
Abstract
The appearance of lipid-rich foam cells is a major feature of vulnerable atherosclerotic plaque formation. The transformation of macrophages into foam cells results from excessive uptake of cholesterol-rich particles by scavenger receptors such as CD68. We cloned a CD68-Fc immunoadhesin, a fusion protein consisting of the extracellular domain of the human CD68 and a human Fc domain, and investigated the function in vitro. Specific binding of CD68-Fc to OxLDL with an affinity of 10 nmol/L was determined by surface plasmon resonance and increased binding to lipid-rich human and ApoE−/− mice plaque tissue. This was confirmed both by immunohistochemical staining of CD68-Fc-treated paraffin sections from human plaques and by ELISA-based quantification of CD68-Fc binding to human atherosclerotic plaque extracts. In an in vitro model of macrophage/foam cell formation, CD68-Fc reduced foam cell formation significantly. This was caused both by interference of CD68-Fc with OxLDL uptake into macrophages and platelets and by the inhibition of platelet/OxLDL phagocytosis. Finally, expression of metalloproteinases by macrophages/foam cells was inhibited by CD68-Fc. In conclusion, CD68-Fc seems to be a promising new tool for preventing macrophage/foam cell formation. Thus, CD68-Fc might offer a novel therapeutic strategy for patients with acute coronary syndrome by modulating the generation of vulnerable plaques.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atherosclerotic changes of arteries arise as a consequence of the deposition and retention of serum lipoproteins in the vascular wall. Recently, it has become evident that atherosclerosis represents an inflammatory response of macrophages and lymphocytes which is induced and triggered by pathogenic lipoproteins deposited in the arterial wall [1, 2]. Macrophages have been shown to express at least six structurally different scavenger receptors involved in the cellular uptake of modified forms of low-density lipoproteins (LDL) and intracellular deposition of cholesterol [3]. Because scavenger receptors are not downregulated by excessive intracellular cholesterol deposition, as occurs with LDL receptors, an unregulated and continuous accumulation of lipids occurs in macrophage/foam cells [4]. Modified LDL has pro-atherogenic properties because it triggers adhesion, endocytosis, phagocytosis, and activates several signal transduction pathways [5]. The earliest visible atherosclerotic lesion, the fatty streak, is the result of the deposition of cholesterol-laden macrophage/foam cells. Thus, lipid uptake into the artery wall represents the prerequisite and initiating step in the pathogenesis of atherosclerosis as it is involved in its subsequent progression and also in changes of the morphology of advanced lesions into vulnerable plaques [2, 6].
The scavenger receptor CD68 belongs to the class D of scavenger receptors and is considered to be a member of the lysosomal-associated membrane protein (lamp) family. CD68 has been detected in macrophages, Langerhans cells, and dendritic cells, and it binds and internalizes modified lipoproteins. Its main role in atherogenesis is the retention of oxidized lipoproteins because of its extensive binding to OxLDL combined with its broad intracellular distribution [7]. In contrast to the scavenger receptors of class A and B, CD68 exclusively binds the atherogenic OxLDL and not other lipoprotein forms, such as protective HDL.
Because of the central role of scavenger receptors in foam cell formation, therapeutic options to inhibit the expression of scavenger receptors of macrophage/foam cells have been favored as promising strategies [8, 9]. Modification of scavenger receptors with PPARγ- and LXR-specific ligands [10], as well as HMG-CoA reductase inhibitors (statins), has been reported [11, 12]. Statins are very effective drugs for lowering total and low-density lipoprotein cholesterol and also act by their pleiotropic effects, including downregulation of the scavenger receptors SR-A [9, 13], CD36, and LOX-1 [14]. However, statins are applied systemically and exert side effects and drug interactions, predominantly in high-risk and elderly patients with poor renal function [15].
For that reason, more specific and directed approaches for altering both lipid deposition and inflammatory responses in macrophages are needed. Our approach is designed to inhibit foam cell formation not by the blockade of a specific class of scavenger receptors but by the binding of their main ligand oxLDL to prevent its scavenger receptor-mediated uptake (Fig. 1a). We cloned and characterized an immunoadhesin which resembles the scavenger receptor CD68 and binds the ligand with high affinity. CD68-Fc serves only as a “catcher” to bind and eliminate the ligand Ox-LDL (Fig. 1a) and does not interfere with Ox-LDL binding scavenger receptors on monocytes/macrophages, such as CD68, CD36, and SR-A.

a Schematic illustration showing inhibition of OxLDL uptake into macrophages and prevention of foam cell formation using a CD68-Fc fusion protein. It is hypothesized that the binding of OxLDL to several different scavenger receptors belonging to class A (SR-A), class B (SR-B), and class D (CD68) could be prevented by the fusion protein CD68-Fc which catches the ligand Ox-LDL. Thus, the specific inhibition of foam cell formation would not be the result of the blockade of a specific class of scavenger receptors but by the binding of their main ligand OxLDL to prevent its scavenger receptor-mediated uptake. b Characterization of the purified fusion protein. CD68-Fc (2.5 µg) and control Fc (2.5 µg) lacking the extracellular CD68 domain underwent SDS-PAGE. Immunoblotting with peroxidase-conjugated goat anti-human Fc antibody was performed. c OxLDL binds with high affinity to immobilized CD68-Fc. The overlay plot of the surface plasmon resonance analysis shows the association phase (0–300 s) of increasing concentrations of OxLDL (75, 50, 25, 10, and 5 nM), LDL (50 nM), and for HDL (150 nM) dissolved in binding buffer. CD68-Fc was linked to the sensor chip surface SD CMD 20L
We provide evidence that CD68-Fc binds to oxLDL with high affinity and specificity and to lipid-rich murine and human atherosclerotic plaque specimens. It inhibits OxLDL uptake and inhibits macrophage/foam cell formation and specific functions in vitro. Thus, the soluble form of the CD68-receptor might be a novel strategy for treatment of patients with vulnerable plaques, including acute coronary syndromes.
Materials and methods
Reagents
For immunofluorescence staining, the following antibodies and reagents were used: mouse anti-human CD68 (clone KP1, DakoCytomation, Hamburg, Germany) in combination with Alexa fluor anti-mouse IgG1 antibody (Invitrogen, Karlsruhe, Germany). Dil-OxLDL (lipoprotein low density, oxidized human) was purchased from Biomedical Technologies (Stoughton, USA).
Cloning of soluble human CD68, generation, and purification of the adenoviral constructs Ad-control Fc and Ad-CD68-Fc
To generate a soluble form of CD68, we fused the extracellular domain of human CD68 to the human Fc-domain by methods already described for a previously cloned GPVI-Fc [16].
The Fc-domain (human IgG1) was generated synthetically (Medigenomix, Martinsried, Germany) based on frequently used codons in CHO cells. The Fc receptor binding site (LLGG234-237AAAA) and the complement binding site (P331S) were mutated. The DNA coding for the optimized Fc was cloned with Kpn/EcoRV in the plasmid pcDNA-FRT (KpnI/XhoI filled up) to get pcDNA Fc opt.
For cloning of the extracellular domain of the human CD68, total RNA from cultured human megakaryocytes was isolated (RNeasy Mini Kit, Qiagen, Hilden, Germany) according to the manufacturer’s recommendations. Reverse transcription was performed (Omniscript RT Kit; Qiagen) using 2 µg RNA at 37°C overnight. One hundred nanograms of the cDNA was used as a template in PCR amplification of the human CD68 using the Expand High Fidelity PCR System (Roche Molecular Biochemicals, Mannheim, Germany). The PCR fragment was cloned with Kpn/NotI into the plasmid pcDNA Fc opt (KpnI/NotI), generating a plasmid encoding the human extracellular CD68 fused to the human Fc-domain, including a specific hinge region. For cloning of a soluble Fc control fragment lacking the extracellular domain of CD68, the leader peptide of CD40 was amplified from a human heart cDNA library (Clontech, Palo Alto, CA, USA). The PCR fragment was cloned in the plasmid pcDNA Fc opt with KpnI/NotI. The accuracy of plasmids was confirmed by sequencing (MediGenomix).
Generation of stable CD68-Fc-expressing CHO cells
Flp-InTM-CHO cells (Invitrogen) were co-transfected with a 9:1 ratio of the plasmids pOG44/pcDNA5-FRT CD68-Fc-opt (Invitrogen) or plasmids pOG44/pcDNA5-FRT Fc-opt at a confluency of 70%. Twenty-four hours after transfection, the cells were washed and fresh medium was added. Forty-eight hours after transfection, the cells were split 1 to 20 into fresh medium containing 500 µg/mL hygromycin. Hygromycin-resistant foci were isolated, expanded, and tested for the expression of CD68-Fc by Western blot.
Purification of control-Fc and CD68-Fc
The culture supernatant of stable CHO cells was collected, centrifuged (3,800×g, 30 min, 4°C), and filtrated (0.2 µm). Soluble control Fc or human CD68 was precipitated by addition of 1.2 vol ammonium sulfate (761 g/L) and stirring overnight at 4°C. The proteins were pelleted by centrifugation (3,800×g, 30 min, 4°C), dissolved in 0.1 vol phosphate-buffered saline (PBS), and dialyzed in PBS overnight at 4°C. The protein solution was clarified by centrifugation (14,000×g, 30 min, 4°C), filtrated (0.2 µm), and loaded on Protein A-columns (HiTrapTM Protein A HP, Amersham Pharmacia Biotech AB, Uppsala, Sweden), equilibrated with binding buffer (20 mM sodium phosphate buffer, pH 7.0, 0.02% NaN3). The column was washed with binding buffer until OD280 < 0.01 and eluted with elution buffer (100 mM glycine, pH 2.7). The eluted fractions (900 µL) were neutralized with 100 µL neutralization buffer (1 M Tris/HCl, pH 9.0, 0.02% NaN3), pooled, dialyzed in PBS overnight at 4°C, aliquoted, and frozen at 20°C. The column was neutralized by binding buffer and washed with 20% (v/v) ethanol.
Surface plasmon resonance
Binding of the different lipoproteins to CD68-Fc was measured by surface plasmon resonance. The Ibis dual-channel optical measuring system, the biosensor chip SD CMD 20L, and the amine coupling kit (EDC/NHS) were from Xan Tec Bioanalytics GmbH, Münster, Germany. CD68-Fc was immobilized on a carboxymethyl dextran gel sensor chip with an average molecular mass of 20 kDa, brush confirmation, and low density (SD CMD 20L). For covalent binding between the carboxyl groups of the chip surface and the amine groups of the protein, an amine coupling kit from Xan Tec Bioanalytics was used. Fc linked to the sensor chip was used as control; Fc did not significantly bind any of the used lipoproteins (data not shown). For the determination of the dissociation constant K d, different concentrations of OxLDL in binding buffer (10 mM MOPS, 0.3 mM EDTA, 1.43 mM β-mercaptoethanol, 150 mM NaCl, 0.001% (v/v) NP40 (Igepal CA-630)), adjusted to pH 7.0, were applied to the detector cell at 20°C. The interaction was analyzed for 300 s. After termination of a binding experiment, the sensor surface was regenerated by incubation with 1 M NaCl. The data were analyzed with kinetic evaluation software, and the kinetic parameters of a single-phase association were determined by nonlinear regression of the respective data points.
Assessment of CD68-Fc binding to human atheromatous plaque material
Atherosclerotic tissue specimens were obtained from patients who underwent surgery for high-grade carotid artery stenosis. Patient consent was obtained as approved by the Ethics Committee of the Faculty of Medicine of the University of Munich. The investigation conforms with the principles outlined in the Declaration of Helsinki. The carotid plaque tissue was removed by a technique of intraoperative endarterectomy that preserved the plaque structure. Specimens containing lipid-rich soft plaques were collected. Parts of the specimens were fixed with 4% paraformaldehyde, embedded in paraffin, and sectioned. Semi-thin sections were incubated with CD68-Fc or unspecific human control-Fc. Specific binding was detected using an antibody against the human Fc fragment and a peroxidase-labeled secondary antibody. Further plaque samples were weighed, homogenized, and stored at −80°C. For ELISA, the atheromatous plaque concentration was adjusted to 50 mg/mL wet weight, and the measured protein concentrations ranged between 0.5 and 2.5 mg/mL. Cell culture plates (96 wells) were coated overnight at 4°C with 30 µL of diluted plaque (10 mg/mL). Plates were washed with 100 µL/well PBS/0.05% Tween 20 (PBST), blocked with Roti-Block (Roth, Karlsruhe, Germany) overnight, and washed again twice with PBST. Then, 20 µg/mL CD68-Fc or an equimolar amount of control Fc was added, incubated for 1 h in PBST at room temperature, and washed five times. A peroxidase-conjugated goat anti-human IgG Fc fragment-specific antibody (Dianova, Hamburg, Germany) was added in a dilution of 1:10,000 and incubated for 1 h at room temperature. After washing with PBST, 100 µL of detection reagent (BM Blue POD Substrate, Roche) was added and incubated for 1 min. The reaction was stopped by the addition of 100 µL 1 M H2SO4, and the plate was measured at 450 nm against the reference wavelength 590 nm. Binding of CD68-Fc was expressed as percent of the control Fc binding.
Cell culture
Human platelets
Human platelets were isolated from venous blood as described before [17]. The platelet isolation procedure revealed a high purity of platelets without any measurable contamination of polymorphonuclear cells or monocytes as verified by the absence of CD14 (flow cytometry) and of myeloperoxidase (ELISA).
Human progenitor cells
Human CD34+ progenitor cells were isolated from human cord blood and cultured as described before [17]. Human cord blood was obtained from healthy women immediately after childbirth with the approval of the local Ethics Committee (project no. 76/2005). The experiments with human CD34+ progenitor cells were performed in accordance to the principles outlined in the Declaration of Helsinki.
Human monocytes
Monocytes were isolated from human venous blood by adherence after Ficoll-Paque purification of peripheral blood mononuclear cells, as described elsewhere [18].
In vitro model to induce foam cell generation and inhibition by CD68-Fc
CD34+ progenitor cells were co-cultured with platelets for 10 days, as described previously [17]. In brief, human CD34+ progenitor cells were isolated from umbilical cord blood and co-incubated (50,000 cells) with isolated human platelets (2 × 108 cells) for up to 10 days and processed for immunofluorescence microscopy and lipid staining [17]. To evaluate the effect of CD68-Fc on the number of foam cells, CD34+ progenitor cells were cultured in the presence or absence of CD68-Fc containing culture supernatants isolated from CHO cells in concentrations as indicated by final cell number counting under a microscope.
In vitro immunofluorescence and lipid staining
CD34+ cells were co-incubated with medium alone or with platelets for 10 days. Cells were fixed with 2% formaldehyde solution for 20 min and incubated with 0.5% Sudan red (Sigma, 20 min). Nuclei were counterstained with hematoxylin solution (Sigma, 5 min). Afterwards, cells were washed with 2% glycine, permeabilized with 0.2% Triton-X100, and incubated with a monoclonal mouse anti-human CD68 antibody (4.7 µg/mL) for 1 h. As secondary antibody, an Alexa Fluor anti-mouse IgG1 antibody (10 µg/mL) was added for 1 h. Unspecific binding was prevented by bovine serum albumin (3%, 1 h). Samples were covered with fluorescent mounting medium (DakoCytomation) and analyzed by standard (Axiovert 200, Zeiss), confocal immunofluorescence (LSM 510, Zeiss and Leica TCS SP, Leica Microsystems) and a field emission scanning electron microscope (JSM-6300F, Jeol Ltd., Tokyo, Japan). Probes were excited at 423 nm and analyzed at an emission wavelength at 565 nm.
Examination of the uptake of modified LDL
Platelets were co-cultivated with CD34+ progenitor cells or monocytes for 3–5 days. Cells were than incubated with either 1 µg/mL Dil-OxLDL (red fluorescence) for 2 h or with platelets which were pre-incubated with Dil-OxLDL (10 µg/mL) for 3 h. Dil-OxLDL accumulation and phagocytosis of lipid-laden platelets by macrophage foam cells was then visualized by fluorescence microscopy.
Measurement of LDL uptake using flow cytometry
Platelets were co-cultured for 3–5 days with CD34+ progenitor cell or monocytes. Dil-OxLDL (1 µg/mL) was pre-incubated in the presence or absence of CD68-Fc (62.5 or 125 nmol/L) for 30 min and added to macrophage/foam cell—platelet co-cultures for 2 h at 37°C. Furthermore, platelets themselves were incubated with or without CD68-Fc for 3 h. Cells were washed, trypsinized, and analyzed on a FACS-Calibur flow cytometer (Becton Dickinson) at the second fluorescence. A total of 1 × 105 cells were analyzed per measurement. The red Dil-OxLDL fluorescence was evaluated at an excitation wavelength at 488 nm and an emission wavelength at 565 nm.
SDS-PAGE and immunoblotting
Electrophoresis and immunoblotting of purified CD68-Fc was performed according to a previously described method [19].
Determination of metalloproteinase activity
MMP-2 and MMP-9 activity was determined in supernatants of foam cells at day 10 of co-cultivation by gelatin zymography, as described previously [17]. SDS gels containing 10% gelatin were used (Invitrogen).
Data presentation and statistics
Comparisons between group means were performed using Student’s t test or ANOVA where applicable. Data are presented as mean ± SEM. A value of p < 0.05 was considered as statistically significant.
Results
A soluble form of human CD68-Fc can be cloned, expressed in cell culture, and purified
To generate a soluble form of human CD68, the extracellular domain of human CD68 was cloned and fused to the human immunoglobin Fc domain. Stable cell lines coding for CD68-Fc or control Fc were then prepared. CD68-Fc and control Fc were expressed as secreted soluble proteins using the CHO cell line to prevent misfolding and non-glycosylation of the expressed proteins. The recombinant proteins could be isolated with high purity. The molecular mass of CD68-Fc was ∼75–80 kDa under reducing conditions in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), as detected by immunoblotting (Fig. 1b, right). A ∼160-kDa protein was identified under non-reducing conditions (Fig. 1b, left), which confirms that CD68-Fc is present as a homodimer. The human Fc fragment signal was found at ∼50 kDa.
CD68-Fc binds to OxLDL with high sensitivity and specificity
OxLDL dissolved in binding buffer specifically binds to CD68-Fc immobilized on a SD CMD 20 L surface plasmon resonance sensor chip. Nonlinear regression of the respective data points with kinetic evaluation software yielded a K d of 10 nM, reflecting a high-affinity interaction between the CD68-Fc protein and the corresponding lipoprotein. The other tested lipoproteins such as LDL and HDL did not show any specific interaction under the chosen experimental conditions (Fig. 1c).
CD68-Fc preferentially binds to lipid-rich structures of human atherosclerotic plaque specimens
To determine whether CD68-Fc binds to human atherosclerotic plaque tissue, we examined the binding properties of CD68-Fc to human carotid plaque specimens ex vivo [20, 21]. Immunohistochemical stainings revealed a high and specific binding of CD68-Fc to lipid-rich plaque structures (Fig. 2a, upper right). No notable binding was observed after incubation of plaque specimens with control Fc (Fig. 2a, upper left). We also obtained a slightly increased amount of CD68-Fc binding to an aortic artery specimen without obvious plaque formation derived from a 60-year-old man (Fig. 2a, lower right).

Binding of soluble CD68-Fc at human atheromatous plaque material and ApoE−/− mice. a Immunohistochemical stainings with an anti-Fc antibody after incubation of paraffin sections with CD68-Fc (312 nmol/L) or control-Fc (312 nmol/L). In contrast to control Fc (left side), a high and specific binding of CD68-Fc to lipid-rich, atheromatous plaque structures is visible (upper right side, brown staining) and weak binding to human aorta without obvious atherosclerotic plaque in a patient with overall vascular disease (lower right side). b Binding of CD68-Fc (312 nmol/L) at plaque material from ApoE−/− mice compared to healthy control mice. c Quantification of CD68-Fc binding by an ELISA assay performed on atheromatous human plaque immobilized onto culture plates. A significantly higher binding capacity of CD68-Fc (20 µg/mL) could be measured compared to control-Fc (20 µg/mL, 2.74 ± 0.29 vs. 1.00 ± 0.10, respectively, p < 0.01, n = 5)
To further investigate the specific binding of CD68-Fc on plaque material, we incubated atheromatous plaques of ApoE−/− mice with and without CD68-Fc. The immunohistochemical stainings of ApoE−/− mice showed an increased binding of soluble CD68-Fc compared to the Fc control (Fig. 2b, upper panel). No CD68-Fc and Fc-binding was found on the aorta of non-atherosclerotic control mice (Fig. 2b, lower panel).
To quantify the binding of CD68-Fc to lipid-rich atheromatous plaque pieces, an ELISA was performed on homogenized and solubilized plaque material immobilized onto culture plates using an anti-human Fc antibody (Fig. 2c). After incubation with CD68-Fc and control Fc, a significantly higher binding capacity of CD68-Fc could be measured compared to control Fc (2.74 ± 0.29 vs. 1.00 ± 0.10, *p < 0.01).
Foam cell formation can be reduced by CD68-Fc in vitro
We then sought to determine whether CD68-Fc, which accumulates at lipid-rich lesions, is able to inhibit foam cell formation in vitro. To test the inhibitory potential of CD68-Fc and to find an effective dosage, an in vitro model of foam cell formation was used, enabling a reproducible induction of foam cell formation after co-cultivation of human CD34+ progenitor cells or human monocytes with platelets [17].
After 3–5 days of co-cultivation, characteristic macrophage/foam cells were observed. These cells were positively stained for CD68 (Fig. 3a). Transmission electron microscopy revealed the presence of multiple vesicles and a high number of intracellular lipid droplets, lipids in vesicles, and multilamelar lipid depositions in the cytoplasm (Fig. 3b), which is characteristic of human foam cells.

In vitro model to generate foam cells. CD34+ cells were co-incubated with human platelets for up to 10 days and foam cell formation was visualized. a Confocal microscopy (×40/1.3 oil, LSM 510, Zeiss) after immunostaining with specific antibodies against the scavenger receptor CD68. The positive green immunostaining indicates a differentiation into the macrophage/monocyte lineage and the expression of scavenger receptor CD68 by foam cells (scale bar, 10 µm). b Transmission electron pictures of a single foam cell. The granular cytoplasm is filled with platelets (plt) and lamellar lipid depositions (lm). c, d Uptake of Dil-Ox-LDL by foam cells. Foam cells were incubated with low (c) and high (d) doses of Dil-Ox-LDL (red labeling) for 1 h. Fluorescence microscopy demonstrates that these foam cells are able to bind and internalize substantial amounts of Dil-OxLDL which is deposited into vesicles inside the cytoplasm
To visualize cellular LDL binding and to prove if modified LDL is bound and taken up, early macrophage foam cells (at days 3–5 of co-cultivation) were incubated with fluorochrome-conjugated OxLDL (Dil-OxLDL). Fluorescence microscopy verified that these cells bound and internalized substantial quantities of modified LDL in a dose-dependent manner (Fig. 3c, d). The cytoplasm of these cells was filled with lipid droplets and vesicles.
CD68-Fc reduces Dil-Ox-LDL uptake by macrophages and platelets
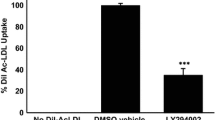
Fluorescence-activated cell sorting (FACS) analysis showed that macrophage foam cells take up Dil-OxLDL dose-dependently (Fig. 4a, upper and middle panels). Additionally, we incubated human macrophages with Dil-OxLDL (1 µg/mL) in a time-dependent manner. Our FACS analysis showed that macrophages bind an increased amount of Dil-OxLDL during 1 and 12 h. To prove if CD68-Fc influences the uptake of modified LDL into macrophages, Dil-OxLDL (200 ng/mL) was pre-incubated for 30 min in the presence or absence of CD68-Fc. According to the time-dependent experiments, we added the pre-incubated Dil-OxLDL and CD68-Fc to monocyte/platelet co-cultures for 2 h. FACS analysis showed that Dil-Ox-LDL uptake could be reduced by CD68-Fc when compared with control cultures treated with PBS- and Fc control-treated macrophages (Fig. 4b). However, we found also an inhibition of the OxLDL uptake into macrophages using the Fc control fusion protein compared to PBS-treated controls (Fig. 4b).

CD68-Fc inhibits Dil-OxLDL uptake by macrophages. Macrophages were generated after a co-incubation of human monocytes with platelets. a Dil-OxLDL is taken up by macrophages in a dose (middle panel) and time (lower panel) dependent fashion which was determined by FACS analysis (n = 3). b Dil-OxLDL was pre-incubated for 30 min in the presence or absence of CD68-Fc (125 nmol/L) and added to monocyte/platelet co-cultures for 2 h. Dil-Ox-LDL uptake was significantly reduced by CD68-Fc when compared with control cultures treated with control-Fc (125 nmol/L, *n = 3)
Furthermore, a substantial uptake of Dil-OxLDL into platelets was observed following prolonged incubation of at least 3 h using at an application dose of 10 µg/mL Dil-OxLDL (Fig. 5a). The comparison of the mean fluorescence after Dil-OxLDL uptake by macrophages and platelets revealed notable differences between both cell types. At the low dose of 200 ng/mL Dil-OxLDL macrophages showed (Fig. 4a) even more uptake than platelets at 10 µg/mL Dil-OxLDL (Fig. 5a).

CD68-Fc inhibits Dil-OxLDL uptake by platelets. a FACS analysis demonstrated that Dil-OxLDL is also taken up by platelets in a dose-dependent manner compared to control (untreated platelets, n = 3). Confocal microscopy picture of dense granules of platelets in green (left picture, green fluorescence) shows the uptake of Dil-OxLDL (10 µg/mL, yellow fluorescence). The middle and right pictures show the binding of Dil-OxLDL (red fluorescence) on the surface of platelets. b Dil-OxLDL (10 µg/mL) was pre-incubated for 30 min in the presence or absence of CD68-Fc (125 nmol/L) and added to freshly isolated platelets for 3 h. In contrast to control-Fc (125 nmol/L), CD68-Fc caused a significant reduction of Dil-OxLDL uptake into platelets (*p < 0.05 vs. Fc, n = 4). c CD68-Fc inhibits phagocytosis of Dil-OxLDL-labeled platelets. Platelets were pre-incubated with Dil-OxLDL (10 µg/mL) for 3 h. Then, platelets loaded with Dil-OxLDL were incubated in the presence or absence of CD68-Fc (125 nmol/L) for 30 min and added to macrophage foam cell cultures (monocyte/platelet co-culture) for further 1 h. Phagocytosis of Dil-OxLDL-loaded platelets was reduced by CD68-Fc treatment vs. control Fc (*p < 0.05, n = 3)
To further prove if Dil-OxLDL is also taken up by platelets, increasing doses of Dil-OxLDL were added to freshly isolated human platelets (Fig. 5a). Confocal microscopy picture detected OxLDL in the dense granules of platelets after incubation of platelets with Dil-OxLDL, while the remaining OxLDL is bound on the surface of platelets (Fig. 5a). Furthermore, FACS analysis showed that Dil-OxLDL at a concentration of 10 µg/mL was also taken up by platelets. Dil-OxLDL (10 µg/mL) was then pre-incubated for 30 min in the presence or absence of CD68-Fc and added to platelets for 3 h. In contrast to control Fc, pre-incubation with CD68-Fc caused a significant reduction in Dil-OxLDL uptake by platelets (mean fluorescence of Dil-OxLDL, 3.15 ± 0.59 vs. 6.47 ± 1.01, respectively, n = 4, p < 0.05; Fig. 5b).
To determine whether CD68-Fc also influences the phagocytosis of modified LDL bound by platelets prior to their incubation with macrophage foam cells, platelets were pre-incubated with Dil-OxLDL (10 µg/mL) for 3 h. Platelets loaded with Dil-OxLDL were then incubated in the presence or absence of CD68-Fc for 30 min and added to macrophage foam cell cultures for a further 1 h. FACS measurements showed that CD68-Fc was also able to significantly inhibit platelet-mediated Dil-OxLDL uptake into macrophage foam cells via phagocytosis when compared to control cultures treated with control Fc (mean fluorescence of Dil-OxLDL, 96.87 ± 5.03 vs. 50.20 ± 10.14, respectively, n = 3, p < 0.05; Fig. 5c).
CD68-Fc reduces foam cell formation and MMP-9 secretion
Finally, we evaluated the effect of CD68-Fc on platelet-mediated foam cell generation from CD34+ progenitor cells. CD68-Fc substantially reduced formation of platelet-mediated foam cells in a dose-dependent manner (Fig. 6 a, b). A more than 73.7% inhibition was measured at a total protein concentration of 125 nmol/L CD68-Fc (CD68-Fc vs. control-Fc, mean ± SEM 30.6 ± 15.1 vs. 115.5 ± 11.0, respectively, p < 0.01) and 59.2% inhibition with 62.5 nmol/L CD68-Fc (CD68-Fc vs. control Fc, mean ± SEM; 36.9 ± 12.5 vs. 90.9 ± 4.2; p < 0.01).

Reduction of foam cell formation and proteolytic function by CD68-Fc. a CD34+ progenitor cells were co-cultivated with freshly isolated platelets. Purified CD68-Fc (62 or 125 nmol/L) and control Fc (62 or 125 nmol/L) protein were added to the CD34+/platelet co-culture in different concentrations and incubated for 10 days. Controls were incubated with medium only. *p < 0.01 significance compared to control Fc-treated cultures (n = 3). b Phase contrast images show foam cells (arrows) after co-cultivation of CD34+ progenitor cells with platelets in the presence or absence (control) of CD68-Fc. c CD34+ cells were co-incubated with freshly isolated platelets and incubated for 10 days with wild-type CHO culture supernatants (wt) or supernatants containing CD68-Fc (CD68-Fc). SDS-PAGE zymography of co-culture supernatants shows a decreased proteolytic activity of MMP-9 after treatment with CD68-Fc (representative of three independent experiments)
To evaluate the effect of CD68-Fc on specific foam cell functions, the activity of matrix metalloproteinases (MMPs) was studied. Zymographical analysis revealed that CD68-Fc substantially reduced MMP-9 activity in the culture supernatant (Fig. 6c).
Discussion
The major findings of the present study are: (1) A soluble recombinant form of the scavenger receptor CD68 can be generated that binds specifically to OxLDL, but not to native LDL or HDL under the chosen experimental conditions. (2) CD68-Fc inhibits binding and internalization of OxLDL into platelets and macrophages and attenuates the formation of macrophages/foam cells in an in vitro model. The findings imply that CD68-Fc might be a promising strategy to treat and prevent formation of vulnerable plaques, thus patients with acute coronary syndromes.
The study provides evidence for a new strategy to reduce foam cell formation in a specific manner via the inhibition of OxLDL-uptake by macrophages and platelets using the immunoadhesin CD68-Fc. Several years ago, the macrophage-derived foam cell was thought to be an elusive target for therapeutic intervention [22]. However, several reports have now provided proof for new possibilities to inhibit lipid accumulation and inflammatory responses in macrophages in a specific manner without influencing native or acquired immune function [9, 23, 24]. Because scavenger receptors are not downregulated by excessive cholesterol accumulation in the cell, they can mediate continuous accumulation of lipids in lesion macrophages [4, 8]. Thus, inhibition of this critical process in atherogenesis may reduce foam cell formation specifically and very efficiently.
Advances in both technology and the development of new generations of antibody-derived reagents have provided new options for prophylactic and therapeutic purposes [23, 25]. The latest therapeutic antibodies or parts of antibodies are genetically engineered human recombinant molecules providing high specificity and efficacy without stimulating the donors immune system [25]. Therapeutic antibodies can function as signaling molecules, activators, target specific cells, or block the action of specific molecules [23]. Other approaches include intravenous injection of immunoglobulins (Ig) to treat autoimmune and systemic inflammatory diseases. The Fc fragment of an Ig may block Fc receptors on phagocytic cells including macrophages. Immune complexes containing OxLDL are deposited in human atherosclerotic lesions during atherogenesis [26]. In vitro, OxLDL–IgG immune complexes were also found to promote foam cell formation [27]. Thus, phagocytosis of OxLDL autoantibody immune complexes by plaque-derived macrophages through an Fc-mediated pathway could represent another uptake mechanism leading to foam cell formation. This putative mechanism might also explain our finding that the Fc protein alone used in our study as control protein for the Fc-CD68 receptor construct reduces Ox-LDL uptake into macrophages, albeit to a much lesser extent than Fc-CD68 (Fig. 4b).
Immunoglobulin therapy has already been shown to suppress atherosclerosis due to Fc receptor-mediated anti-inflammatory action [28]. Another approach using the transfer of polyclonal immunoglobulins containing natural anti-OxLDL antibodies could demonstrate a reduction of atherosclerosis in ApoE−/− mice [29].
Our goal was to generate and characterize a soluble form of the scavenger receptor CD68 to test its ability to bind and accumulate in human lipid-rich atherosclerotic plaques and thus inhibit foam cell formation. The recombinant CD68-Fc consists of two CD68-Fc molecules that are cross-linked by disulfide bonds formed from the Cys in the Fc domain of each molecule. It has been reported for several other immunoglobulin-like receptors, such as GPVI-Fc, that only the dimeric form but not the monomers of the extracellular domain possess a high affinity and binding for the ligand [30]. Such fusion proteins containing IgG1 can be produced in sufficient amounts, with high purity and are relatively stable in blood. The increase in size caused by receptor dimerization and fusion with the Ig heavy chain (Fc region) enhances the avidity and prolongs the circulating half-life in blood compared to recombinant receptors alone, such as CD68 or GPVI [23]. Preliminary pharmacokinetic studies in mice indicate that CD68-Fc is detectable in blood for at least 48 h (unpublished data). Because the Fc region in IgG1-based immunoadhesins retain many functions of the parental isotype, including binding to Fc receptors and complement activation [31], the complement binding and Fc receptor binding loci were knocked out by a mutation enhancing the possibility for a potential application in humans. Due to their exquisite target specificity and pharmacokinetic stability, some immunoadhesins have already been successfully used to treat inflammatory and autoimmune disorders, such as inflammation or sepsis [32], without showing dramatic side effects.
Ramprasad et al. [7] described a specific OxLDL binding function of CD68 and macrosialin, the mouse homolog, in ligand blots using human monocyte-derived macrophages, whereas de Beer et al. [33] did not find a specific binding in murine macrophages. However, de Beer et al. showed also that both MM-LDL and OxLDL upregulated macrosialin mRNA and protein expression two to threefold in resident murine peritoneal macrophages. Thus, although the inhibition through gene silencing did not support a direct role for macrosialin as an OxLDL receptor, the authors admitted that there is “strong circumstantial evidence” for an OxLDL binding function of macrosialin and CD68 based on other studies.
The present in vitro studies with plasmon resonance analysis showed a high affinity of 10 nmol/L and highly specific binding of CD68-Fc to OxLDL. CD68-Fc did not bind to either native LDL or HDL cholesterin under the chosen experimental conditions. Moreover, ex vivo binding studies showed binding of CD68-Fc to atherosclerotic plaque specimens. In human plaques, strong binding was detected, but also slight binding of CD68-Fc was evident in the control arteries without plaque formation. These arteries, however, were from patients with coronary heart diseases and therefore not healthy at all. To determine the specific binding of CD68-Fc to atherogenous plaques compared to non-atherosclerotic arteries, we used plaque material from ApoE−/− mice and compared these to arteries from wild-type mice. The immunohistochemical stainings revealed a specific binding of CD68-Fc on atherogenous plaques of ApoE−/− mice compared to Fc control or non-atherosclerotic arteries of wild-type mice. Furthermore, preliminary autoradiography studies indicate a specific and significantly higher enrichment of 124J-CD68-Fc at lipid-rich lesions in the aortic arches of ApoE−/− mice vs. wild-type mice, which was confirmed by simultaneous lipid stainings with Sudan III. To answer the question what might happen to OxLDL/CD68-Fc aggregates if these are not taken up and degraded by macrophages, small animal PET images were made. They indicate that some 124J-CD68-Fc (bound to OxLDL or unbound) appear as well as in liver, bladder, and thyroids after 48 h (unpublished data).
These data provide evidence that soluble CD68-Fc has a high affinity to OxLDL and lipid-rich lesions and accumulates at the sites where scavenger receptor-mediated uptake of modified lipoproteins and consequent foam cell formation play a crucial role. Furthermore, in our in vitro model, foam cell formation could be induced reproducibly in vitro after several days co-incubation of human CD34+ progenitor cells or monocytes with activated platelets [17]. These foam cells are characterized by their typical morphology, the expression of the scavenger receptor CD68, and intracellular lipid deposition. They are able to take up Dil-OxLDL and accumulate lipids in their cytoplasm. Therefore, this is a suitable in vitro model to study agents influencing foam cell formation.
By the addition of CD68-Fc, platelet-induced foam cell generation derived from CD34+ progenitor cells or monocytes could be reduced dose-dependently and to comparable extent, as we have already described for statins [17]. The fact that the control protein human Fc also showed a slight inhibitory effect on Dil-OxLDL uptake compared to untreated control cultures may be due to the overlying blockade by the Fc domain interacting with macrophage Fc receptors, e.g., Fcγ which mediates to some extent foam cell formation by the uptake of OxLDL immune complexes. To elucidate the underlying mechanisms, FACS analysis was performed. We found a dose- and time-dependent uptake of Dil-OxLDL into macrophage foam cells. Furthermore, CD68-Fc was also able to inhibit the uptake of Dil-OxLDL into platelets. Interestingly, the platelet-mediated “transport” of modified LDL into macrophage foam cells via phagocytosis could also be reduced by CD68-Fc. If the ligand Ox-LDL is bound by the fusion protein CD68-Fc, the binding of OxLDL to several different scavenger receptors might be reduced belonging to class A (SR-A), class B (SR-B), and class D (CD68) of scavenger receptors (Fig. 1a). Our data also provide some proof for a new mechanism of LDL uptake into the arterial vessel wall and consequent foam cell formation. We propose that platelets represent a major “player” in this process as adhering platelets take up modified LDL via scavenger receptors, such as CD36 or LOX-1 [14], further contributing to the oxidation of lipoproteins through the substantial release of ROS (unpublished data) and transfer of modified LDL into macrophages after phagocytosis.
In addition, CD68-Fc also reduced the activity of MMP-9, a protease that is critically involved in foam cell generation, atheroprogression, and plaque rupture. The weakening of lipid-rich atheromatous plaques by the MMP-dependent degradation of the fibrous cap contributes to the occurrence of acute coronary events. Thus, inhibition of MMP activity is of major importance regarding the prevention of unstable angina and myocardial infarction [34].
Other approaches developed to inhibit foam cell formation were directed against single classes of human macrophage scavenger receptors, such as the type SR-AI. Laukkanen et al. [35] cloned a secreted “decoy” macrophage scavenger receptor (sMSR) that contains an extracellular portion of the human MSR type AI and constructed an adenoviral vector that directs high-level expression of sMSR in macrophages under the control of the human CD68 promoter. However, the concept of inhibiting a single class of scavenger receptors is not very efficient and not very specific because other functions, e.g., host defense functions, are also influenced. Furthermore, it may be difficult to obtain long-lasting expression of the transgene in the target tissue and to avoid potential immunological problems related to repeated adenoviral gene transfer [36].
The strategy to block the ligand, and not a single receptor, with antibody-like proteins [25] in a way it cannot interact with its transmembrane receptor is already successfully used as therapeutic concept for several other diseases, such as tumors or rheumatic diseases [23, 32, 37]. Some transmembrane receptors have a soluble counterpart which is involved in the regulation of growth hormone or cytokine effects [38]. Secreted proteins have also been used in therapeutic studies to inhibit the activity of the native transmembrane receptor [32, 37]. Thus, secreted decoy receptors offer potential new tools to influence pathological processes in vivo.
In conclusion, our in vitro and ex vivo data suggest that CD68-Fc may be a useful therapeutic protein capable of accumulating at lipid-rich lesions and vulnerable plaques, which could therefore enable the treatment of atherosclerotic lesions before the first symptoms in patients occur.
References
Badimon L, Martinez-Gonzalez J, Llorente-Cortes V, Rodriguez C, Padro T (2006) Cell biology and lipoproteins in atherosclerosis. Curr Mol Med 6:439–456
Stoll G, Bendszus M (2006) Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke 37:1923–1932
Moore KJ, Freeman MW (2006) Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol 26:1702–1711
Brown MS, Goldstein JL (1983) Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem 52:223–261
Gough PJ, Gordon S, Greaves DR (2001) The use of human CD68 transcriptional regulatory sequences to direct high-level expression of class A scavenger receptor in macrophages in vitro and in vivo. Immunology 103:351–361
Falk E (2006) Pathogenesis of atherosclerosis. J Am Coll Cardiol 47:C7–12
Ramprasad MP, Terpstra V, Kondratenko N, Quehenberger O, Steinberg D (1996) Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc Natl Acad Sci USA 93:14833–14838
Greaves DR, Gordon S (2005) Thematic review series: the immune system and atherogenesis. Recent insights into the biology of macrophage scavenger receptors. J Lipid Res 46:11–20
Li AC, Glass CK (2002) The macrophage foam cell as a target for therapeutic intervention. Nat Med 8:1235–1242
Llaverias G, Rebollo A, Pou J, Vazquez-Carrera M, Sanchez RM, Laguna JC, Alegret M (2006) Effects of rosiglitazone and atorvastatin on the expression of genes that control cholesterol homeostasis in differentiating monocytes. Biochem Pharmacol 71:605–614
Han J, Zhou X, Yokoyama T, Hajjar DP, Gotto AM Jr, Nicholson AC (2004) Pitavastatin downregulates expression of the macrophage type B scavenger receptor, CD36. Circulation 109:790–796
Van Berkel TJ, Out R, Hoekstra M, Kuiper J, Biessen E, Van Eck M (2005) Scavenger receptors: friend or foe in atherosclerosis? Curr Opin Lipidol 16:525–535
Fuhrman B, Koren L, Volkova N, Keidar S, Hayek T, Aviram M (2002) Atorvastatin therapy in hypercholesterolemic patients suppresses cellular uptake of oxidized-LDL by differentiating monocytes. Atherosclerosis 164:179–185
Bruni F, Pasqui AL, Pastorelli M, Bova G, Cercignani M, Palazzuoli A, Sawamura T, Gioffre WR, Auteri A, Puccetti L (2005) Different effect of statins on platelet oxidized-LDL receptor (CD36 and LOX-1) expression in hypercholesterolemic subjects. Clin Appl Thromb Hemost 11:417–428
Bays H (2006) Statin safety: an overview and assessment of the data—2005. Am J Cardiol 97:6C–26C
Massberg S, Konrad I, Bültmann A, Schulz C, Münch G, Peluso M, Lorenz M, Schneider S, Besta F, Müller I, Hu B, Langer H, Kremmer E, Rudelius M, Heinzmann U, Ungerer M, Gawaz M (2004) Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J 18:397–399
Daub K, Langer H, Seizer P, Stellos K, May AE, Goyal P, Bigalke B, Schönberger T, Geisler T, Siegel-Axel D, Oostendorp RA, Lindemann S, Gawaz M (2006) Platelets induce differentiation of human CD34+ progenitor cells into foam cells and endothelial cells. FASEB J 20:2559–2561
Schmidt R, Bültmann A, Ungerer M, Joghetaei N, Bulbul O, Thieme S, Chavakis T, Toole BP, Gawaz M, Schömig A, May AE (2006) Extracellular matrix metalloproteinase inducer regulates matrix metalloproteinase activity in cardiovascular cells: implications in acute myocardial infarction. Circulation 113:834–841
Axel DI, Frigge A, Dittmann J, Runge H, Spyridopoulos I, Riessen R, Viebahn R, Karsch KR (2001) All-trans retinoic acid regulates proliferation, migration, differentiation, and extracellular matrix turnover of human arterial smooth muscle cells. Cardiovasc Res 49:851–862
Schönberger T, Siegel-Axel D, Bussl R, Richter S, Judenhofer MS, Haubner R, Reischl G, Klingel K, Münch G, Seizer P, Pichler BJ, Gawaz M (2008) The immunoadhesin glycoprotein VI-Fc regulates arterial remodelling after mechanical injury in ApoE−/− mice. Cardiovasc Res 80:131–137
Schulz C, Penz S, Hoffmann C, Langer H, Gillitzer A, Schneider S, Brandl R, Seidl S, Massberg S, Pichler B, Kremmer E, Stellos K, Schönberger T, Siess W, Gawaz M (2008) Platelet GPVI binds to collagenous structures in the core region of human atheromatous plaque and is critical for atheroprogression in vivo. Basic Res Cardiol 103:356–367
Brewer HB Jr (2000) The lipid-laden foam cell: an elusive target for therapeutic intervention. J Clin Invest 105:703–705
Brekke OH, Sandlie I (2003) Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Discov 2:52–62
Choudhury RP, Lee JM, Greaves DR (2005) Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med 2:309–315
Stockwin LH, Holmes S (2003) The role of therapeutic antibodies in drug discovery. Biochem Soc Trans 31:433–436
Oksjoki R, Kovanen PT, Lindstedt KA, Jansson B, Pentikainen MO (2006) OxLDL–IgG immune complexes induce survival of human monocytes. Arterioscler Thromb Vasc Biol 26:576–583
Griffith RL, Virella GT, Stevenson HC, Lopes-Virella MF (1988) Low density lipoprotein metabolism by human macrophages activated with low density lipoprotein immune complexes. A possible mechanism of foam cell formation. J Exp Med 168:1041–1059
Yuan ZY, Liu Y, Kishimoto C, Shioji K, Yokode M, Liu ZQ (2003) The Fc region of immunoglobulin suppresses atherosclerosis in apolipoprotein E knockout mice. Zhonghua Yi Xue Za Zhi 83:489–493
Nicoletti A, Paulsson G, Caligiuri G, Zhou X, Hansson GK (2000) Induction of neonatal tolerance to oxidized lipoprotein reduces atherosclerosis in ApoE knockout mice. Mol Med 6:283–290
Miura Y, Takahashi T, Jung SM, Moroi M (2002) Analysis of the interaction of platelet collagen receptor glycoprotein VI (GPVI) with collagen. A dimeric form of GPVI, but not the monomeric form, shows affinity to fibrous collagen. J Biol Chem 277:46197–46204
Ashkenazi A, Chamow SM (1997) Immunoadhesins as research tools and therapeutic agents. Curr Opin Immunol 9:195–200
Yung RL (2001) Etanercept Immunex. Curr Opin Investig Drugs 2:216–221
de Beer MC, Zhao Z, Webb NR, van der Westhuyzen DR, de Villiers WJ (2003) Lack of a direct role for macrosialin in oxidized LDL metabolism. J Lipid Res 44:674–685
Rouis M (2005) Matrix metalloproteinases: a potential therapeutic target in atherosclerosis. Curr Drug Targets Cardiovasc Haematol Disord 5:541–548
Laukkanen J, Lehtolainen P, Gough PJ, Greaves DR, Gordon S, Yla-Herttuala S (2000) Adenovirus-mediated gene transfer of a secreted form of human macrophage scavenger receptor inhibits modified low-density lipoprotein degradation and foam-cell formation in macrophages. Circulation 101:1091–1096
Jalkanen J, Leppanen P, Narvanen O, Greaves DR, Yla-Herttuala S (2003) Adenovirus-mediated gene transfer of a secreted decoy human macrophage scavenger receptor (SR-AI) in LDL receptor knock-out mice. Atherosclerosis 169:95–103
Levine SJ (2004) Mechanisms of soluble cytokine receptor generation. J Immunol 173:5343–5348
Rose-John S, Heinrich PC (1994) Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J 300(Pt 2):281–290
Acknowledgments
We acknowledge the excellent technical assistance of Jadwiga Kwiatkowska, Christina Neff, Birgit Fehrenbacher, and Barbara Proksch. We also thank Maria Lissner and Lydia Kotchoubey of the Dept. of Transfusion Medicine for providing blood samples and Richard Brandl, Vascular Surgery, Krankenhaus München-Schwabing, Germany for providing specimens of carotid atherectomies.
Funding
The study was supported by grants of the Deutsche Forschungsgemeinschaft (Graduiertenkolleg [GK 794] and [GRK 438]) to M.G., K.D. and S.P. and Transregio-SFB-19 “Inflammatorische Kardiomyopathie”) and the Bundesministerium für Bildung und Forschung (BMBF) to D.S., M.G. and C.L.). M.S. was supported by the Deutsche Forschungsgemeinschaft (Sch 897/3, SFB-773 Z2).
Author information
Authors and Affiliations
Corresponding author
Additional information
Karin Daub, Dorothea Siegel-Axel, and Tanja Schönberger shared first authorship.
Rights and permissions
About this article
Cite this article
Daub, K., Siegel-Axel, D., Schönberger, T. et al. Inhibition of foam cell formation using a soluble CD68-Fc fusion protein. J Mol Med 88, 909–920 (2010). https://doi.org/10.1007/s00109-010-0629-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-010-0629-y