
Abstract
The carotid body is a sensory organ that detects acute changes in arterial blood oxygen (O2) levels and reflexly mediates systemic cardiac, vascular, and respiratory responses to hypoxia. This article provides a brief update of the roles of gas messengers as well as redox homeostasis by hypoxia-inducible factors (HIFs) in hypoxic sensing by the carotid body. Carbon monoxide (CO) and nitric oxide (NO), generated by heme oxygenase-2 (HO-2) and neuronal nitric oxide synthase (nNOS), respectively, inhibit carotid body activity. Molecular O2 is a required substrate for the enzymatic activities of HO-2 and nNOS. Stimulation of carotid body activity by hypoxia may reflect reduced formation of CO and NO. Glomus cells, the site of O2 sensing in the carotid body, express cystathionine γ-lyase (CSE), an H2S generating enzyme. Cth −/− mice, which lack CSE, exhibit severely impaired hypoxia-induced H2S generation, sensory excitation, and stimulation of breathing in response to low O2. Hypoxia-evoked H2S generation in the carotid body requires the interaction of CSE with HO-2, which generates CO. Carotid bodies from Hif1a +/− mice with partial HIF-1α deficiency do not respond to hypoxia, whereas carotid bodies from mice with partial HIF-2α deficiency are hyper-responsive to hypoxia. The opposing roles of HIF-1α and HIF-2α in the carotid body have provided novel insight into molecular mechanisms of redox homeostasis and its role in hypoxia sensing. Heightened carotid body activity has been implicated in the pathogenesis of autonomic morbidities associated with sleep-disordered breathing, congestive heart failure, and essential hypertension. The enzymes that generate gas messengers and redox regulation by HIFs represent potential therapeutic targets for normalizing carotid body function and downstream autonomic output in these disease states.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oxygen (O2) is essential for survival of mammalian cells and hypoxia (reduced O2 availability) profoundly impacts cells, organs, and physiological systems. The carotid bodies are bilateral sensory organs located at the bifurcation of the common carotid artery that monitor arterial blood O2 concentrations and relay sensory information to brainstem neurons, which regulate vital functions including breathing, heart rate, and blood pressure [1, 2]. The following features distinguish O2 sensing by the carotid body from other tissues. First, carotid bodies are exquisitely sensitive to hypoxia and respond to even modest decreases in arterial PO2 (e.g., from ∼100 to 80 mmHg). Second, the sensory response to low PO2 is fast and occurs within seconds after the onset of hypoxia. The remarkable sensitivity and the speed with which they respond to hypoxia makes the carotid body O2 sensing unique compared to other tissues [3]. Reflexes arising from carotid bodies are of critical importance in conferring physiological adaptations to high altitude and play central roles in evoking autonomic morbidities under sleep-disordered breathing and congestive heart failure [1].
Since their discovery as sensory organs, much attention has been focused on understanding how carotid bodies sense and respond to hypoxia [1, 2, 4]. Based on similarities between the stimulus–response of the carotid body and the binding characteristics of O2 to hemoglobin, it was proposed that oxy → deoxy conformational transition of heme prosthetic group(s) is important for hypoxic sensing by the carotid body [1, 2]. Like O2, nitric oxide (NO) and carbon monoxide (CO) are gases that bind to heme [5, 6]and mediate biological effects through the activation of heme-containing proteins (e.g., soluble guanylate cyclase) [7]. Hydrogen sulfide (H2S) is emerging as another physiologically important gas messenger. Chemically, H2S is a reducing agent and, like hypoxia, generates a reduced cellular milieu. The discovery of the transcriptional activator hypoxia-inducible factor 1 (HIF-1), and subsequent identification of additional HIF family members, has led to important insights into the molecular underpinnings of O2 and redox homeostasis [8]. Recent studies have revealed that HIFs regulate carotid body responses to hypoxia by determining the levels of reactive oxygen species (ROS), which are metabolites of O2. This article reviews the effects of NO, CO, H2S, and ROS on hypoxic sensing by the carotid body.
Gas messengers and carotid body O2 sensing
Nitric oxide (NO)
Nitric oxide synthase (NOS) catalyzes the conversion of arginine + O2 to citrulline + NO [4]. Neuronal (nNOS/ NOS1), endothelial (eNOS/NOS3), and inducible (iNOS/NOS2) isoforms of NOS have been identified. Neuronal and endothelial NOS are constitutively expressed and require Ca2+ for their activity, whereas iNOS expression is induced in response to a variety of stimuli, including hypoxia, and does not require Ca2+ for its activity. NOS isoforms contain a heme prosthetic group that binds to O2. nNOS has a high K m for O2 relative to the other NOS isoforms, suggesting that even modest reductions in O2 concentration lead to significant loss of enzyme activity [9]. Indeed, there is a linear relationship between O2 concentration and nNOS activity over the entire physiological range [10].
The effect of hypoxia on NOS activity in the carotid body was determined by measuring conversion of 3H-arginine to 3H-citrulline [11]. Substantial NOS activity was observed under normoxic conditions (PO2 ∼140 mmHg) and chelation of extracellular Ca2+ abolished the enzymatic activity, suggesting that it arises from constitutively expressed NOS isoforms [11]. NOS inhibitors (L-NNA or L-NAME) stimulate, and NO donors inhibit, carotid body activity [11–14]. These findings suggest that O2-dependent generation of NO exerts an inhibitory influence on the carotid body activity.
Glomus cells are the primary site of O2 sensing in the carotid body and they are innervated by sensory fibers originating from the petrosal ganglion [2]. Glomus cells do not express nNOS, which is instead produced by nerve fibers as well as neurons of the petrosal ganglion. Besides its sensory innervation, the carotid body receives an efferent nerve supply from the autonomic nervous system. Electrical stimulation of the cut end of the carotid sinus nerve causes depression of carotid body sensory nerve activity [15–17]. This finding implied that the carotid body response to hypoxia is regulated by an efferent inhibitory pathway. Indeed, the carotid body response to hypoxia is augmented in the absence of “efferent inhibition” [18]. Until recently, mechanisms mediating the “efferent inhibition” remained uncertain. The finding that NOS activity increases in response to electrical stimulation of the carotid sinus nerve (as evidenced by elevated 3H-citrulline production) and that carotid sinus nerve fibers express nNOS led to the suggestion that NO mediates efferent inhibition [14]. Studies by Campanucci and Nurse provided evidence that NO is an important mediator of efferent inhibition of the carotid body [19]. These investigators found that nNOS-expressing nerve fibers originate from neurons located in paraganglia along the glossopharyngeal and carotid sinus nerves (glossopharyngeal neurons or GPNs). It was proposed that NO released from efferent nerves regulates the excitability of glomus cells, resulting in decreased afferent nerve activity [19]. Glomus cells express a variety of K+ channels and hypoxia inhibits some of these K+ channels [4, 20]. Ca2+ influx via voltage gated Ca2+ channels is an obligatory step in producing sensory excitation by hypoxia [1]. NO donors enhance K+ conductance [21] and inhibit L-type voltage-activated Ca2+ channel activity in glomus cells [22] suggesting that NO depresses the glomus cell excitability. Kline et al. [23] reported that nNOS deficient (Nos1 −/−) mice exhibit an enhanced ventilatory response to hypoxia (Table 1), which is a hallmark reflex initiated by the carotid body, and irregular breathing during hypoxia, an effect that was attributed to enhanced carotid body sensitivity to low O2. These findings suggest an absence of efferent inhibition of the carotid body in nNOS deficient mice. Figure 1 illustrates the role of nNOS-derived NO in efferent inhibition of carotid body activity.

NO-mediated efferent inhibitory pathway in the carotid body. During normoxia, O2-dependent NO generation by neuronal nitric oxide synthase (nNOS) in glossopharyngeal neurons (GPN) mediates efferent inhibition by activating K+ channels and inhibiting L-type Ca2+ channels in glomus cells, which results in decreased sensory activity. Under hypoxic conditions, NO production is reduced, which contributes to sensory excitation
Endothelial NOS (eNOS) is expressed in the blood vessels that perfuse the carotid body [24]. eNOS knockout mice exhibit attenuated peripheral chemoreceptor sensitivity, reduced hypoxic ventilatory response, and hypertension [25]. The decreased sensitivity to hypoxia was associated with absence of eNOS expression in carotid body blood vessels. How might NO from eNOS affect the carotid body activity? Carotid body is a highly vascular organ and receives highest blood flow per gram of tissue [1]. NO derived from eNOS is a potent vasodilator and regulate the blood flow to the carotid body. The attenuated hypoxic sensitivity in eNOS knockout mice was attributed to persistent vasoconstriction resulting in reduced blood flow and the ensuing chronic tissue hypoxia of the carotid body. Consistent with this possibility, carotid bodies from eNOS knockout mice displayed hyperplasia of glomus cells [25], similar to that seen with chronic hypoxia [1].
The biological actions of NO are mediated by multiple pathways, which include activation of heme-containing guanylate cyclase and subsequent elevation of cyclic guanosine monophosphate (cGMP) levels [7]. NOS inhibitors reduce, and NO donors increase, cGMP levels in the carotid body [14]. Besides the cGMP pathway, effects of NO also involve the formation of S-nitrosothiols, whose actions depend on the redox environment of the cell [26, 27]. Summers et al. [22] reported that NO-induced inhibition of L-type Ca2+ currents in rabbit glomus cells is prevented by N-ethylmalemide, which blocks nitrosylation of proteins by NO, implicating S-nitrosothiol signaling. These studies, albeit limited, indicate that both cGMP-dependent and cGMP-independent signaling pathways mediate the effects of NO in glomus cells.
Carbon monoxide (CO)
CO was thought to be a physiologically inert gas like N2 and H2. Over the decades, respiratory physiologists employed CO as a tool to understand the mechanisms of O2 transport and the control of breathing by hypoxia. For instance, at the beginning of this century, Haldane and Krogh used CO to test whether O2 passively diffuses or is actively secreted from the lungs [28]. In the late 1960s, Lloyd, Cunningham, and co-workers [29] reported that brief inhalation of CO gas for several breaths eliminated hypoxia-evoked hyperventilation in human subjects. Because of the high affinity of CO for hemoglobin, they proposed that the deoxy conformation of a heme prosthetic group triggered hypoxic signaling by the carotid body [29].
Sjostrand [30] was one of the first to suggest that CO is formed during the breakdown of heme. It is now well established that CO is generated in mammalian cells during heme degradation by the enzyme heme oxygenase (HO), with NADPH and cytochrome P-450 reductase as co-factors [31]. Enzymatic generation of CO requires molecular oxygen. The affinity of the heme–HO complex for O2 is 30–90-fold greater than myoglobin [6]. Substrates for HO include the α and β chains of hemoglobin, denatured myoglobin, met-hemoglobin, and proteolytic products of cytochrome c; in contrast, oxy-hemoglobin and intact myoglobin or cytochrome b are not substrates for HO [32].
Inducible (HO-1) and constitutively expressed (HO-2) isoforms of HO have been identified [31], and the latter isoform is predominantly expressed in neuronal cells [33]. Carotid body glomus (type I) cells express HO-2 and its expression is not evident in either nerve fibers or type II supporting cells [34]. The K m of HO-1 and HO-2 for O2 are in the range of 30–80 μM [6].
Zinc-protoporphyrin-9 (ZnPP9), a potent HO inhibitor, stimulated carotid body activity in a dose-dependent manner, whereas CuPP9, which has negligible effects on HO activity, had no effect on carotid body activity [34]. Exogenous administration of CO reversed ZnPP9-induced sensory excitation of the carotid body. Furthermore, administration of an HO inhibitor augmented the hypoxic ventilatory response, and bilateral sectioning of carotid sinus nerves or exogenous CO abolished this effect [33]. These observations provided evidence that CO generated by HO-2 is an important gas messenger contributing to O2 sensing by the carotid body.
Once generated, CO might act back on the same glomus cell (autocrine action) and/or nearby glomus cells (paracrine action) and/or afferent nerve endings. Since elevation of cytosolic Ca2+ in glomus cells is an obligatory step in evoking sensory excitation in response to hypoxia [2], the effects of HO inhibitors on Ca2+ responses of glomus cells were tested. ZnPP9 elevated [Ca2+]i in glomus cells and this response was eliminated by exogenous administration of CO [35]. ZnPP9-evoked elevation of [Ca2+]i was absent after removing extracellular Ca2+, suggesting that this response requires activation of Ca2+ channels. Indeed, ZnPP9 augmented Ca2+ currents [35].
A recent study reported that hypoxia-evoked glomus cell secretion of catecholamines was not altered in Hmox2 −/− mice, which lack HO-2 expression [36]. Previous studies reported that glomus cell secretory response often does not coincide with sensory excitation of the carotid body by hypoxia [37]. Therefore, further studies are needed to determine whether the carotid body sensory response to hypoxia is altered in Hmox2 −/− mice. Not withstanding this limitation, pharmacological studies in intact rats suggest that endogenously generated CO inhibits carotid body sensory activity. Because HO-2 requires O2 for its activity, stimulation of carotid body activity by hypoxia may reflect, in part, reduced formation of CO [38]. Recent studies have provided interesting insights into the mechanism(s) by which CO exerts its inhibitory influence on carotid body activity (see below).
Hydrogen sulfide (H2S)
Cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS) are two major enzymes that catalyze the formation of H2S. While CBS is more abundant in the central nervous system, CSE is the predominant enzyme in peripheral tissues [39]. CSE immunoreactivity was seen in glomus cells of rat and mouse carotid bodies as demonstrated by co-localization with tyrosine hydroxylase, an established marker of glomus cells [40]. CBS expression was also reported in murine and cat carotid bodies [41, 42].
Peng et al. [40] examined the contribution of CSE to the carotid body response to hypoxia using genetic and pharmacological approaches. In Cth −/− mice, which lack CSE, basal H2S levels in the carotid bodies were reduced by one half compared to wild-type mice. The residual H2S must arise from sources other than CSE, presumably CBS. Cth −/− mice exhibited a severely impaired hypoxic sensory response (Table 1). A similar absence of the hypoxic sensory response was also seen in rats treated with DL-propargylglycine (PAG), an inhibitor of CSE [43, 44]. In contrast, the carotid body response to CO2 was unaffected in Cth −/− mice and was even augmented in rats treated with PAG, suggesting that impaired CSE function selectively affects the hypoxic sensory response. The ventilatory response to hypoxia was also severely impaired in Cth −/− mice and in PAG-treated rats. These findings demonstrate that CSE contributes to hypoxic sensing by catalyzing H2S formation.
Li et al. [41] reported that amino oxyacetic acid (AOAA) and hydroxylamine, which are inhibitors of CBS, impaired the mouse carotid body response to hypoxia and ventilatory stimulation by hypoxia. These results suggest that CBS-catalyzed H2S production may also play a role in sensory excitation by hypoxia.
Exogenous application of the H2S donor NaHS stimulates carotid body sensory activity in mice and rats in a concentration-dependent manner [40, 41]. NaHS-evoked sensory excitation, like hypoxia, was rapid in onset, occurred within seconds after its application, and returned to baseline after termination of the stimulus [40, 41]. NaHS also stimulated carotid body sensory activity in Cth −/− mice, suggesting that H2S is a downstream signaling molecule. It has been proposed that H2S also mediates O2 sensing by trout gill chemoreceptors, indicating that it is a conserved system across vertebrate phyla [45].
KATP channels are the major targets of H2S in blood vessels [46]. KATP channels are expressed in glomus cells [47]. However, glibenclamide, a potent inhibitor of KATP channels, was ineffective in preventing carotid body stimulation by NaHS or hypoxia [40]. The excitatory effects of H2S on carotid body activity seem to require Ca2+ [40, 41] and may involve enhanced excitability of glomus cells resulting from inhibition of Ca2+-activated K+ currents [41, 48].
Acetylcholine (ACh) and adenosine triphosphate (ATP) are putative excitatory transmitters implicated in the sensory transmission of the hypoxic stimulus at the carotid body [49]. Li et al. [41] reported that pyridoxal phosphate-6-azophenyl-2′,4′-disulfonic acid, which is an inhibitor of purinergic receptors, and hexamethonium, a blocker of nicotinic cholinergic receptors, prevented NaHS-evoked sensory excitation of the mouse carotid body. Based on these observations, Li et al. [41] suggested that H2S-induced sensory excitation is coupled to the release of ATP/ACh from glomus cells. However, Fitzgerald et al. [42] reported that Na2S, which is another H2S donor, inhibited ACh release and had variable effects on ATP release from cat carotid bodies. Pinacidil, an opener of KATP channels, mimicked the effects of Na2S on ACh and ATP release [42]. These findings are intriguing because H2S donors stimulate carotid body sensory activity, yet they seem to inhibit the release of ACh and ATP, two neurotransmitters that are implicated in sensory excitation by hypoxia.
Hypoxia increases H2S generation: evidence for interaction of CSE with HO-2
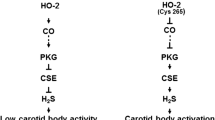
Hypoxia increased H2S levels in both mouse and rat carotid bodies in a stimulus-dependent manner and this response was absent in Cth −/− mice as well as in rats treated with PAG [40]. These findings suggest that CSE is a major source of H2S generation in response to hypoxia. How might hypoxia increase H2S generation? HO-2 catalyzes the formation of CO, which inhibits carotid body activity [34]. Since HO-2 generation of CO is O2 dependent, it is likely that the low levels of H2S under normoxia are due to the inhibitory influence of CO on CSE. This hypothesis is supported by the finding that an HO-2 inhibitor markedly elevated basal H2S levels in the carotid body under normoxia and increased baseline sensory activity in wild-type mice. Remarkably, these effects were absent in Cth −/− mice [40]. Furthermore, a CO donor inhibited hypoxia-evoked H2S generation in the carotid body [40]. These findings suggest that CO physiologically inhibits the CSE-dependent generation of H2S and that hypoxia reduces HO-2 activity to reverse the inhibition and augment H2S formation. It was proposed that interacting proteins working in concert as a “chemosome” mediate hypoxic sensing by the carotid body [3]. It is likely that interactions between HO-2 and CSE constitute an important component of the “chemosome”. The potential interaction between HO-2/CO and CSE/H2S and their contribution to the carotid body response to hypoxia are illustrated in Fig. 2. A recent study by Morikawa et al [50] reported that interaction between HO2/CO and CBS/H2S is critical for hypoxia evoked cerebral vasodilatation. However, unlike CBS, CSE is not a heme containing enzyme. Therefore, further studies are needed to delineate the precise molecular mechanisms by which CO inhibits H2S generation from CSE in the carotid body.

Potential interaction between heme oxygenase-2 (HO-2) and cystathionine γ-lyase (CSE) in glomus cells of the carotid body. O2-dependent CO generation from HO-2 inhibits CSE activity resulting in low levels of H2S and sensory activity. During hypoxia CO generation from HO-2 is reduced, resulting in removal of CO inhibition on CSE, leading to elevated H2S levels, which contribute to sensory excitation
Role of hypoxia-inducible factors in O2 sensing by the carotid body
Hypoxia-inducible factors (HIFs) play essential roles as master regulators of O2 and redox homeostasis [8]. HIF-1 and HIF-2 are heterodimeric proteins, which consist of a constitutively expressed HIF-1β subunit and an O2-regulated HIF-1α or HIF-2α subunit, respectively. Hif1a −/− mice, which have a complete deficiency of HIF-1α, die at midgestation with defects in the heart, blood vessels, and red blood cell production, indicating that all three components of the circulatory system are dependent upon HIF-1 for their normal development [51–53]. Hif1a +/− mice, which have a partial deficiency of HIF-1α, develop normally but have impaired responses to hypoxia and ischemia [54–63].
Glomus cells express low levels of HIF-1α and relatively high levels of HIF-2α [1]. Carotid bodies from Hif1a +/− mice appear histologically normal but do not respond to hypoxia [59] (Table 1). In contrast to Hif1a +/− mice, carotid bodies from Epas1 +/− mice, which have a partial deficiency of HIF-2α, show augmented responses to hypoxia (Table 1) and Epas1 +/− mice manifest hypertension and respiratory instability, two hallmarks of carotid body hyperactivity [64]. HIF-2α is required for the production of anti-oxidant enzymes [60] and the expression of SOD2 mRNA, which encodes manganese superoxide dismutase, is significantly reduced in the carotid bodies of Epas1 +/− mice [64]. Daily treatment of Epas1 +/− mice for 2 weeks with manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride, a potent membrane permeable anti-oxidant, corrected the hyperactive carotid body response to hypoxia, hypertension, and respiratory instability [64]. In contrast, Hif1a +/− mice have impaired carotid body expression of NOX2 mRNA, which encodes NADPH oxidase, a pro-oxidant enzyme that generates superoxide [65]. Taken together, these data suggest that a balance between HIF-1α and HIF-2α is required for redox homeostasis in the carotid body, which in turn is required for the maintenance of respiratory and cardiovascular homeostasis.
Perspective, physiological and clinical implications
Reflexes from the carotid bodies are important for physiological adaptations to chronic hypoxia. For instance, ventilatory acclimatization to hypoxia (VAH), a hallmark adaptation to high altitude, is manifested by progressive increase in ventilation. VAH is absent in carotid body denervated animals and in mice deficient in HIF-1α [1]. Chronic hypoxia lasting several days to months lead to desensitization of the carotid body response to hypoxia and blunted hypoxic ventilatory response, which was attributed to upregulation of iNOS expression and the resulting increase in NO [1]. The roles of gaseous messengers other than NO in the chemoreflex function during chronic hypoxia have not been investigated.
Carotid body reflexes are implicated in the pathogenesis of a variety of cardiovascular diseases associated with autonomic dysfunction [66]. For instance, a heightened carotid body reflex may underlie the increased sympathetic nervous system activity seen in patients with sleep-disordered breathing due to obstructive sleep apnea, congestive heart failure (CHF), and neurogenic hypertension. It is likely that altered levels of gas messengers as well as alterations in redox homeostasis by HIFs contribute to heightened carotid body reflexes under these conditions. Recent studies involving an experimental model of CHF lend partial support to this hypothesis. Rabbits with CHF exhibit enhanced carotid body sensitivity to hypoxia and augmented sympathetic activity, and these effects were associated with reduced nNOS expression in the carotid body [21]. Overexpression of nNOS in the carotid body using adenoviral vectors partially normalized the carotid body response to hypoxia and sympathetic nerve activity in rabbits with CHF [21]. In a mouse model of obstructive sleep apnea, increased expression of HIF-1α and decreased expression of HIF-2α leading to increased ROS levels (Fig. 3) has been shown to play a critical role in the dysregulation of autonomic control of blood pressure and breathing [61, 67]. These findings suggest that enzymes generating gas messengers and redox regulation by HIFs represent potential therapeutic targets in CHF, sleep-disordered breathing, and neurogenic hypertension.

Pathogenesis of hypertension in patients with obstructive sleep apnea. Airway obstruction results in hypoxemia, awakening, airway clearing, and reoxygenation. This cycle of intermittent hypoxia is repeated dozens of times per night. In the carotid body, chronic intermittent hypoxia results in increased ROS levels, which induce increased synthesis and decreased degradation of HIF-1α as well as increased degradation of HIF-2α. Increased HIF-1α-dependent NOX2 expression and decreased HIF-2α-dependent SOD2 expression result in further increases in ROS levels, which trigger glomus cell depolarization, leading to sympathetic activation and systemic hypertension
Besides carotid bodies, aortic bodies located at the aortic arch represent another O2-sensing chemo-sensory organs [1, 2]. However, compared to carotid body, the role of O2 sensing by the aortic body in health and disease is less well established. Also, little is known on the cellular mechanisms and the role of gaseous messengers in O2 sensing by the aortic bodies.
References
Kumar P, Prabhakar NR (2012) Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol 2:141–219
Prabhakar NR (2000) Oxygen sensing by the carotid body chemoreceptors. J Appl Physiol 88:2287–2295
Prabhakar NR (2006) O2 sensing at the mammalian carotid body: why multiple O2 sensors and multiple transmitters? Exp Physiol 91:17–23
Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL (2005) Acute oxygen-sensing mechanisms. N Engl J Med 353:2042–2055
Abu-Soud HM, Rousseau DL, Stuehr DJ (1996) Nitric oxide binding to the heme of neuronal nitric-oxide synthase links its activity to changes in oxygen tension. J Biol Chem 271:32515–32518
Migita CT, Matera KM, Ikeda-Saito M, Olson JS, Fujii H, Yoshimura T, Zhou H, Yoshida T (1998) The oxygen and carbon monoxide reactions of heme oxygenase. J Biol Chem 273:945–949
Snyder SH (1992) Nitric oxide: first in a new class of neurotransmitters. Science 257:494–496
Semenza GL (2011) Oxygen sensing, homeostasis, and disease. N Engl J Med 365:537–547
Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S (2004) Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem 279:36167–36170
Elayan IM, Axley MJ, Prasad PV, Ahlers ST, Auker CR (2000) Effect of hyperbaric oxygen treatment on nitric oxide and oxygen free radicals in rat brain. J Neurophysiol 83:2022–2029
Prabhakar NR, Kumar GK, Chang CH, Agani FH, Haxhiu MA (1993) Nitric oxide in the sensory function of the carotid body. Brain Res 625:16–22
Chugh DK, Katayama M, Mokashi A, Bebout DE, Ray DK, Lahiri S (1994) Nitric oxide-related inhibition of carotid chemosensory nerve activity in the cat. Respir Physiol 97:147–156
Trzebski A, Sato Y, Suzuki A, Sato A (1995) Inhibition of nitric oxide synthesis potentiates the responsiveness of carotid chemoreceptors to systemic hypoxia in the rat. Neurosci Lett 190:29–32
Wang ZZ, Stensaas LJ, Dinger BG, Fidone SJ (1995) Nitric oxide mediates chemoreceptor inhibition in the cat carotid body. Neuroscience 65:217–229
Fidone SJ, Sato A (1970) Efferent inhibition and antidromic depression of chemoreceptor A-fibers from the cat carotid body. Brain Res 22:181–193
Neil E, O'Regan RG (1971) Efferent and afferent impulse activity recorded from few-fibre preparations of otherwise intact sinus and aortic nerves. J Physiol 215:33–47
Neil E, O’Regan RG (1971) The effects of electrical stimulation of the distal end of the cut sinus and aortic nerves on peripheral arterial chemoreceptor activity in the cat. J Physiol 215:15–32
Lahiri S, Smatresk N, Pokorski M, Barnard P, Mokashi A (1983) Efferent inhibition of carotid body chemoreception in chronically hypoxic cats. Am J Physiol 245:R678–R683
Campanucci VA, Nurse CA (2007) Autonomic innervation of the carotid body: role in efferent inhibition. Respir Physiol Neurobiol 157:83–92
Peers C, Wyatt CN, Evans AM (2010) Mechanisms for acute oxygen sensing in the carotid body. Respir Physiol Neurobiol 174:292–298
Schultz HD, Li YL (2007) Carotid body function in heart failure. Respir Physiol Neurobiol 157:171–185
Summers BA, Overholt JL, Prabhakar NR (1999) Nitric oxide inhibits L-type Ca2+ current in glomus cells of the rabbit carotid body via a cGMP-independent mechanism. J Neurophysiol 81:1449–1457
Kline DD, Yang T, Huang PL, Prabhakar NR (1998) Altered respiratory responses to hypoxia in mutant mice deficient in neuronal nitric oxide synthase. J Physiol 511(Pt 1):273–287
Wang ZZ, Stensaas LJ, Bredt DS, Dinger B, Fidone SJ (1994) Localization and actions of nitric oxide in the cat carotid body. Neuroscience 60:275–286
Kline DD, Yang T, Premkumar DR, Thomas AJ, Prabhakar NR (2000) Blunted respiratory responses to hypoxia in mutant mice deficient in nitric oxide synthase-3. J Appl Physiol 88:1496–1508
Campbell DL, Stamler JS, Strauss HC (1996) Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J Gen Physiol 108:277–293
Stamler JS (1994) Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell 78:931–936
Torrance RW (1996) Carbon monoxide excretion, not oxygen secretion? Adv Exp Med Biol 410:335–339
Lloyd BB, Cunningham DJC, Goode RC (1968) Depression of hypoxic hyperventilation in man by sudden inspiration of carbon monoxide. In: Torrance RW (ed) Arterial Chemoreceptors Oxford, Blackwell, pp 145–150
Sjostrand T (1949) Endogenous formation of carbon monoxide in man. Nature 164:580
Maines MD (1997) The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 37:517–554
Kutty RK, Maines MD (1981) Purification and characterization of biliverdin reductase from rat liver. J Biol Chem 256:3956–3962
Prabhakar NR (1998) Endogenous carbon monoxide in control of respiration. Respir Physiol 114:57–64
Prabhakar NR, Dinerman JL, Agani FH, Snyder SH (1995) Carbon monoxide: a role in carotid body chemoreception. Proc Natl Acad Sci U S A 92:1994–1997
Overholt JL, Bright GR, Prabhakar NR (1996) Carbon monoxide and carotid body chemoreception. Adv Exp Med Biol 410:341–344
Ortega-Saenz P, Pascual A, Gomez-Diaz R, Lopez-Barneo J (2006) Acute oxygen sensing in heme oxygenase-2 null mice. J Gen Physiol 128:405–411
Donnelly DF (1996) Chemoreceptor nerve excitation may not be proportional to catecholamine secretion. J Appl Physiol 81:657–664
Prabhakar NR (1999) NO and CO as second messengers in oxygen sensing in the carotid body. Respir Physiol 115:161–168
Gadalla MM, Snyder SH (2010) Hydrogen sulfide as a gasotransmitter. J Neurochem 113:14–26
Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR (2010) H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci U S A 107:10719–10724
Li Q, Sun B, Wang X, Jin Z, Zhou Y, Dong L, Jiang LH, Rong W (2010) A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid Redox Signal 12:1179–1189
Fitzgerald RS, Shirahata M, Chang I, Kostuk E, Kiihl S (2011) The impact of hydrogen sulfide (H(2)S) on neurotransmitter release from the cat carotid body. Respir Physiol Neurobiol 176:80–89
Abeles RH, Walsh CT (1973) Acetylenic enzyme inactivators. Inactivation of gamma-cystathionase, in vitro and in vivo, by propargylglycine. J Am Chem Soc 95:6124–6125
Washtien W, Abeles RH (1977) Mechanism of inactivation of gamma-cystathionase by the acetylenic substrate analogue propargylglycine. Biochemistry 16:2485–2491
Olson KR, Healy MJ, Qin Z, Skovgaard N, Vulesevic B, Duff DW, Whitfield NL, Yang G, Wang R, Perry SF (2008) Hydrogen sulfide as an oxygen sensor in trout gill chemoreceptors. Am J Physiol Regul Integr Comp Physiol 295:R669–R680
Zhao W, Zhang J, Lu Y, Wang R (2001) The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J 20:6008–6016
Kim D, Kim I, Papreck JR, Donnelly DF, Carroll JL (2011) Characterization of an ATP-sensitive K(+) channel in rat carotid body glomus cells. Respir Physiol Neurobiol 177:247–255
Telezhkin V, Brazier SP, Cayzac SH, Wilkinson WJ, Riccardi D, Kemp PJ (2010) Mechanism of inhibition by hydrogen sulfide of native and recombinant BKCa channels. Respir Physiol Neurobiol 172:169–178
Nurse CA (2010) Neurotransmitter and neuromodulatory mechanisms at peripheral arterial chemoreceptors. Exp Physiol 95:657–667
Morikawa T, Kajimura M, Nakamura T, Hishiki T, Nakanishi T, Yukutake Y, Nagahata Y, Ishikawa M, Hattori K, Takenouchi T et al (2012) Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc Natl Acad Sci U S A. doi:10.1073/pnas.1119658109
Compernolle V, Brusselmans K, Franco D, Moorman A, Dewerchin M, Collen D, Carmeliet P (2003) Cardia bifida, defective heart development and abnormal neural crest migration in embryos lacking hypoxia-inducible factor-1alpha. Cardiovasc Res 60:569–579
Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY et al (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 12:149–162
Yoon D, Pastore YD, Divoky V, Liu E, Mlodnicka AE, Rainey K, Ponka P, Semenza GL, Schumacher A, Prchal JT (2006) Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J Biol Chem 281:25703–25711
Bosch-Marce M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, Zhang H, Strazza M, Rey S, Savino L et al (2007) Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ Res 101:1310–1318
Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL, Semenza GL (2003) Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia–reperfusion injury. Circulation 108:79–85
Cai Z, Zhong H, Bosch-Marce M, Fox-Talbot K, Wang L, Wei C, Trush MA, Semenza GL (2008) Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1 alpha. Cardiovasc Res 77:463–470
Feinman R, Deitch EA, Watkins AC, Abungu B, Colorado I, Kannan KB, Sheth SU, Caputo FJ, Lu Q, Ramanathan M et al (2010) HIF-1 mediates pathogenic inflammatory responses to intestinal ischemia–reperfusion injury. Am J Physiol Gastrointest Liver Physiol 299:G833–G843
Kannan KB, Colorado I, Reino D, Palange D, Lu Q, Qin X, Abungu B, Watkins A, Caputo FJ, Xu DZ et al (2011) Hypoxia-inducible factor plays a gut-injurious role in intestinal ischemia reperfusion injury. Am J Physiol Gastrointest Liver Physiol 300:G853–G861
Kline DD, Peng YJ, Manalo DJ, Semenza GL, Prabhakar NR (2002) Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. Proc Natl Acad Sci U S A 99:821–826
Li J, Bosch-Marce M, Nanayakkara A, Savransky V, Fried SK, Semenza GL, Polotsky VY (2006) Altered metabolic responses to intermittent hypoxia in mice with partial deficiency of hypoxia-inducible factor-1alpha. Physiol Genomics 25:450–457
Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR (2006) Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol 577:705–716
Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT et al (1999) Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest 103:691–696
Zhang X, Liu L, Wei X, Tan YS, Tong L, Chang R, Ghanamah MS, Reinblatt M, Marti GP, Harmon JW et al (2010) Impaired angiogenesis and mobilization of circulating angiogenic cells in HIF-1alpha heterozygous-null mice after burn wounding. Wound Repair Regen 18:193–201
Peng YJ, Nanduri J, Khan SA, Yuan G, Wang N, Kinsman B, Vaddi DR, Kumar GK, Garcia JA, Semenza GL et al (2011) Hypoxia-inducible factor 2alpha (HIF-2alpha) heterozygous-null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc Natl Acad Sci U S A 108:3065–3070
Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR (2011) Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol 226:2925–2933
Prabhakar NR, Peng YJ (2004) Peripheral chemoreceptors in health and disease. J Appl Physiol 96:359–366
Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR (2009) Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci U S A 106:1199–1204
Acknowledgments
This work was supported by the National Institutes of Health grants HL-76537, HL-90554, and HL-86493 (N.R.P) and the Johns Hopkins Institute for Cell Engineering (G.L.S).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Prabhakar, N.R., Semenza, G.L. Gaseous messengers in oxygen sensing. J Mol Med 90, 265–272 (2012). https://doi.org/10.1007/s00109-012-0876-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-012-0876-1