
Abstract
Bortezomib represents the first proteasome inhibitor (PI) with demonstrated antitumor activity in the clinical setting, particularly for treatment of hematological malignancies. At the preclinical level, its action is shown to be mediated by induction of growth arrest and apoptosis in many tumor types, including androgen-dependent (AD) and androgen-independent (AI) prostate cancer (PCa) cells. Hypoxia-inducible factor-1α (HIF-1α), which is directly involved in tumor growth, is one of the most studied and promising molecular targets for anti-cancer therapy and is often overexpressed in PCa. Bortezomib has been reported to impair tumor growth by also inhibiting HIF-1α. In this study, we investigated the effect of bortezomib on the expression, activity and localization of HIF-1α in LNCaP (AD) and PC3 (AI) PCa cells. First, we show that hypoxic upregulation of HIF-1α protein levels and activity involves both the PI3K/Akt/mTOR and p44/42 MAPK pathways. Second, bortezomib inhibits expression of HIF-1α protein under both normoxic and hypoxic conditions, represses HIF-1 transcriptional activity and attenuates the release of vascular endothelial growth factor. These effects correlate with the ability of bortezomib to cause dephosphorylation of phospho-Akt, phospho-p70S6K, and phospho-S6RP, thus inactivating a pathway known to be required for HIF-1α protein expression at the translational level. Furthermore, bortezomib also abrogates p44/42 MAPK phosphorylation, which results to reduced nuclear translocation of HIF-1α. Taken together, these results suggest that bortezomib inhibits HIF-1α protein synthesis and its nuclear targeting through suppression of PI3K/Akt/mTOR and MAPK pathways, respectively, in both AD and AI PCa cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bortezomib (PS-341, Velcade®) is a specific and reversible boronate inhibitor of the chymotryptic activity of the 20S proteasome currently approved for the treatment of multiple myeloma and mantle cell lymphoma [1]. Bortezomib has been studied extensively in vitro and in vivo, and anticancer activity was observed in cellular and animal models of several solid tumors, including androgen-dependent (AD) and androgen-independent (AI) prostate cancer (PCa) cells [2]. Bortezomib indirectly inhibits angiogenesis in multiple myeloma and endothelial cells by decreasing vascular endothelial growth factor (VEGF) levels [3], and these effects involve the repression of HIF-1[4].
The transcriptional activator HIF-1 plays a crucial role in adaptation to low oxygen conditions and tumor progression by switching on genes that are involved in angiogenesis, cell metabolism, apoptosis, invasion, or/and metastasis. HIF-1 is a heterodimer composed of two subunits, HIF-1α and HIF-1β (also known as ARNT). HIF-1α is the oxygen-regulated subunit that determines HIF-1 activity. Under normoxic conditions, HIF-1α is hydroxylated by HIF-1 prolyl hydroxylases (PHDs), and subsequently targeted by the von Hippel–Lindau protein, ubiquitinated, and finally degraded by the 26S proteasome. In addition, HIF-1α is hydroxylated at Asn803 by the factor inhibiting HIF-1 (FIH), which represses HIF-1α transcriptional activity by blocking recruitment of the p300 coactivator. Under hypoxic conditions, HIF-1α is stabilized and translocates into the nucleus, where it heterodimerizes with HIF-1β, thereby activating transcription of several target genes, including VEGF [5].
In addition to oxygen-dependent regulation, HIF-1α activation is controlled by multiple oncogenic pathways, which involve loss of tumor suppressor genes, such as PTEN and growth factor signaling. PTEN negatively regulates downstream effectors of phosphatidyl inositol-3 kinase (PI3K), such as Akt, which has been reported to increase HIF-1α protein synthesis and mediate VEGF induction under hypoxia [6]. On the other hand, enhanced MAPK signaling is a common event in neoplasia as constitutively active MAPK signaling transforms benign mammalian cells to malignant [7]. p44/42 (also known as ERK1/2) refers to two serine/threonine kinases of the MAPK signaling pathway that have been implicated in HIF-1 activation by promoting the formation and modulating the transactivation activity of the HIF-p300/CBP complex [8]. MAPK activation also increases HIF-1 transcriptional activity via direct phosphorylation of HIF-1α [9]. More recent studies have shown that p44/42-mediated modification of HIF-1α at residues Ser641 and Ser643 causes activation of HIF-1 by inhibiting the CRM1-dependent nuclear export of HIF-1α [10–12].
The underlying mechanism of bortezomib-mediated inhibition of HIF-1α function is not fully understood, and published data are controversial. Previous studies have shown that bortezomib inhibits the hypoxic response by stimulating the interaction between the C-terminal activation domain (CAD) of HIF-1α and the FIH, thus impairing recruitment of p300 coactivator to HIF-1 [13]. However, it has also been reported that CAD inhibition by bortezomib is FIH independent [14, 15]. More recently, bortezomib was suggested to control HIF-1α expression via phosphorylation of eIF2α, which caused inhibition of HIF-1α protein translation in PCa cells [16].
In this work, we have analysed several levels of HIF-1α regulation in AD (LNCaP) and AI (PC3) PCa cells exposed to the proteasome inhibitor bortezomib. Our results show that bortezomib inhibits HIF-1α protein synthesis by impairing the PI3K/Akt/mTOR pathway. Furthermore, we show for the first time that bortezomib interferes with the p44/42 MAPK (ERK1/2) pathway in PCa cells, causing, thereby, efficient repression of HIF-1α nuclear accumulation and transcriptional activity. These data suggest a multi-potent role for bortezomib, at least, in PCa, and offer novel therapeutic perspectives for its clinical application.
Materials and methods
Cell lines and culture
Human LNCaP and PC3 PCa cells lines were purchased from the European Collection of Animal Cell Cultures (ECACC, Health Protection Agency, Salisbury, UK). Cells were cultured in RPMI 1640 (Euroclone, UK)) supplemented with 10% heat-inactivated fetal bovine serum (FBS; (Gibco, UK), 5% l-glutamine (Gibco, UK), 100 U/ml penicillin/streptomycin solution (Euroclone, UK). All cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. For all experiments, NEP-specific activity was measured, and low-passage LNCaP cells were used [17]. For hypoxic treatment, cells were exposed for the indicated times to 1% O2, 94% N2 and 5% CO2 in an IN VIVO2 200 hypoxia work-station (RUSKINN Life Sciences). All other incubation protocols were as stated in figure legends.
Reagents and antibodies
The PI bortezomib (VELCADE®) was purchased from Janssen-Cilag Pharmaceuticals, Greece. TransPassTM D2 Transfection Reagent was from New England Biolabs Inc., Beverly, MA, USA. Antibodies were obtained from the following commercial sources: human HIF-1α and HIF-1β/ARNT (BD Biosciences Transduction Laboratories, New Jersey, USA); phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), p44/42 MAPK (Erk1/2), phospho-Akt (Ser473), total Akt; phospho-p70 S6 kinase (Thr389); phospho-S6 ribosomal protein (Ser235/236), total eIF2a (Cell Signaling Technology, Inc., Danvers, USA); Histone 2B (Abcam, Cambridge, UK). Horseradish peroxidise-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology, California, USA. The kinase inhibitors, LY294002 (PI3 Kinase Inhibitor), rapamycin (FRAP/mTOR Inhibitor), and PD98059 (MEK1 Inhibitor) were from Cell Signaling Technology, Inc.
Western blot and immunofluorescence
Fractionation of cells, analysis of fractions, or total cellular proteins by sodium dodecyl sulfate polyacrylamide gel electrophoresis, immunoblotting, and immunofluorescent microscopy were carried out as previously described [10–12]. All experiments were performed in triplicate and representative results are shown.
Luciferase reporter assays
To examine the transcriptional activity of HIF-1, LNCaP and PC3 cells were cotransfected with the firefly luciferase reporter plasmid pGL3–5HRE-VEGF and the Renilla luciferase expressing plasmid PCIRenilla, under the control of an autologous promoter (pGL3) as previously described [10, 11]. Luciferase activity was measured using the dual-luciferase assay system (Promega, Wisconsin, USA) with a luminometer (TD20/20, Turner Designs).
Quantification of VEGF by ELISA
LNCaP and PC3 cells were plated in six-well plates and were incubated with indicated chemicals for 24 h under normoxic or hypoxic conditions (1% O2). Media were then collected, and VEGF levels were determined using Quantikine ELISA kits (R&D Systems, Inc.). Results were expressed as VEGF concentrations (pg/μg protein).
20S Proteasome enzymatic activity assay
Total protein cell lysates were prepared using a 0.5% CHAPS buffer, which did not affect proteasomal enzymatic activity. Chymotryptic activity of the 20S proteasome was measured in total cell lysates as previously described [17].
Statistical analysis
The Graph Pad Instat Statistical package for Windows was used. Data are expressed as mean ± standard deviation (SD). The one-way analysis of variance (ANOVA) with the Bonferroni post-test was used for the comparison of data, and the statistical significance limit was set at p < 0.05.
Results
Hypoxic induction of HIF-1α protein and activity in PCa cells involves the PI3K/Akt/mTOR and MAPK pathways
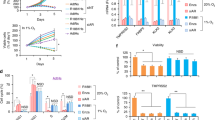
To assess the pattern of HIF-1α protein expression after treatment with hypoxia in PC3 and LNCaP cells, we performed a kinetic analysis. As shown in Fig. 1a, LNCaP cells do not express HIF-1α in normoxia, whereas a basal HIF-1α level was detected in normoxic PC3 cells. Under hypoxic conditions, HIF-1α was upregulated at 2 h and HIF-1α protein levels remained high up to 48 h in both LNCaP and PC3 cells.

Effect of PI3K/Akt, MAPK, and mTOR pathways inhibition on HIF-1α induction and transcriptional activity under hypoxia in LNCaP and PC3 cells. a Kinetics of HIF-1α accumulation in LNCaP and PC3 cells. Cells were exposed to hypoxia (1% O2), for the indicated times, and HIF-1α was measured by immunoblotting. Actin served as a loading control. b Inhibition of the PI3K/Akt/mTOR pathway but not of the MAPK pathway prevents HIF-1α protein induction by hypoxia in LNCaP and PC3 cells. Cells were pretreated for 20 min with the indicated concentrations of PD98059 or LY294002 or rapamycin. In all cases, cells were exposed to hypoxia after pre-treatment and incubation was continued for 16 h. HIF-1α, p-MAPK, MAPK, p-Akt, Akt, and p-S6 were measured by immunoblotting. Actin served as a loading control. The lines divide images from different parts of the same gel. c Inhibition of PI3K/Akt and MAPK signaling represses the hypoxia-induced upregulation of HIF-1α transcriptional activity. LNCaP and PC3 cells were treated with the indicated concentrations of PD98059 or LY294002 or both for 20 min, followed by culturing at normoxic or hypoxic conditions for 16 h. HIF-1α transcriptional activities were measured using an HRE-driven firefly luciferase expression construct and a renilla luciferase construct as an internal normalization control. Results are shown as fold increase in relation to the corresponding normoxic conditions and represent the mean of three independent experiments performed in triplicate (±SEM) (*p < 0.001; no inhibitors versus inhibitors treated)
We then investigated the role of PI3K pathway in HIF-1α induction under hypoxia in LNCaP and PC3 cells. The PI3K pathway was active in both cell lines under both normoxia and hypoxia, as evidenced by high levels of phosphorylated Akt, the downstream effector of PI3K. Treatment of LNCaP and PC3 cells with the PI3K inhibitor LY294002 (Fig. 1b, left panels), which as expected reduced the levels of phospho-Akt(Ser473), decreased the expression of HIF-1α, suggesting that induction of HIF-1α by hypoxia requires activation of the PI3K pathway. To confirm this finding, we used rapamycin, an inhibitor of mTOR, acting downstream of both PI3K and Akt. Treatment of cells with rapamycin, which as expected blocked S6 ribosomal protein phosphorylation(Ser235/236) (an established downstream target of mTOR), also impaired the induction of HIF-1α by hypoxia (Fig. 1b, right panels).
Analysis of the MAPK pathway under the same conditions revealed that hypoxia induced p44/42 MAPK phosphorylation in both LNCaP and PC3 cells. However, treatment with the MAPK pathway inhibitor PD98059, which reduced the levels of p44/42 MAPK phosphorylation, had no detectable effect on HIF-1α protein levels under hypoxia (Fig. 1b, left panels).
We then examined the effect of the PI3K and the MAPK inhibitors on the hypoxia-induced transcriptional activity of HIF-1. As measured by luciferase reporter gene assays in transfected cells, hypoxic treatment led to an approximately 70-fold increase in HIF-1 transcriptional activity in LNCaP cells and a 10-fold increase in PC3 cells, compared to untreated cells (Fig. 1c). Addition of each inhibitor alone or in combination significantly reduced the hypoxia-stimulated HIF-1α transcriptional activity in both cell lines, although this effect was more pronounced in LNCaP compared to PC3 cells. The survival of LNCaP and PC3 cells was tested after treatment with LY294002 and PD98059 under hypoxia for 16 h, and no significant loss of viability was detected in either cell line (Supplementary Fig. 1). Thus, under hypoxia inhibition of the PI3K pathway impairs both protein and activity levels of HIF-1α, whereas inhibition of the MAPK pathway impairs the activity of HIF-1α without affecting its protein expression levels.
Bortezomib attenuates both HIF-1α expression and transcriptional activity in PCa cells under hypoxia
The effects of bortezomib on HIF-1α expression were analysed in PC3 and LNCaP cells under both normoxic and hypoxic conditions. In normoxia, a basal amount of HIF-1α protein was detected only in PC3 cells, and this was almost completely abrogated after incubation with either 10 or 100 nM of bortezomib whereas a lower dose (1 nM) of bortezomib had no significant effect. After 16 h exposure to hypoxia, HIF-1α levels were significantly increased in both cell lines, but the addition of 10 or 100 nM bortezomib significantly reversed this effect, while 1 nM bortezomib remained ineffective (Fig. 2a).

Effect of bortezomib on HIF-1α induction and VEGF expression in LNCaP and PC3 cells. a Bortezomib down-regulates HIF-1α protein level. Cells were exposed to increasing concentrations of bortezomib (1, 10, and 100 nM) under normoxic or hypoxic conditions for 16 h and HIF-1α was measured by immunoblotting. Actin served as a loading control. Densitometric analysis of the bands in blots was performed with the public domain software for image analysis ‘ImageJ’ (National Institute of Health, USA). b Bortezomib down-regulates HIF-1α transcriptional activity. LNCaP and PC3 cells were treated with bortezomib (1, 10, and 100 nM) under normoxic or hypoxic conditions for 16 h. HIF-1a transcriptional activities were measured using an HRE-driven firefly luciferase expression construct and a renilla luciferase construct as an internal normalization control. Results are shown as fold increase in relation to the corresponding normoxic conditions and represent the mean of three independent experiments performed in triplicate (±SEM) (*p < 0.001; no bortezomib versus bortezomib-treated). c Bortezomib inhibits VEGF expression. LNCaP and PC3 cells were treated with bortezomib (1, 10, and 100 nM) under normoxic or hypoxic conditions for 16 h. VEGF expression was measured by VEGF ELISA (R&D Systems) in conditioned medium. (# p < 0.001; normoxia versus hypoxia, *p < 0.001; no bortezomib versus bortezomib-treated). d Bortezomib inhibits proteasome activity. LNCaP and PC3 cells were treated with bortezomib (1, 10, and 100 nM) under normoxic or hypoxic conditions for 16 h. The proteasome activity was measured by a fluorogenic assay kit (Chemicon International) in total cell lysates. Data represent the mean (±SEM) of three independent experiments performed in triplicate and are expressed as percent of the initial value of proteasome activity in normoxia (*p < 0.001; no bortezomib versus bortezomib-treated)
We then examined the effect of bortezomib on HIF-1 transcriptional activity using a reporter gene assay. Treatment of hypoxic cells with either 10 or 100 nM of bortezomib resulted in a significant reduction of transcriptional activity of HIF-1α in both LNCaP and PC3 cells (Fig. 2b). Interestingly, the inhibition of HIF-1 transcriptional activity by 100 nM bortezomib was much more pronounced than its effects on HIF-1α protein expression levels (compare Fig. 2a, b), indicating a dual mode of inhibitory action for bortezomib in PCa cells.
Since bortezomib could down-regulate both HIF-1α protein levels and transcriptional activity, we tested the impact of bortezomib on production of VEGF, a known target of HIF-1, by PC3 and LNCaP cells. As expected, hypoxia upregulated VEGF protein secretion (Fig. 2c) by up to 1.8-fold in LNCaP and 1.5-fold in PC3. However, this effect was inhibited by bortezomib in a dose-dependent manner (Fig. 2c). Baseline VEGF secretion levels of both cell lines in normoxia were also decreased after treatment with bortezomib (Fig. 2c).
We then measured 20S proteasome activity of PC3 and LNCaP cells under both normoxic and hypoxic conditions in the presence of bortezomib (1, 10, and 100 nM). There was no significant change in proteasome activity after addition of 1 nM bortezomib, whereas treatment of normoxic or hypoxic cells with either 10 or 100 nM of bortezomib resulted in significant reduction of proteasome activity in both cell lines, in a dose-dependent manner (Fig. 2d).
Bortezomib inhibits the PI3K/Akt and MAPK signaling pathways in PCa cells under hypoxia
To test whether inhibition of HIF-1α expression by bortezomib is associated with modulation of PI3K/Akt or/and MAPK signaling, we examined the effect of bortezomib on these pathways in PC3 and LNCaP cells. As shown in Fig. 3a, bortezomib down-regulated phospho-Akt(Ser473) in hypoxia in both cell types in a dose-dependent manner, whereas protein levels of total Akt were not altered. Furthermore, bortezomib partially inhibited phosphorylation of the downstream targets of Akt, p70S6K(Thr389) and S6RP(Ser235/236), in both cell lines and in a way that parallels the decrease in HIF-1α protein levels (Fig. 3b). Phosphorylation of p44/42 MAPK was increased in hypoxia but was markedly reduced in a dose-dependent manner after treatment with bortezomib, while the total amount of p44/42 MAPK remained unchanged under the same conditions (Fig. 3c). Since both PI3K/Akt and MAPK pathways are required for HIF-1α protein expression and activity under hypoxia, their effective inhibition by bortezomib explains its negative effects on HIF-1 activity in PCa cells.

Effect of bortezomib on HIF-1α translation regulation. a Bortezomib inhibits Akt activation. Cells were exposed to increasing concentrations of bortezomib (1, 10, and 100 nM) under hypoxic conditions for 16 h and phosphorylated Akt levels, and total Akt levels were measured by immunoblotting. b Bortezomib down-regulates phospho-p70 S6 kinase and phospho-S6 ribosomal protein. Cells were treated as above under hypoxic conditions and HIF-1α, phosphorylated p70 S6 kinase, and phosphorylated S6 ribosomal protein were measured by immunoblotting. Actin served as a loading control. c Bortezomib inhibits p44/42 MAPK activation. Cells were treated as above under hypoxic conditions, and phosphorylated MAPK and total MAPK were measured by immunoblotting
Bortezomib impairs HIF-1α nuclear accumulation via p44/42 MAPK inactivation
As already mentioned above, the inhibitory effect of bortezomib on HIF-1 transcriptional activity was more pronounced compared to its negative effect on HIF-1α protein expression levels, suggesting that residual HIF-1α in cells treated with bortezomib might be devoid of activity (Fig. 2a, b). To investigate this phenomenon, we analyzed the effect of bortezomib on the intracellular localization of HIF-1α by indirect immunofluorescence microscopy. In normoxia, HIF-1α could be barely detected in LNCaP or PC3 cells, while a strong nuclear signal of HIF-1α was readily visible under hypoxic conditions (Fig. 4a). However, in the presence of bortezomib, the signal became weaker and, more importantly, HIF-1α was re-distributed in both nucleus and cytoplasm. These effects were dose-dependent and observed in both the AD (LNCaP) and the AI (PC3) cell lines (Fig. 4a).

Effect of bortezomib on HIF-1α intracellular localization and p44/42 MAPK phosphorylation. a Bortezomib impairs HIF-1α nuclear accumulation. LNCaP and PC3 cells were incubated under hypoxic conditions (1% O2) in the presence of the indicated concentrations of bortezomib for 16 h before processing for immunofluorescence microscopy. Nuclei are stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI). Magnification bars = 10 μm. b Bortezomib impairs p44/42 MAPK activation. Western blot analysis of cytoplasmic (left panels) and nuclear (right panels) fractions of LNCaP and PC3 cells incubated as in a. Actin and eIF2α served as cytoplasmic markers while ARNT and histone 2B served as nuclear markers
To confirm these data, the subcellular distribution of HIF-1α was also analysed in PC3 and LNCaP cells by biochemical fractionation, which was monitored using actin and eIF2α as cytoplasmic markers while ARNT and Histone 2B as nuclear markers. HIF-1α was found almost exclusively in the nuclear fraction after exposure to hypoxia for 16 h (Fig. 4b). Treatment with increasing concentrations of bortezomib resulted in reduced nuclear HIF-1α levels, while, at the same time, cytoplasmic HIF-1α was correspondingly increased (Fig. 4b). These changes were accompanied by changes in the phosphorylation status of p44/42 MAPK. As shown in Fig. 4b, p44/42 MAPK phosphorylation was evident in nuclear extracts but absent in cytoplasmic extracts of both cell lines. Bortezomib inhibited nuclear MAPK phosphorylation and subsequent activation in a dose-dependent manner.
Discussion
Given the important role of the ubiquitin-proteasome system (UPS) in cellular homeostasis and its involvement in several carcinogenesis-related processes in the prostate, it comes as no surprise that UPS inhibition has antitumor effects in the prostate and has been proposed as prostate cancer treatment [2]. The antitumor action of the PI bortezomib in PCa has been proposed to be mediated via upregulation of pro-apoptotic proteins, including p53 and p21 [18], combined with deactivation of anti-apoptotic and pro-survival proteins, particularly NF-κB and its targets, such as Bcl-2 [19].
The HIF pathway has been previously implicated in PCa progression to androgen independence [20]. HIF-1α is overexpressed in primary PCa and bone metastases [21]. Androgen independence in PCa is a critical event hallmarking a switch to a more aggressive phenotype. Exposure of AD PCa cells to hypoxia has a similar effect which has been explained by a high ratio of HIF-1-dependent: p53-dependent transcription [22]. In the AD state, modeled by LNCaP cells, androgen-mediated induction of HIF-1α has been reported to emerge through post-transcriptional and post-translational modulations of HIF-1α activity [23] as well as androgen-regulated autocrine tyrosine kinase receptor/PI3K/Akt/mTOR signaling [24]. Reversibly, hypoxia enhances through HIF-1α the transcriptional activity of androgen receptor in a low-androgen environment [25]. Another important mechanism of PCa development and progression is centered upon the MAPK pathway [26], which has been previously shown to be responsible for HIF-1 nuclear targeting and transcriptional activation in other types of cancer cells [10, 11].
In our study, we have shown that hypoxia-mediated HIF-1α induction and transcriptional activity are PI3K/Akt/mTOR- and p44/42 MAPK-dependent processes in both AD and AI PC cells. We have also shown that bortezomib downregulates HIF-1α expression and activity by interfering with both of these pathways in a dose-dependent manner.
First, our data support that bortezomib down-regulates the hypoxia-induced HIF-1α protein expression by inhibiting the Akt/mTOR/p70S6K/S6RP pathway. Indeed, Akt-dependent activation of p70S6K and its downstream target S6RP, known to lead to stimulation of HIF-1α expression [6], were markedly reduced after treatment with bortezomib, which in parallel decreased HIF-1α protein level in both PCa cell lines. This is in line with a previous report suggesting that bortezomib mediates inhibition of HIF-1α translation in hepatocellular carcinoma cells [27]. Translational control is known to involve, apart from the Akt/mTOR pathway, the eukaryotic translation initiation factor 2α (eIF2α), phosphorylation of which has been also implicated in translational repression of HIF-1α by bortezomib in PCa cells [16]. Thus, both our and previous data suggest that bortezomib exerts, at least, part of its effect at the level of HIF-1α protein synthesis.
Second, our findings highlight for the first time the role of the MAPK pathway in the effects mediated by bortezomib under hypoxia in PCa cells. We observed a decrease in HIF-1α nuclear accumulation after treatment with bortezomib, which coincided with impaired p44/42 MAPK phosphorylation and an increased cytoplasmic fraction of HIF-1α under hypoxic conditions. Furthermore, HIF-1α transcriptional activity was down-regulated when cells were treated with either bortezomib or the MAPK-pathway inhibitor PD98059, with the latter not affecting HIF-1α protein expression levels. In the literature, the effect of bortezomib on MAPK signaling has been previously studied in different tumor types, such as breast cancer cells [28], plasma cell leukemias [29], and multiple myeloma cells [30], but the results were controversial. In agreement with this report, we have previously shown in non-PCa cells that phosphorylation of HIF-1α by p44/42 MAPK enhances HIF-1 transcriptional activity by promoting HIF-1α nuclear accumulation [12, 31], whereas inhibition of HIF-1α phosphorylation leads to HIF-1α inactivation by CRM1-dependent export to the cytoplasm [10, 11]. More importantly, this MAPK-dependent regulation of HIF-1α has often been shown to be independent of HIF-1α protein levels [9, 10, 31, 32].
Our findings can, therefore, explain the previously reported inhibitory effects of bortezomib on HIF-1α transcriptional activity even when HIF-1α protein was shown to accumulate [14–16]. Our findings further suggest that the previously reported effect of bortezomib on the recruitment of p300 onto HIF-1α, either via a FIH-dependent [13] or a FIH-independent [14, 15] mechanism might be less important or even irrelevant for HIF-1α function when HIF-1α nuclear accumulation is blocked. By demonstrating that nuclear concentration of HIF-1α is dramatically reduced after bortezomib-mediated MAPK inactivation, our work offers a new insight into the hierarchy of effects precipitated by bortezomib in hypoxic AD and AI PCa cells. If bortezomib-mediated reduction of proteasome activity was responsible for inhibition of HIF-1α degradation, we would expect an increase in HIF-1α protein levels, but instead, we observed down-regulation of HIF-1α expression as shown in Fig. 2a. Consequently, it can be deduced that the mode of action of bortezomib entails an effect on HIF-1α, which is independent of HIF-1α protein stability. This is indeed supported by our data showing the interference of bortezomib with the PI3K and MAPK pathways, which are required for HIF-1α expression and activity, respectively, in PCa cells. It is presently not possible to tell whether bortezomib blocks these pathways directly (e.g., as a kinase inhibitor) or indirectly through some unknown proteasome substrates.
Comparing the AD and AI state, low but detectable levels of HIF-1α protein were present in the normoxic PC3 but not in the LNCaP cells. One possible reason may be the fact that PC3 are advanced prostate cancer cells with a high proliferative rate [33] and, unlike the less aggressive AD LNCap cells, have an “active hypoxic response pathway” even under normoxic conditions. This “constitutive” activation may blunt further activation of HIF-1α transcriptional activity and VEGF secretion under hypoxia. Indeed, the AD LNCaP cell line displayed higher levels of HIF-1α transcriptional activity and VEGF secretion in response to hypoxia. On the other hand, this may, at least partially, be attributed to an androgen-mediated activation of the PI3K/Akt/HIF-1α cascade. This is in agreement with clinical results showing significant correlation between HIF-1α immunohistochemical expression and androgen receptor expression and indicating that androgens may regulate VEGF levels through the activation of HIF-1α in androgen-sensitive tumors [34]. Furthermore, treatment with bortezomib as well as the PI3K and MAPK inhibitors was less effective in inhibiting the hypoxia-induced HIF-1 transcriptional activity in PC3 compared to LNCaP cells. This may also be attributed to the constitutive expression of HIF-1α in PC3 cells, which may be one of the several molecular changes that occur upon progression of prostate cancer to AI state. Therefore, in this state HIF-1α expression and activity appear to be less dependent on the Akt and MAPK pathways. Nevertheless, both signaling pathways as well as the hypoxia-induced HIF-1α expression, nuclear accumulation, and activity were effectively inhibited by 100 nM bortezomib pointing to its efficiency as an antitumor agent in both types of PCa.
In conclusion, we have revealed a dual role of the proteasome inhibitor bortezomib with regard to its biological activity in LNCaP and PC3 PCa cells. Bortezomib inhibits kinases involved in both the PI3K/Akt and MAPK pathways (Fig. 5). This is a significant finding in view of the fact that the Akt/mTOR and MAPK signaling pathways are often coordinately deregulated during prostate cancer progression in humans. Targeting both Akt/mTOR and MAPK signaling pathways may be an effective treatment for patients with advanced prostate cancer, particularly those with hormone-refractory disease [35]. As a result of its dual role, bortezomib exerts a double down-regulating effect on HIF-1α, since it simultaneously “turns off” its protein synthesis and prevents its MAPK-dependent nuclear accumulation. The latter effect may allow bortezomib to block HIF-1 function and subsequent tolerance to hypoxia, regardless of the cellular HIF-1α protein levels. This novel biological concept contributes to the understanding of the antitumor effects of bortezomib and supports its further clinical application to PCa treatment.

Schematic model of the possible mechanisms of HIF-1α downregulation by bortezomib. Bortezomib represses HIF-1α protein synthesis and nuclear accumulation in PCa cell lines by inhibiting the PI3K/Akt/TOR and MAPK pathways, respectively
References
Adams J, Kauffman M (2004) Development of the proteasome inhibitor Velcade (Bortezomib). Cancer Invest 22:304–411
Papandreou CN, Logothetis CJ (2004) Bortezomib as a potential treatment for prostate cancer. Cancer Res 64:5036–5043
Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, Shiraishi N, Ribatti D, Nico B, Vacca A et al (2006) Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res 66:184–191
Veschini L, Belloni D, Foglieni C, Cangi MG, Ferrarini M, Caligaris-Cappio F, Ferrero E (2007) Hypoxia-inducible transcription factor-1 alpha determines sensitivity of endothelial cells to the proteosome inhibitor bortezomib. Blood 109:2565–2570
Semenza GL (2009) Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol 19:12–16
Jiang BH, Liu LZ (2009) PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv Cancer Res 102:19–65
Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG (1994) Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265:966–970
Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J (2003) MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem 278:14013–14019
Richard DE, Berra E, Gothié E, Roux D, Pouysségur J (1999) p42/p44 MAP kinases phosphorylate hypoxia inducible factor 1 alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem 274:32631–32638
Mylonis I, Chachami G, Samiotaki M, Panayotou G, Paraskeva E, Kalousi A, Georgatsou E, Bonanou S, Simos G (2006) Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem 281:33095–33106
Mylonis I, Chachami G, Paraskeva E, Simos G (2008) Atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1alpha by MAPK. J Biol Chem 283:27620–27627
Triantafyllou A, Mylonis I, Simos G, Bonanou S, Tsakalof A (2008) Flavonoids induce HIF-1alpha but impair its nuclear accumulation and activity. Free Radic Biol Med 44:657–670
Shin DH, Chun YS, Lee DS, Huang LE, Park JW (2008) Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia inducible factor-1. Blood 111:3131–3136
Kaluz S, Kaluzova M, Stanbridge EJ (2006) Proteasomal inhibition attenuates transcriptional activity of hypoxia-inducible factor 1 (HIF-1) via specific effect on the HIF-1a C-terminal activation domain. Mol Cell Biol 26:5895–5907
Birle DC, Hedley DW (2007) Suppression of the hypoxia-inducible factor-1 response in cervical carcinoma xenografts by proteasome inhibitors. Cancer Res 67:1735–1743
Zhu K, Chan W, Heymach J, Wilkinson M, McConkey DJ (2009) Control of HIF-1alpha expression by eIF2 alpha phosphorylation-mediated translational repression. Cancer Res 69:1836–1843
Patrikidou A, Vlachostergios PJ, Voutsadakis IA, Hatzidaki E, Valeri RM, Destouni C, Apostolou E, Daliani D, Papandreou CN (2011) Inverse baseline expression pattern of the NEP/neuropeptides and NFκB/proteasome pathways in androgen-dependent and androgen-independent prostate cancer cells. Cancer Cell Int 11:13
Ikezoe T, Yang Y, Saito T, Koeffler HP, Taguchi H (2004) Proteasome inhibitor PS-341 down-regulates prostate-specific antigen (PSA) and induces growth arrest and apoptosis of androgen-dependent human prostate cancer LNCaP cells. Cancer Sci 95:271–275
Fahy BN, Schlieman MG, Mortenson MM, Virudachalam S, Bold RJ (2005) Targeting BCL-2 overexpression in various human malignancies through NF-kappaB inhibition by the proteasome inhibitor bortezomib. Cancer Chemother Pharmacol 56:46–54
Kimbro KS, Simons JW (2006) Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr Relat Cancer 13:739–749
Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW (1999) Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 59:5830–5835
Salnikow K, Costa M, Figg WD, Blagosklonny MV (2000) Hyperinducibility of hypoxia-responsive genes without p53/p21-dependent checkpoint in aggressive prostate cancer. Cancer Res 60:5630–5634
Sheflin LG, Zou AP, Spaulding SW (2004) Androgens regulate the binding of endogenous HuR to the AU-rich 3'UTRs of HIF-1alpha and EGF mRNA. Biochem Biophys Res Commun 322:644–651
Mabjeesh NJ, Willard MT, Frederickson CE, Zhong H, Simons JW (2003) Androgens stimulate hypoxia-inducible factor 1 activation via autocrine loop of tyrosine kinase receptor/phosphatidylinositol 3'-kinase/protein kinase B in prostate cancer cells. Clin Cancer Res 9:2416–2425
Mitani T, Yamaji R, Higashimura Y, Harada N, Nakano Y, Inui H (2011) Hypoxia enhances transcriptional activity of androgen receptor through hypoxia-inducible factor-1α in a low androgen environment. J Steroid Biochem Mol Biol 123:58–64
Papatsoris AG, Karamouzis MV, Papavassiliou AG (2007) The power and promise of "rewiring" the mitogen-activated protein kinase network in prostate cancer therapeutics. Mol Cancer Ther 6:811–819
Chen KF, Yeh PY, Yeh KH, Lu YS, Huang SY, Cheng AL (2008) Down-regulation of phospho-Akt is a major molecular determinant of bortezomib-induced apoptosis in hepatocellular carcinoma cells. Cancer Res 68:6698–6707
Codony-Servat J, Tapia MA, Bosch M et al (2006) Differential cellular and molecular effects of bortezomib, a proteasome inhibitor, in human breast cancer cells. Mol Cancer Ther 5:665–675
Esparís-Ogando A, Alegre A, Aguado B, Mateo G, Gutiérrez N, Bladé J, Schenkein D, Pandiella A, San Miguel JF (2005) Bortezomib is an efficient agent in plasma cell leukemias. Int J Cancer 114:665–667
Hideshima T, Chauhan D, Hayashi T, Akiyama M, Mitsiades N, Mitsiades C, Podar K, Munshi NC, Richardson PG, Anderson KC (2003) Proteasome inhibitor PS-341 abrogates IL-6 triggered signaling cascades via caspase-dependent downregulation of gp130 in multiple myeloma. Oncogene 22:8386–8393
Mylonis I, Lakka A, Tsakalof A, Simos G (2010) The dietary flavonoid kaempferol effectively inhibits HIF-1 activity and hepatoma cancer cell viability under hypoxic conditions. Biochem Biophys Res Commun 398:74–78
Hur E, Chang KY, Lee E, Lee SK, Park H (2001) Mitogen-activated protein kinase kinase inhibitor PD98059 blocks the trans-activation but not the stabilization or DNA binding ability of hypoxia-inducible factor-1alpha. Mol Pharmacol 59:1216–1224
Zhong H, Semenza GL, Simons JW, De Marzo AM (2004) Up-regulation of hypoxia-inducible factor 1a is an early event in prostate carcinogenesis. Cancer Detection and Prevention 28:88–93
Boddy JL, Fox SB, Han C, Campo L, Turley H, Kanga S, Malone PR, Harris AL (2005) The androgen receptor is significantly associated with vascular endothelial growth factor and hypoxia sensing via hypoxia-inducible factors HIF-1a, HIF-2a, and the prolyl hydroxylases in human prostate cancer. Clin Cancer Res 11:7658–7663
Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C, Abate-Shen C (2008) Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest 118:3051–3064
Acknowledgments
We would wish to thank Dr. A. J. Giaccia (Stanford University) and Dr. M. U. Muckenthaler (University of Heidelberg, Germany) for providing us with the firefly luciferase reporter plasmid pGL3–5HRE-VEGF and the Renilla luciferase expressing plasmid PCIRenilla, respectively. We also thank Professor A. Molyvdas and Dr E. Paraskeva (Department of Physiology, University of Thessaly School of Medicine, Biopolis, Larissa, Greece) for use of research equipments in their laboratories.
Disclosure statement
The authors report no potential conflict of interest relevant to this article.
Author information
Authors and Affiliations
Corresponding authors
Additional information
C. N. Papandreou and P. Liakos have equal contributions.
An erratum to this article is available at http://dx.doi.org/10.1007/s00109-013-1030-4.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 335 kb)
Rights and permissions
About this article
Cite this article
Befani, C.D., Vlachostergios, P.J., Hatzidaki, E. et al. Bortezomib represses HIF-1α protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J Mol Med 90, 45–54 (2012). https://doi.org/10.1007/s00109-011-0805-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-011-0805-8