeostatic synaptic plasticity. Trends Mol Med 12:458–460
Vlachos A (2012) Synaptopodin and the spine apparatus organelle-regulators of different forms of synaptic plasticity? Ann Anat 194:317–320
Vlachos A, Maggio N, Segal M (2008) Lack of correlation between synaptopodin expression and the ability to induce LTP in the rat dorsal and ventral hippocampus. Hippocampus 18:1–4
Vlachos A, Korkotian E, Schonfeld E, Copanaki E, Deller T, Segal M (2009) Synaptopodin regulates plasticity of dendritic spines in hippocampal neurons. J Neurosci 29:1017–1033
Vlachos A, Orth CB, Schneider G, Deller T (2012) Time-lapse imaging of granule cells in mouse entorhinohippocampal slice cultures reveals changes in spine stability after entorhinal denervation. J Comp Neurol 520:1891–1902
Vlachos A, Ikenberg B, Lenz M, Becker D, Reifenberg K, Bas-Orth C, Deller T (2013) Synaptopodin regulates denervation-induced homeostatic synaptic plasticity. Proc Natl Acad Sci U S A 110:8242–8247
Wall AM, Mukandala G, Greig NH, O’Connor JJ (2015) Tumor necrosis factor-α potentiates long-term potentiation in the rat dentate gyrus after acute hypoxia. J Neurosci Res 93:815–829
Funding
This work was supported by German-Israeli-Foundation (GIF G-1317-418.13/2015).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All experiments were approved by the Institutional Animal Care and Use Committee of The Chaim Sheba Medical Center (Tel HaShomer, Israel), which adheres to the Israeli law on the use of laboratory animals and NIH rules. Experimental procedures were performed also according to the German animal welfare legislation as approved by the animal welfare officer at Albert-Ludwigs-University Freiburg, Faculty of Medicine. Human material was not employed in this study.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Maggio, N., Vlachos, A. Tumor necrosis factor (TNF) modulates synaptic plasticity in a concentration-dependent manner through intracellular calcium stores. J Mol Med 96, 1039–1047 (2018). https://doi.org/10.1007/s00109-018-1674-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-018-1674-1