
Abstract
A comparative analysis of in vitro antiviral activity (in terms of the concentration of semi-maximal inhibition, IC50) against influenza virus A/PR/8/34 (H1N1) of a large series of parent 1,2,3-triazolyl nucleoside analogues (with uracil, thymine, 6-methyluracil, quinazoline-2,4-dione moieties as nucleic bases) and their prodrug forms with masked 5ʹ-phosphate groups (diethyl phosphate, diphenyl phosphate, phosphoramidate) and negatively charged H-phosphonate and monophosphate groups was carried out. Obtained structure-activity relationships were interpreted based on the assumption that the synthesized parent 1,2,3-triazolyl nucleoside analogues and their prodrug forms, by analogy with the literature data, are metabolized by cellular kinases to their active 5ʹ-triphosphate forms that inhibit the activity of viral RNA-dependent RNA polymerase (RdRp). A correlation was found between the experimental values of IC50 and the theoretical values of the binding energies of 5ʹ-triphosphate derivatives of the parent 1,2,3-triazolyl nucleoside analogues in the active site of RdRp.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Antiviral nucleoside analogues are known to be metabolized by cellular kinases to their 5ʹ-triphosphate derivatives when entering the cell [1]. They are the active forms of nucleoside analogues which inhibit the synthesis of viral RNA, thus preventing the replication of viruses [2,3,4]. However, intracellular metabolism of nucleoside analogues does not always proceed successfully and completely to their active 5ʹ-triphosphate derivatives. Quite often, intracellular metabolism of nucleoside analogues either does not occur at all or stops at the stage of formation of 5ʹ-monophosphates [5,6,7]. The reason is that nucleoside analogues differ in structure from the structure of native nucleosides, and some differ very much and cellular kinases do not recognize them well [5,6,7].
It is very likely that it is the failure of kinases to convert nucleoside analogues into their active 5ʹ-triphosphate derivatives that causes poor antiviral activity or lack thereof in many nucleoside analogues. To overcome the problem of intracellular phosphorylation, it seemed logical to use as antiviral agents not the nucleoside analogues themselves, but their 5ʹ-mono, di- or triphosphate derivatives. The literature provides examples of the synthesis of such compounds [8, 9], however, there is negligible data on the evaluation of their antiviral activity. We are aware of only one case when 5ʹ-triphosphates were added to infected cells [10], in most cases experiments were carried out with individual viral RNA-dependent RNA polymerases (RdRp) [11, 12].The probable reason is the fear of chemists and biologists that nucleoside analogues with a phosphate substituent at position C5ʹ (nucleotide analogues) due to their negative charge cannot penetrate into the cell through the lipid-rich cell membrane [13, 14].
To overcome the problem of penetration into the cell of negatively charged 5ʹ-phosphate analogues of nucleosides (nucleotides), A.Mongometri in 1961 proposed using their esters which could pass through lipid membranes and, as a result of hydrolysis by cellular esterases, turn into the original 5ʹ-phosphate analogues of nucleosides [13]. In the early 1990s, K. McGuigan proposed masking a negatively charged phosphate substituent in the 5ʹth position with an aryl group and an amino acid ester residue [15]. Since both of these approaches replacing (masking) the phosphate group in nucleotide analogues with dialkyl (diaryl) phosphate or phosphoramidate moieties were applied to nucleoside analogues approved by the U.S. Food and Drug Administration (FDA) as drugs for the treatment of infections caused by HIV, hepatitis B and C viruses, they were called prodrug approaches, and derivatives of nucleoside analogues having dialkyl(diaryl) phosphate or phosphoramidate substituent in the 5ʹth position have been called their prodrug forms. The literature provides many examples of prodrugs of such structure being much more active than their parent nucleoside analogues as inhibitors of replication of HIV [16], hepatitis B [17], hepatitis C [18, 19], dengue [20] viruses, as well as SARS-CoV-2 [21]. Despite numerous data on the antiviral activity of both various nucleoside analogues [2, 22,23,24] and their prodrug forms of various structures, including 5ʹ-triphosphates [10,11,12, 16,17,18,19,20,21], there is no systematic comparative analysis of them in the literature.
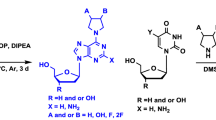
Herein, for the first time, we carried out such a comparative analysis of antiviral activity against influenza virus A/PR/8/34 (H1N1) of a large series of parent 1,2,3-triazolyl nucleoside analogues and their prodrug forms with the popular in the literature [9] masked phosphate substituents in the 5ʹth position (diethyl phosphate, diphenyl phosphate, phosphoramidate) and negatively charged H-phosphonate and monophosphate groups to check whether negatively charged analogues of 1,2,3-triazolyl nucleosides will be active or not. This series consists of two parts: 1.2.3-triazolyl nucleoside analogues with uracil and quinazoline-2,4-dione moieties as nucleic bases and their prodrug forms which were synthesized by us earlier [25, 26] and 1.2.3-triazolyl nucleoside analogues with 5ʹ-methyluracil (thymine) and 6-methyluracil moieties as nucleic bases and their prodrug forms whose synthesis and antiviral activity against influenza virus A/PR/8/34 (H1N1) are described for the first time in this paper.
Results and discussion
Chemistry
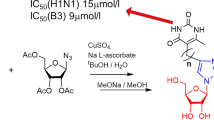
The synthesis of 1.2.3-triazolyl nucleoside analogues with 6-methyluracil and 5-methyluracil (thymine) moieties as nucleic bases and their prodrug forms was carried out according to a methodology we developed earlier [25]. Alkynyl derivatives of 6-methyluracil 1a,b and thymine 2a,b prepared by analogy with the methods previously described [26] were involved in the copper-catalyzed reaction of azide-alkyne cycloaddition (CuAAC) with 2,3,5-tri-O-acetyl-β-D-ribofuranosylazide 3 [25] to obtain 1,2,3-triazolyl nucleoside analogues 4a,b and 5a,b with protected hydroxyl groups. (Scheme 1). The formation of the 1,2,3-triazole ring was confirmed by the appearance in the 1H NMR spectra of compounds 4a,b and 5a,b a signal of the triazolyl proton at the C5ʹʹ atom within the range of 7.50-7.97 ppm. Triazole carbon atoms C4ʹʹ in the 13C NMR spectra of compounds 4a,b and 5a,b resonated within the range of 142.85-147.68 ppm, and signals of triazole carbon atoms C5ʹʹ were observed within the range of 120.10-123.54 ppm. All these facts fully corresponded to the characteristic features of the 1H and 13C NMR spectra of 1,2,3-triazoles have been described in the literature [27,28,29]. The anomeric protons of acetylated D-ribofuranose residues in the 1H NMR spectra of compounds 4a,b and 5a,b were represented by single doublets within the range of 5.98-6.13 ppm with vicinal constants within the range of 3.5–4.2 Hz. This indicated that compounds 4a,b and 5a,b were obtained in the form of individual β-isomers. Then the O-acetyl protective groups of compounds 4a,b and 5a,b were removed with a 0.1 N MeONa/MeOH solution and the target 1,2,3-triazolyl nucleoside analogues with free hydroxyl groups 6a,b and 7a,b (Scheme 1) were obtained with good yields (85–98%).

Synthesis of 1,2,3-triazolyl nucleoside analogues
For further phosphorylation of compounds 6a,b and 7a,b at the C5ʹ position by reactive phosphorus-containing reagents, it was necessary to selectively protect the hydroxyl groups at the C2ʹ and C3ʹ atoms, leaving the hydroxyl group free at the C5ʹ atom. For this purpose, nucleoside analogues 6a,b and 7a,b by the reaction with 2,2-dimethoxypropane in acetone in the presence of p-toluene sulphonic acid (TsOH) were converted into 1,2,3-triazolyl nucleoside analogues 8a,b, 9a,b with isopropylidene protection of hydroxyl groups at the C2ʹ and C3ʹ atoms and the free hydroxyl group at the C5ʹ atom (Scheme 1).
According to the already known procedure [25], nucleoside analogues 6a,b, 7a,b were involved in the reaction with diethyl phosphorocloridate and diphenyl phosphorocloridate in pyridine at room temperature to obtain 5ʹ-diethylphosphates 10a,b, 11a,b and 5ʹ-diphenylphosphates 12a,b, 13a,b, respectively (Scheme 2). The listed nucleotide analogues were isolated by flash chromatography on silica gel with 11-83% yields, respectively. The presence of a diethyl phosphate group in compounds 10a,b, 11a,b was indicated by 31P NMR spectra, in which a single signal presented within the range from -1.2 to -1.6 ppm. In the 1H NMR spectra of these compounds, a triplet was observed at 1.22–1.56 ppm and a quartet at 4.02–4.07 ppm with vicinal coupling constants within the range of 7.05–7.15 Hz corresponding to the resonance of the ethyl groups. In addition, the signals of methylene protons at the C5ʹ atom in compounds 10a,b, 11a,b were shifted downfield and resonated as multiplets within the range of 4.13-4.34 ppm. A similar spectral pattern was observed in the NMR spectra of compounds 12a,b, 13a,b. In their 31P NMR spectra, a singlet corresponding to the phosphorus atom of the phosphate group was observed at -12.2 ppm. In the 1H NMR spectra the signals of protons of aromatic rings of the phosphate group appeared as a multiplet at 7.09–7.32 ppm. In addition, two doublet of doublets corresponding to the resonance of the methylene protons at the C5ʹ atom observed in the 1H NMR spectra of the initial compounds 6a,b, 7a,b in the spectra of the products 12a,b, 13a,b were shifted downfield as a complex multiplet at 4.32-4.46 ppm.

Synthesis of 5ʹ-diethyl and 5ʹ-diphenyl derivatives of 1.2.3-triazolyl nucleoside analogues
By analogy with the method already used [25], the synthesis of 5ʹ-phosphoramidate derivatives of 1,2,3-triazolyl nucleoside analogues 6a,b, 7a,b was carried out in four stages (Scheme 3). Note that according to the 31P NMR spectra in which two signals of approximately equal intensity were observed within the region of 3.61–4.9 ppm, phosphoramidates 17a,b, 18a,b, 19a,b, 20a,b were isolated as a mixture of two diastereomers. The absolute configurations of diastereomeric phosphoramidate derivatives 19a,b, 20a,b were not determined because, as it appeared later, they were inactive and therefore the configuration of their chiral centres had no significance for us in this study.

Synthesis of 5ʹ-phosporamidates of 1,2,3-triazolyl nucleoside analogues
Synthesis of derivatives of 1,2,3-triazolyl nucleoside analogues 6a,b, 7a,b with a H-phosphonate substituent at the 5ʹth position was carried out in 4 stages in accordance with the procedure described earlier [25] (Scheme 4). At the first stage, the interaction of salicylic acid 21 with phosphorus trichloride produced salicyl phosphorochloridate 22 which was then involved in the reaction with 2ʹ,3ʹ-O-isopropylidene-protected 1,2,3-triazolyl nucleoside analogues 8a,b, 9a,b to afford their 5ʹ-salicylphosphite derivatives 23a,b, 24a,b as a mixture of two diastereomers which appeared in the 31P {1H} NMR spectra as two singlets at 125 ppm corresponding to the phosphorus (III) atom. In the 31P NMR spectra these signals were observed as triplets with 3JPH constants within the range of 8.8–9.2 Hz. Salicylphosphites 23a,b, 24a,b were hydrolyzed in situ with aqueous triethylamine to obtain 2ʹ,3ʹ-O-isopropylidene-protected 5ʹ-H-phosphonate nucleoside analogues 25a,b, 26a,b isolated by flash chromatography on silica gel in 62-72% yields (Scheme 4). A single signal within the range of 4.02–4.14 ppm in the 31P NMR spectra of compounds 25a,b, 26a,b confirmed the presence of the 5ʹ-H-phosphonate group. In the 1H NMR spectra the formation of 5ʹ-H-phosphonates was evidenced by a change in a multiplicity and a downfield shift of the proton signals at the C5ʹ atom, as well as the appearance of a proton signal at the phosphorus atom which resonated as a doublet at 6.6 ppm with a spin-coupling constant of 620–622 Hz. In addition, the spectra contained signals of triethylammonium protons with integral intensities corresponding to two triethylammonium molecules per nucleotide anion molecule. At the last stage, the 2ʹ,3ʹ-O-isopropylidene protection of the D-ribofuranose residue was removed with aqueous trifluoroacetic acid and targeted 1,2,3-triazolyl nucleoside analogues 27a,b, 28a,b with the H-phosphonate group at the atom C5ʹ were obtained in 60-95% yields (Scheme 4). The removal of 2ʹ,3ʹ-O-isopropylidene protection was indicated by the absence of two singlets in the 1H NMR spectra at 1.38 and 1.57 ppm corresponding to the resonance of methyl groups of the isopropylidene protection.

Synthesis of 5ʹ-H-phosphonates of 1,2,3-triazolyl analogues
Synthesis of 5ʹ-monophosphate derivatives of 1,2,3-triazolyl nucleoside analogues was carried out in 4 stages (Scheme 5) by analogy with the previously described procedure [25]. First, the hydrogen atom at the phosphorus in 5ʹ-H-phosphonates 25a,b, 26a,b was replaced with a trimethylsilyl group. The resulting trimethylsilyl esters 29a,b, 30a,b were in situ oxidized by molecular iodine in the presence of triethylamine to afford 5ʹ-iodophosphates 31a,b, 32a,b in turn hydrolyzed in situ to obtaine 2ʹ,3ʹ-O-isopropylidene-protected phosphates 33a,b, 34a,b isolated by flash chromatography on silica gel in 19-48% yields (Scheme 5). The formation of the 5ʹ-phosphate group was indicated by the 31P NMR spectra in which the signal of the phosphorus atom of the 5ʹ-H-phosphonate group within the range of 4.02-4.14 ppm disappeared and a single signal within the range of 0.63–1.22 ppm corresponding to the phosphorus atom of the 5ʹ-phosphate group was observed. In the 1H NMR spectra of phosphates 33a,b, 34a,b there was no doublet of the proton at the phosphorus atom of the 5ʹ-H-phosphonate group at 6.6 ppm with a spin-coupling constant of 620–622 Hz. Besides, the protons at the C5ʹ atom, having undergone a downfield shift, resonated as a multiplet within the range of 3.89-3.91 ppm, whereas in the initial H-phosphonates 25a,b, 26a,b they resonated at 3.85–3.86 ppm. The listed spectral facts testified to the successful oxidation of the 5ʹ-H-phosphonate group to the phosphate one. In addition, the formation of phosphates 33a,b, 34a,b was confirmed by ESI mass spectrometry. At the last stage, the 2ʹ,3ʹ-O-isopropylidene protection of the D-ribofuranose residue was removed with aqueous trifluoroacetic acid and targeted 1,2,3-triazolyl nucleotide analogues 35a,b, 36a,b with the monophosphate group in the 5ʹth position were obtained in 55–76% yields.

Synthesis of 5ʹ-monophosphates of 1,2,3-triazolyl nucleoside analogues
Antiviral evaluation
The in vitro antiviral activity of synthesized 5ʹ-phosphorylated 1,2,3-triazolyl analogues of nucleosides 10a,b-13a,b, 19a,b, 20a,b, 27a,b, 28a,b, 35a,b, 36a,b was evaluated against influenza virus A/Puerto Rico/8/34 (H1N1). The results expressed as a concentration causing 50% inhibition of virus replication (IC50), a concentration causing the death of 50% of cells infected with the virus (CC50), and a selectivity index (SI) equal to the CC50/IC50 ratio are presented in Table 1. For a systematic analysis of the antiviral activity of the entire series of synthesized 5ʹ-phosphorylated 1,2,3-triazolyl nucleoside analogues and their parent compounds, Table 1 also presents data for previously synthesized 5ʹ-phosphorylated 1,2,3-triazolyl nucleoside analogues 37a,b-46a,b [25], as well as parent 1,2,3-triazolyl nucleoside analogues 47a,b-50a,b [26].
Before proceeding to the analysis of the antiviral activity of the mentioned series of compounds, it is necessary to make the following explanation. Since the most active compounds in the studied series showed moderate (one can say low) antiviral activity (the IC50 values within the range of 18-74 μM), we did not carry out serious intracellular experiments such as studying the metabolism of parent compounds 47a,b-50a,b and their prodrug forms 10a,b-13a,b, 19a,b, 20a,b, 27a,b, 28a,b, 35a,b, 36a,b-46a,b by cellular kinases. We considered it quite sufficient to rely on numerous literature data [2,3,4] that both nucleoside analogues and their prodrug forms are metabolized by cellular kinases to their 5ʹ-triphosphate derivatives. It is these 5ʹ-triphosphate derivatives (and not the nucleoside analogues themselves or their prodrug forms) that inhibit a viral replication [2,3,4]. Hence it follows that the experimental IC50 values do not refer to the parent nucleoside analogues and their prodrug forms mentioned above, but to the 5ʹ-triphosphate derivatives of the parent nucleoside analogues. Therefore, it can be assumed that the IC50 values correlate with the efficiency of intracellular metabolism of the mentioned parent nucleoside analogues or their prodrug forms up to 5ʹ-triphosphate derivatives.
Using the above assumptions based on the literature data [2,3,4], the analysis of Table 1 allows us to draw four important conclusions. First, the parent 1,2,3-triazolyl nucleoside analogues possessing the uracil (compounds 47a,b) and thymine (compounds 49a,b) moieties were inactive. Of all the parent 1,2,3-triazolyl nucleoside analogues, only compounds with the 6-methyluracil (compound 48b) and quinazoline-2,4-dione (compounds 50a,b) moieties instead of natural nucleic bases showed activity. It was moderate activity with the IC50 values within the range of 30-48 μM.
Secondly. Approximately the same moderate activity (the IC50 values within the range of 18-74 μM) was shown by 5ʹ-phosphorylated derivatives of the same parent nucleoside analogues 48b, 50a,b, namely 5ʹ-diethyl phosphate 10b and 5ʹ-diphenyl phosphates 12a, 43a,b. Taking into account that the synthesis of viral RNA is inhibited not by nucleoside analogues but by their 5ʹ-triphosphate derivatives [2,3,4], the data obtained show that intracellular metabolism of both parent nucleoside analogues 48b, 50a,b and their 5ʹ-phosphorylated derivatives 10b, 12a, 43a,b proceeds with the same efficiency.
Thirdly. The introduction in the 5ʹth position of inactive 1,2,3-triazolyl nucleoside analogues 47a with the uracil (IC50 > 880 μM) and 49b with the thymine (IC50 = 278 μM) moieties of a phosphoramidate and diphenyl phosphate substituents, respectively, allowed them to penetrate into the cell and be metabolized to the corresponding 5ʹ-triphosphates. Indeed, 5ʹ-phosphoramidate 39a with the uracil and 5ʹ-diphenyl phosphate 13b with the thymine moieties, that is, according to the literature data [2,3,4], the corresponding 5ʹ-triphosphates showed moderate activity with the IC50 values of 25 and 18 μM, respectively.
Fourthly. The negatively charged 5ʹ-H-phosphonates 27b with the 6-methyluracil and 28b with thymine moieties showed moderate activity with the IC50 values of 74 and 25 μM, respectively. This seems surprising since nucleoside analogues with a phosphate substituent at the position C5ʹ (nucleotide analogues) cannot penetrate the cell through the lipid-rich cell membrane due to their negative charge [13, 14]. However, the literature provides at least one example when a negatively charged nucleotide analogue showed high antiviral activity [30]. This is 3ʹ-azido-3ʹ-deoxythymidine-5ʹ-yl H-phosphonate which has been registered in the Russian Federation as an antiviral agent used as a part of combined antiretroviral therapy for HIV infection and called Nikavir [9, 30]. One can assume that negatively charged Nicavir and 5ʹ-H-phosphonates 27b and 28b do not penetrate into the cell themselves but inhibit the functions of external glycoproteins of viruses preventing the penetration of viruses into the cell.
Summing up the analysis of the data of Table 1, one can note that only 11 compounds, namely, 10b, 12a, 13b, 27b, 28b, 39a, 43a,b, 48b, 50a,b, showed moderate antiviral activity against influenza virus A/Puerto Rico/8/34 (H1N1) with the IC50 values within the range of 18-74 μM. The remaining compounds were completely inactive.
Molecular docking
We carried out a molecular docking study to test the ability of the synthesized compounds to inhibit some well-known viral target proteins. Nucleoside analogues can inhibit influenza A virus replication by interacting with a limited set of targets only. Among them the influenza viral RNA-dependent RNA polymerase (RdRp) plays a special role. Important structural unit of RdRp is the acidic polymerase, namely, its N-terminal cation-dependent domain (PA-Nter, PDB code 4AWK [31]). This active site is characterized by low selectivity for the structure of an inhibitor molecule. PA-Nter is highly conserved in all RNA viruses [32, 33], including strains of influenza A virus [34, 35]. Based on these literature data, we chose the active site of PA-Nter RdRp of influenza A virus (PDB code 4AWK) as a drug target.
It was already mentioned above that based on the literature data [2,3,4], we assumed that the parent compounds 47a,b-50a,b and their prodrug forms 10a,b-13a,b, 19a,b, 20a,b, 27a,b, 28a,b, 35a,b, 36a,b-46a,b are metabolized by cellular kinases to their active 5ʹ-triphosphate derivatives 51-57 (presented in Table 2) and it is 51-57 that inhibit a viral replication.Therefore, we carried out molecular docking of 5ʹ-triphosphates 51-57 into the active site of the selected drug target. The binding energies of 5ʹ-triphosphates 52-57 which are derivatives of the active compounds 10b, 12a, 13b, 39a, 43a,b, 48b, 50a,b into the active site of the protein PA-Nter endonuclease domain (PDB code 4AWK), as well as, for comparison, the binding energy of the hypothetical 5ʹ-triphosphate 51 having no active precursors are presented in Table 2. Triphosphates 55, 56, 57 containing the quinazolin-2,4-dione and uracil moieties demonstrate the highest binding energies (8.9–9.8 kcal/mol). But compound 51 also with uracil moiety has the lowest binding energy (7.8 kcal/mol) in the PA-Nter active site. Consider the position of the ligands in the protein cavity (Fig. 1) to find an explanation for this discrepancy. According to molecular docking calculations, compounds 57, 55, 51 are located approximately in the same region of space and their triphosphate fragments are localized deep in the cavity of the PA-Nter active site. On the contrary, compound 56 is localized in such a way that its triphosphate fragment is located at the mouth of this cavity. This arrangement of the compounds under study seems surprising because compound 51 with the uracil moiety is not located in the same area of the active site in which the lead compound 56 also having the uracil moiety is located, but in the area in which the leaders 57 and 55 with the quinazoline-2,4-dion moiety are located. A significant difference in the structure of compounds 56 and 51 can serve as a possible explanation for this phenomenon. In the lead compound 56, the uracil moiety is attached to the 1,2,3-triazolylribofuranosyl fragment via the methylene bridge, whereas in compound 51 via the butylene chain, that certainly increased the size of compound 51. This led to the fact that compound 51 could not locate in the same area of space as compound 56 and appeared to be in the same place where the lead compounds 57 and 55 with quinazoline-2,4-dione moieties are located. However, once in this region of the cavity of the PA-Nter active site, compound 51 did not find amino acid residues with which it could form hydrogen bonds. As a result, compound 51 is retained in the active site only due to van der Waals interactions and shows the lowest binding energy among the studied compounds (7.8 kcal/mol, Table 2). The lead compounds 57 and 55 are retained in the cavity of the active site by hydrogen bonding of their triphosphate fragments with the amino acid residue Val122 and the π-π stacking interaction of their quinazoline-2,4-dione moiety with the amino acid residue Tyr24.

Molecular docking simulations of the optimized docking model of compounds 51, 55, 56, 57 in the PA-Nter (PDB code 4AWK) active site of the RdRp of influenza virus A (H1N1) obtained in the lowest-energy conformations
In contrary to compound 55, 5ʹ-triphosphate 57 is additionally hydrogen-bonded with the Lys134 residue in the active site of the acidic polymerase, which explains the higher binding energy of compound 57 (9.8 kcal/mol) compared to compound 55 (8.9 kcal/mol, Table 2).
The lead compound 56, being localized in another region of the cavity of the PA-Nter active site, is nevertheless well retained in it due to the hydrogen bonding of its triphosphate fragment with the amino acid residues Arg84 and Trp88 and shows the high binding energy of 9.1 kcal/mol (Table 2).
It has already been noted above that as according to the literature data [2,3,4], it is 5ʹ-triphosphate derivatives that inhibit viral replication, the observed experimental values of IC50 (Table 1) can be attributed not to the parent nucleoside analogues 47a,b-50a,b and their prodrug forms 10a,b-13a,b, 19a,b, 20a,b, 27a,b, 28a,b, 35a,b, 36a,b-46a,b but to their corresponding 5ʹ-triphosphate derivatives 51-57. Therefore the IC50 values presented in Table 1 can be correlated with the binding energies of triphosphates 51-57 in the PA-Nter active site (Table 2). Indeed, comparing Table 1 and Table 2, one can see rather good correlation between antiviral activity of the parent 1,2,3-triazolyl nucleoside analogues 48b, 50a,b, their prodrug forms 10b, 12a, 43a,b, that is, the IC50 values, and the binding energies of their corresponding 5ʹ-triphosphate derivatives 52-57 in the PA-Nter active site. For example, a decrease in IC50 values, that is, an increase in antiviral activity, during the transition from compound 12a (IC50 = 62 μM) to compound 43a (IC50 = 17.9 μM) correlates with an increase in the binding energy during the transition from 5ʹ-triphosphate 52 (8.3 kcal/mol) to 5ʹ-triphosphate 57 (9.8 kcal/mole). Or another example. A decrease in IC50 values (an increase in activity) during the transition from compound 10b (IC50 = 64 μM) to compound 43a (IC50 = 17.9 μM) correlates with an increase in the binding energy during the transition from 5ʹ-triphosphate 54 (8.5 kcal/mol) to 5ʹ-triphosphate 57 (9.8 kcal/mol). This can be considered indirect evidence that the prodrug forms of 1,2,3-triazolyl nucleoside analogues (in this case, 5ʹ-diphenylphosphates 12a, 43a, 5ʹ-diethylphosphate 10b) exhibit antiviral activity against influenza A virus (H1N1) in the form of their 5ʹ-triphosphate derivatives. The following fact also testifies in favour of this conclusion. The parent 1,2,3-triazolyl nucleoside analogue 47a having the uracil moiety is completely inactive (IC50 > 880 μM, Table 1), even though its 5ʹ-triphosphate derivative 56 has the high (9.1 kcal/mol) binding energy in the PA-Nter active site (Table 2). At the same time, the prodrug form of compound 47a (5ʹ-phosphoramidate 39a) showed moderate antiviral activity with the IC50 value of 25 μM (Table 1).
The reason for the observed difference will become clear if we assume (as we did above) that the IC50 values in Table 1 assigned by us to the 5ʹ-triphosphates 51-57 correlate with the efficiency of intracellular metabolism of parent nucleoside analogues 47a,b-50a,b or their prodrug forms 10a,b-13a,b, 19a,b, 20a,b, 27a,b, 28a,b, 35a,b, 36a,b-46a,b to their corresponding 5ʹ-triphosphate derivatives 51-57. The parent compound 47a either failed to penetrate into the cell or was not converted by cellular enzymes into its active 5ʹ-triphosphate form 56, whereas the prodrug form of compound 47a, 5ʹ-phosphoramidate 39a, was able to penetrate into the cell and was converted by cellular enzymes into 5ʹ-triphosphate 56 whose antiviral activity, according to the literature data [2,3,4] was observed experimentally with the IC50 value of 25 μM. The data in Table 1 allow us to draw another important conclusion. Since compounds 12a, 43a and 10b, 43a, on the one hand, and 5ʹ-triphosphates 52, 57 and 54, 57, on the other hand, differ in the heterocyclic moiety, one can conclud that exactly the heterocyclic moiety of both prodrug forms 12a, 10b, 43a and 5ʹ-triphosphates 52, 54, 57 is responsible for the difference in their antiviral activity. It follows from this that the quinazoline-2,4-dione moiety in 1,2,3-triazolyl nucleoside analogues provides greater antiviral activity than the 6-methyluracyl moiety.
Above, we assumed that the antiviral activity of negatively charged 5ʹ-H-phosphonate 27b with the 6-methyluracil moiety and 28b with the thymine moiety is the result of their interaction with the surface glycoproteins hemagglutinin H1 and neuraminidase N1 of influenza A virus (H1N1) which prevent the penetration of influenza virus A into the cell. The effectiveness of this interaction we verified by molecular docking simulations.
The glycoprotein hemagglutinin, which ensures the attachment and fusion of influenza virus with the cell membrane, is a well-studied drug target, and its interaction with antiviral drugs is well modeled in silico [36,37,38]. We used the structure of the hemagglutinin H1 of influenza virus A/Puerto Rico/8/1934/H1N1 obtained by X-ray diffraction (PDB: 1RU7) [39] and downloaded from the RCSB Protein Data Bank database [40]. The binding energies and ligand-protein interactions of active 5ʹ-H-phosphonates 27b and 28b in the active site of hemagglutinin H1 and, for comparison, of inactive 5ʹ-H-phosphonates 27a and 28a obtained by molecular docking simulations together with their IC50 values are presented in Table 3. One can see that for 5ʹ-H-phosphonate 27b there is a good correlation of its activity (the IC50 value) with the binding energy. The binding energy of 5ʹ-H-phosphonate 27b (8.9 kcal/mol) which showed antiviral activity with the IC50 value of 74 μM is greater than the binding energy of inactive (IC50 > 746 μM) 5ʹ-H-phosphonate 27a (8.1 kcal/mol). At the same time, there is no such pronounced correlation for 5ʹ-H-phosphonate 28b. With a huge difference in the IC50 values of 5ʹ-H-phosphonates 28a and 28b, their binding energies differ by only 0.1 kcal/mol. Nevertheless, the binding energies of compounds 27b and 28b in the active site of the protein hemagglutinin H1 are comparable to the binding energy of the lead compound 54 in the active site of the PA-Nter protein (Table 2).
The surface glycoprotein neuraminidase, together with the glycoprotein hemagglutinin, ensures the penetration of influenza virus into the cell. The glycoprotein neuraminidase removes terminal residues of sialic acid from the silylated receptors of the cell membrane, thus preparing them for the attack of the glycoprotein hemagglutinin [41, 42]. The glycoprotein neuraminidase is a well-studied drug target, and its interaction with antiviral drugs is well modeled in silico [43,44,45]. The structure of the glycoprotein neuraminidase N1 of influenza A (H1N1) virus obtained by X-ray diffraction (PDB:4B7Q) [46] which was downloaded from the RCSB Protein Data Bank database [40] was used for the molecular docking calculations. The binding energies and ligand-protein interactions of active 5ʹ-H-phosphonates 27b and 28b and, for comparison, inactive 5ʹ-H-phosphonates 27a and 28a in the active site of the neuraminidase N1 obtained by molecular docking simulations together with their IC50 values are presented in Table 4. One can see that the binding energies of compounds 27b and 28b in the neuraminidase N1 active site (8.3 and 8.1 kcal/mol, respectively, Table 4) are slightly lower than the binding energies of these compounds in the hemagglutinin H1 active site (8.9 and 8.7 kcal/mol, respectively, Table 3). However, based only on these data, it is premature to conclude which namely surface glycoprotein of influenza A (H1N1) virus was inhibited by 5ʹ-H-phosphonates 27b and 28b. Special biological experiments on individual the hemagglutinin H1 and neuraminidase N1 of influenza A (H1N1) virus are needed.
Cytotoxicity evaluation
Table 1 shows that several 5′-phosphorylated 1,2,3-triazolyl nucleoside analogues from the herein synthesized series, namely 10b, 13b, 27b, 28b, appeared to be cytotoxic against MDCK cells used to evaluate antiviral activity. It seemed interesting to check whether herein synthesized compounds are cytotoxic against human cancer cell lines. For this purpose, 5′-phosphorylated 1,2,3-triazolyl nucleoside analogues 10a,b-13a, 19a,b, 20a,b, 27b, 28a,b 35a,b, 36a,b were also evaluated for in vitro cytotoxicity against eight human cancer cell lines: cervical epitheloid carcinoma M-HeLa; duodenal adenocarcinoma HuTu-80; prostate adenocarcinoma PC3, breast adenocarcinoma MCF-7, hepatoblastoma HepG2, pancreatic carcinoma PANC-1, pulmonary adenocarcinoma A549, melanoma А-375 as well as a diploid human cell strain WI-38 composed of fibroblasts. The resulting data expressed as concentrations causing the inhibition of the growth of 50% of cells in the experimental population (IC50) are presented in Table 5. As one can see in Table 5, practically all tested compounds demostrated moderate cytotoxicity against both human cancer cell lines and normal WI-38 cells of the lung embryo. Thus, the tested compounds do not represent a scaffold for the search for new anti-cancer agents, with the possible exception of 5′-phosphate 35a which was the only compound which appeared to be not cytotoxic against normal WI-38 cells (IC50 = 129 μM).
Conclusions
The comparative analysis of antiviral activity against influenza virus A/PR/8/34 (H1N1) of a large series of parent 1,2,3-triazolyl nucleoside analogues and their prodrug forms with masked phosphate groups (diethyl phosphate, diphenyl phosphate, phosphoramidate) and negatively charged H-phosphonate and monophosphate groups was carried out. This series consists of two parts: 1.2.3-triazolyl nucleoside analogues with uracil and quinazoline-2,4-dione moieties as nucleic bases and their prodrug forms that were synthesized by us earlier [25, 26] and 1.2.3-triazolyl nucleoside analogues with 5-methyluracil (thymine) and 6-methyluracil moieties as nucleic bases and their prodrug forms whose synthesis and antiviral activity against influenza virus A/PR/8/34 (H1N1) are described for the first time in this paper. When analyzing the obtained data on antiviral activity (IC50 values), we relied on the literature data [2,3,4] that nucleoside analogues and their prodrug forms, having penetrated into the cell, are metabolized by cellular kinases to their 5ʹ-triphosphate derivatives and it is 5ʹ-triphosphate derivatives that inhibit viral replication. Therefore, we always kept in mind that the experimental values of IC50 in Table 1 actually refer to 5ʹ-triphosphates 52-57 and therefore correlated them with the efficiency of intracellular metabolism converting parent 1,2,3-triazolyl nucleoside analogues 47a,b-50a,b and their prodrug forms 10a,b-13a,b, 19a,b, 20a,b, 27a,b, 28a,b, 35a,b, 36a,b-46a,b into their corresponding 5ʹ-triphosphate derivatives 51-57. In addition, we correlated the experimental IC50 values with the binding energies of the mentioned 5ʹ-triphosphates 51-57 in the active site of a popular in the literature [31,32,33,34,35] a drug target of RNA viruses, including influenza A (H1N1) virus, namely the PA-Nter active site of the viral RNA-dependent RNA polymerase (RdRp) of influenza A (H1N1) virus (PDB code 4AWK). Using the above assumptions based on the literature data [2,3,4], we have drawn the following important conclusions.
Among all the analysed parent compounds, only 1,2,3-triazolyl nucleoside analogues having 6-methyluracil and quinazoline-2,4-dione moieties instead of native nucleic bases (compounds 48b, 50a,b) showed moderate antiviral activity, whereas 1,2,3-triazolyl nucleoside analogues with uracil and thymine moieties (compounds 47a,b, 49a,b) appeared to be completely inactive. Consequently, according to the literature data [2,3,4], they were not converted by cellular kinases into their active 5′-triphosphate derivatives. However, their prodrug forms, namely 5′-diphenyl phosphate and 5′-phosphoramidate derivatives (compounds 13b, 39a) showed moderate activity, therefore, according to the literature data [2,3,4] intracellular kinases were able to convert them into active 5′-triphosphate derivatives. Thus, the introduction of phosphorus-containing substituents into the 5′th position of inactive 1,2,3-triazolyl nucleoside analogues gives them antiviral activity. Approximately the same moderate antiviral activity was demonstrated by both the parent 1,2,3-triazolyl nucleoside analogues 48b, 50a,b with the 6-methylural and quinazoline-2,4-dione moieties and their prodrug forms 10b, 12a, 43a,b. This indicates that they are equally efficiently metabolized by intracellular enzymes to active 5′-triphosphate derivatives of the parent 1,2,3-triazolyl nucleoside analogues 48b, 50a,b. It is interesting to note that an increase in antiviral activity, i.e. a decrease in the IC50 values in the studied series of compounds from 74 to 18 μM (Table 1), approximately correlates with an increase of binding energy of corresponding 5′-triphosphate derivatives in the PA-Nter active site of the RNA-dependent RNA polymerase (RdRp) of influenza A (H1N1) virus (Table 2). For example, a decrease in the IC50 values, that is, an increase in antiviral activity, during the transition from compound 12a (IC50 = 62 μM) to compound 43a (IC50 = 17.9 μM) correlates with an increase in the binding energy during the transition from 5ʹ-triphosphate 52 (8.3 kcal/mol) to 5ʹ-triphosphate 57 (9.8 kcal/mole). This indirectly confirms our assumption that the active compounds 10b, 12a, 13b, 39a, 43a,b, 48b, 50a,b inhibited the replication of influenza virus A (H1N1) in the form of their 5′-triphosphate derivatives 52-57. Moderate antiviral activity was demonstrated by negatively charged 5′-H-phosphonates 27b and 28b (the IC50 values of 74 and 25 μM, respectively). Since negatively charged nucleoside analogues cannot penetrate the lipid-rich cell membrane [13, 14], we assumed that 5′-H-phosphonates 27b and 28b interact with the surface glycoproteins hemagglutinin H1 and neuraminidase N1 of influenza A (H1N1) virus. Molecular docking study indirectly confirmed this proposition by showing that the binding energies of 5′-H-phosphonates 27b and 28b in the active sites of the glycoprotein’s hemagglutinin H1 and neuraminidase N1 are in the range of 8.1–8.9 kcal/mol that approximately corresponds to the binding energies of 5′-triphosphate derivatives 55, 56, 57.
Material and methods
Chemistry
Instrumentations and chemicals
1H NMR spectra were recorded on 400 MHz, 500 MHz and 600 MHz Brucker Advance. 13C NMR spectra were obtained in the above instrument operating at 100.6 MHz. Mass spectra (MALDI) were recorded in a positive ion mode on a Bruker Ultraflex III TOF/TOF mass spectrometer for 10-3 mg/mL solutions in MeOH. The ESI MS measurements were performed using an AmazonX ion trap mass spectrometer (Bruker Daltonic GmbH, Germany) in the positive or negative mode in the mass range of 70–3000.The capillary voltage was 3500 V, nitrogen drying gas 10 L·min-1, desolvation temperature 250°С. A methanol/water solution (70:30) was used as a mobile phase at a flow rate of 0.2 mL/min by binary pump (Agilent 1260 chromatograph, USA). The sample was dissolved in methanol to a concentration of 10-6 g·L-1. Flash chromatography was performed on silica gel 60 (40-63 μm, Buchi, Sepacore). Thin-layer chromatography was carried out on plates with silica gel (Sorbfil, Russia). Spots of compounds were visualized by using ultraviolent fluorescence under a short wavelength (254 nm) followed by heating the plates (at ca. 150 °C) after immersion in a solution of 5% H2SO4 and 95% H2O. All reactions sensitive to air and/or moisture were carried out under argon atmosphere with anhydrous solvents. Anhydrous solvents were purified and dried (where appropriate) according to standard procedures.
Compounds 1a,b, 2a,b, 4a,b, 5a,b, 6a,b, 7a,b were prepared as described earlier [26] and compounds 14, 16, 22 were synthesized as reported in [25]. Their spectral data were in keeping with published ones [25, 26]. Compounds 8a,b, 9a,b were synthesized by analogy with the procedure previously described [25]. Their spectral data are presented below.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose (8a)
A white foam, 72% yield. 1H NMR (CDCl3, 400 MHz): δ 10.03 (s, 1H, NH), 8.28 (s, 1H, H-5′′), 6.16 (d, 1H, J = 2.7 Hz, H-1′), 5.57 (s, 1H, H-5), 5.12–4.96 (m, 4H, H-2′, H-3′, H-7), 4.53-4.48 (m, 1H, H-4′), 4.03-3.96 (m, 1H, OH), 3.87-3.80 (m, 1H, H-5a′), 3.73 - 3.65 (m, 1H, H-5b′), 2.54 (d, 3H, J = 0.7 Hz, CH3), 1.59 (s, 3H, H-7′), 1.35 (s, 3H, H-8′). 13C NMR (CDCl3, 100 MHz): δ 162.78 (s, C = O, C-4), 154.17 (s, C = O, C-2), 152.36 (s, C-6), 144.33 (s, C-4′′), 124.19 (s, C-5′′), 113.85 (s, C-6′), 102.60 (s, C-5), 95.53 (s, C-1′), 88.43 (s, C-4′), 85.92 (s, C-3′), 81.60 (s, C-2′), 62.99 (s, C-5′), 39.25 (s, C-7), 27.09, 25.17 (s, C-7′, C-8′), 20.51 (s, CH3). MALDI MS m/z: calcd. for C16H21N5O6 [M+Na]+ 402.36, found [M+Na]+ 402.20. Analysis calcd. for C16H21N5O6, %: C, 50.66; H, 5.58; N, 18.46. Found, %: C, 50.31; H, 5.43; N, 18.39.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose (8b)
A white foam, 78% yield. 1H NMR (CDCl3, 600 MHz): δ 9.33 (s, 1H, NH), 7.62 (s, 1H, H-5′′), 6.08 (d, 1H, J = 2.2 Hz, H-1′), 5.52 (s, 1H, H-5), 5.27 (dd, 1H, J = 5.9, 2.2 Hz, H-2′), 5.01 (dd, 1H, J = 6.0, 1.7 Hz, H-3′), 4.51-4.49 (m, 1H, H-4′), 3.91-3.86(m, 1H, OH), 3.81-3.74 (m, 3H, H-5a′, H-7), 3.64–3.58 (m, 1H, H-5b′), 2.77 (t, 2H, J = 7.1 Hz, H-10), 2.24 (s, 3H, CH3), 1.77-1.64 (m, 4H, H-8, H-9), 1.58 (s, 3H, H-7′), 1.36 (s, 3H, H-8′). 13C NMR (CDCl3, 100 MHz): δ 162.85 (s, C = O, C-4), 153.66 (s, C = O, C-2), 151.75 (s, C-6), 147.15 (s, C-4′′), 121.28 (s, C-5′′), 113.57 (s, C-6′), 102.20 (s, C-5), 94.79 (s, C-1′), 88.74 (s, C-4′), 85.33 (s, C-3′), 81.77 (s, C-2′), 62.98 (s, C-5′), 43.93 (s, C-7), 27.79, 26.93, 26.01, 25.06, 24.58 (s, C-8, C-9, C-10, C-7′, C-8′), 19.84 (s, CH3). ESI MS m/z: calcd. for C19H27N5O6 [M + H]+ 422.45, [M+Na]+ 444.44, [M + K]+ 460.55; found [M + H]+ 422.06, [M+Na]+ 444.08, [M + K]+ 460.06. Analysis calcd. for C19H27N5O6, %: C, 54.15; H, 6.46; N, 16.62. Found, %: C, 54.19; H, 6.41; N, 16.59.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose (9a)
A white foam, 74% yield. 1H NMR (CDCl3, 600 MHz): δ 10.12 (s, 1H, NH), 8.19 (s, 1H, H-5′′), 7.33 (d, 1H, J = 1.1 Hz, H-6), 6.16 (d, 1H, J = 2.5 Hz, H-1′), 5.13(dd, 1H, J = 6.0, 2.2 Hz, H-2′), 4.99-4.87 (m, 3H, H-3′, H-7), 4.51-4.48 (m, 1H, H-4′), 3.99-3.94 (m, 1H, OH), 3.82-3.77 (m, 1H, H-5a′), 3.69 - 3.64 (m, 1H, H-5b′), 1.87 (s, 3H, CH3),1.57 (s, 3H, H-7′), 1.34(s, 3H, H-8′). 13C NMR (CDCl3, 100 MHz): δ 162.29 (s, C = O, C-4), 151.44 (s, C = O, C-2), 142.01 (s, C-4′′), 140.25 (s, C-6), 123.84 (s, C-5′′), 113.85 (s, C-6′), 111.57 (s, C-5), 95.50 (s, C-1′), 88.62 (s, C-4′), 85.81 (s, C-3′), 81.66 (s, C-2′), 63.02 (s, C-5′), 43.27 (s, C-7), 27.05, 25.14 (s, C-7′, C-8′), 12.22 (s, CH3). ESI MS m/z: calcd. for C16H21N5O6 [M+Na]+ 402.36, [M + K]+ 418.47; found [M+Na]+ 402.01, [M + K]+ 417.98. Analysis calcd. for C16H21N5O6, %: C, 50.66; H, 5.58; N, 18.46. Found, %: C, 50.59; H, 5.49; N, 18.40.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose (9b)
A white foam, 83% yield. 1H NMR (CD3OD, 600 MHz): δ 7.96 (s, 1H, H-5′′), 7.40 (d, 1H, J = 1.4 Hz, H-6), 6.17 (d, 1H, J = 1.9 Hz, H-1′), 5.35 (dd, 1H, J = 6.2, 1.9 Hz, H-2′), 4.93 (dd, 1H, J = 6.0, 2.1 Hz, H-3′),4.35-4.32 (m, 1H, H-4′), 3.73 (t, 2H, J = 6.8 Hz, H-7), 3.52-3.44 (m, 2H, H-5a′, H-5b′), 2.75 (t, 2H, J = 6.9 Hz, H-10), 1.85 (d, 3H, J = 1.1 Hz, CH3), 1.73-1.67 (m, 4H, H-8, H-9), 1.56 (s, 3H, H-7′), 1.38 (s, 3H, H-8′). 13C NMR (DMSO-d6, 100 MHz): δ 164.28 (s, C = O, C-4), 150.90 (s, C = O, C-2), 146.98 (s, C-4′′), 141.37 (s, C-6), 121.46 (s, C-5′′),112.74 (s, C-6′), 108.48 (s, C-5), 92.76 (s, C-1′), 87.66 (s, C-4′), 83.86 (s, C-3′), 81.63 (s, C-2′), 61.16 (s, C-5′), 46.83 (s, C-7), 28.01, 26.76, 25.74, 25.00, 24.47 (s, C-8, C-9, C-10, C-7′, C-8′), 11.89 (s, CH3). MALDI MS m/z: calcd. for C19H27N5O6 [M+Na]+ 444.44; found [M+Na]+ 444.20. Analysis calcd. for C19H27N5O6, %: C, 54.15; H, 6.46; N, 16.62. Found, %: C, 54.13; H, 6.44; N, 16.65.
General method for the synthesis of 5′-diethyl and 5′-diphenyl phosphates of 1′′,2′′,3′′-triazolyl nucleoside analogues
Starting nucleoside analogue 6a,b or 7a,b (1 eq) was heated in vacuo (60 °C, 0.05 Torr) for 30 min, then the flask was filled with argon, and this operation was repeated 2 more times. The dried nucleoside analogue was dissolved in 3–5 mL of freshly distilled (over CaH2) pyridine, a flask was equipped with a rubber septum, argon inlet tube and cooled with a water bath (18–20 °C). Then 2.2–2.5 eq of the corresponding chlorophosphate (diethyl or diphenylchlorophosphate) was added with a syringe under an argon atmosphere. The flask was tightly closed using a PTFE seal and left to stir for 12-24 h at room temperature. Upon completion of the reaction (TLC and 31P NMR control), 1 mL of dry MeOH was added to the resulting mixture and left to stir for another 30 min, then all volatile components were removed using a water jet pump and dried in vacuo (40 °C, 0.05 Torr). The residue was purified by flash chromatography to isolate the target product (eluent - a mixture of chloroform: ethanol 10: 1).
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl diethyl phosphate (10a)
A sticky solid, 11% yield. 1H NMR (CDCl3, 400 MHz): δ 8.02 (s, 1H, H-5′′), 6.07 (d, 1H, J = 2.7 Hz, H-1′), 5.51 (s, 1H, H-5), 5.02 (s, 2H, H-7), 4.60-4.54 (m, 1H, H-2′), 4.50-4.45 (m, 1H, H-3′), 4.30 – 3.98 (m, 7H, H-5′, H-4′, H-8), 2.44 (s, 3H, CH3), 1.27 – 1.21 (m, 6H, H-9). 13C NMR (CDCl3, 100 MHz): δ 163.52 (s, C = O, C-4), 154.63 (s, C = O, C-2), 151.98 (s, C-6), 142.31 (s, C-4′′), 123.63 (s, C-5′′), 102.17 (s, C-5), 92.54 (s, C-1′), 83.00 (d, J = 6.9 Hz, C-4′), 75.06 (s, C-3′), 70.60 (s, C-2′), 67.04 (s, C-8), 66.99 (s, C-8), 64.32 (dd, J = 6.1, 2.4 Hz, C-5′), 39.10 (s, C-7), 20.29 (s, CH3). 15.98 (s, C-9), 15.91 (s, C-9). 31P NMR (CDCl3, 100 MHz) δ, м.д.: -1.57. MALDI MS m/z: calcd. for C17H26N5O9P [M+Na]+ 498.14; found [M+Na]+ 498.10. Analysis calcd. for C17H26N5O9P, %: C, 42.95; H, 5.51; N, 14.73; P, 6.52. Found, %: C, 42.91; H, 5.54; N, 14.69; P, 6.48.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl diethyl phosphate (10b)
A sticky solid, 27% yield. 1H NMR (CDCl3, 400 MHz): δ 9.95 (s, 1H, NH), 7.74 (s, 1H, H-5′′), 6.07 (d, 1H, J = 2.6 Hz, H-1′), 5.52 (s, 1H, H-5), 4.61–4.55 (m, 1H, H-2′), 4.54-4.48 (m, 1H, H-3′), 4.34-4.26 (m. 2H, H-7), 4.24-4.17 (m, 1H, H-4′), 4.14–4.03 (m, 4H, H-11), 3.87-3.71 (m. 2H, H-5′), 2.82-2.69 (m, 2H, H-10), 2.24 (s, 3H, CH3), 1.78-1.60 (m, 4H, H-8, H-9), 1.32-1.24 (m, 6H, H-12). 13C NMR (CDCl3, 100 MHz): δ 163.40 (s, C = O, C-4), 154.12 (s, C = O, C-2), 151.75 (s, C-6), 146.88 (s, C-4′′), 120.68 (s, C-5′′), 101.95 (s, C-5), 92.59 (s, C-1′), 82.69 (d, J = 7.7 Hz, C-4’), 75.24 (s, C-3′), 70.35 (s, C-2′), 66.85 (s, 1H, C-11), 66.80 (s, 1H, C-11), 64.19 (dd, J = 5.9, 2.6 Hz C-5′), 43.84 (s, C-7), 27.68, 25.85, 24.48 (s, C-8, C-9, C-10), 19.80 (s, CH3), 15.98 (s, C-12), 15.92 (s, C-12). 31P NMR (CDCl3, 100 MHz): δ -1.37. MALDI MS m/z: calcd. for C20H32N5O9P [M + H]+ 518.48, [M+Na]+ 540.47, [M + K]+ 556.57; found [M + H]+ 518.1, [M+Na]+ 540.1, [M + K]+ 556.1. Analysis calcd. for C20H32N5O9P, %: C, 46.42; H, 6.23; N, 13.53; P, 5.99. Found, %: C, 46.40; H, 6.26; N, 13.49; P, 5.97.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl diethyl phosphate (11a)
A sticky solid, 64% yield. 1H NMR (CD3OD, 400 MHz):δ 8.13 (s, 1H, H-5′′), 7.54 (d, 1H, J = 1.2 Hz, H-6), 6.02 (d, 1H, J = 3.3 Hz, H-1′), 4.99 (s, 2H, C-7), 4.55 (dd, 1H, J = 4.9, 3.3 Hz, H-2′), 4.41 (t, 1H, J = 5.2 Hz, H-3′), 4.29-4.22 (m, 2H, H-5′), 4.2–4.12 (m, 1H, H-4′), 4.11–4.00 (m, 4H, H-8), 1.85 (d, 3H, J = 1.0 Hz, CH3), 1.3–1.24 (m, 6H, H-9). 13C NMR (CD3OD, 100 MHz): δ 166.73 (s, C = O, C-4), 152.61 (s, C = O, C-2), 144.14 (s, C-4′′), 142.60 (s, C-6), 124.29 (s, C-5′′), 111.54 (s, C-5), 94.13 (s, C-1′), 84.36 (d, J = 7.7 Hz, C-4′), 76.29 (s, C-3′), 71.64 (s, C-2′), 68.07 (s, C-8), 68.01 (s, C-8), 65.50 (dd, J = 5.9, 2.6 Hz, C-5′), 43.64 (s, C-7), 16.39 (s, C-9), 16.34 (s, C-9), 12.23 (s, CH3). 31P NMR (CDCl3, 100 MHz): δ -1.54. MALDI MS m/z: calcd. for C17H26N5O9P [M + H]+ 476.40, [M+Na]+ 498.38; found [M + H]+ 476.03, [M+Na]+ 498.05. Analysis calcd. for C17H26N5O9P, %: C, 42.95; H, 5.51; N, 14.73; P, 6.52. Found, %: C, 42.91; H, 5.54; N, 14.79; P, 6.49.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl diethyl phosphate (11b)
A sticky solid, 67% yield. 1H NMR (CDCl3, 400 MHz):δ 9.42 (s, 1H, NH), 7.66 (s, 1H, H-5′′), 7.01 (d, 1H, J = 1.2 Hz, H-6), 6.02 (d, 1H, J = 2.4 Hz, H-1′), 4.60 (dd, 1H, J = 5.1, 2.7 Hz, H-2′), 4.52 (t, 1H, J = 5.4 Hz, H-3′), 4.34–4.27 (m. 2H, H-7), 4.24-4.15 (m, 1H, H-4′), 4.14–4.04 (m, 4H, H-11), 3.74-3.67 (m. 2H, H-5′), 2.77-2.71 (m, 2H, H-10), 1.89 (s, 3H, CH3), 1.74-1.68 (m, 4H, H-8, H-9), 1.34–1.25 (m, 6H, H-12). 3C NMR (CDCl3, 100 MHz): δ 164.54 (s, C = O, C-4), 151.20 (s, C = O, C-2), 147.03 (s, C-4′′), 140.55 (s, C-6), 120.66 (s, C-5′′), 110.76 (s, C-5), 92.62 (s, C-1′), 82.81 (d, J = 7.3 Hz, C-4′), 75.31 (s, C-3′), 70.48 (s, C-2′), 66.88 (s, C-8), 66.84 (s, C-8), 64.27 (dd, J = 5.4, 3.3 Hz, C-5′), 47.99 (s, C-7), 28.12, 25.69, 24.61, (s, C-8, C-9, C-10), 16.05 (s, C-9), 15.99 (s, C-9), 12.22 (s, CH3). 31P NMR (CDCl3, 100 MHz): δ -1.54. MALDI MS m/z: calcd. for C20H32N5O9P [M + H]+ 518.48, [M+Na]+ 540.47, [M + K]+ 556.57; found [M + H]+ 518.5, [M+Na]+ 540.5, [M + K]+ 556.6. Analysis calcd. for C20H32N5O9P, %: C, 46.42; H, 6.23; N, 13.53; P, 5.99. Found, %: C, 46.43; H, 6.25; N, 13.51; P, 6.00.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl diphenyl phosphate (12a)
A sticky solid, 19% yield. 1H NMR (CD3OD, 400 MHz): δ 8.06 (s, 1H, H-5′′), 7.36-7.02 (m, 10H, Ar), 6.03 (d, 1H, J = 3.1 Hz, H-1′), 5.51 (d, 1H, J = 0.4 Hz, H-5), 5.05 (s, 2H, H-7), 4.55-4.48 (m, 2H, H-2′, H-3′), 4.45–4.38 (m, 2H, H-5′), 4.34-4.28 (m, 1H, H-4′), 2.36 (s, 3H, CH3). 13C NMR (CD3OD, 100 MHz): δ 165.61 (s, C = O, C-4), 156.73 (s, C = O, C-2), 154.10 (d, J = 6.7 Hz, C-Ar), 153.24 (s, C-6), 151.65 (dd, J = 7.2, 1.6 Hz, C-Ar), 144.34 (s, C-4′′), 131.06, 130.27, 126.90, 124.55 (s, C-Ar), 124.38 (s, C-5′′), 121.34, 121.30, 121.19, 121.14 (s, C-Ar), 102.50 (s, C-5), 94.15 (s, C-1′), 84.12 (d, J = 7.7 Hz, C-4′), 76.20 (s, C-3′), 71.59 (s, C-2′), 69.75 (d, J = 6.2 Hz, C-5′), 40.04 (s, C-7), 20.27 (s, CH3). 31P NMR (CD3OD, 100 MHz): δ -12.07. ESI MS m/z: calcd. for C25H26N5O9P [M + H]+ 572.49, [M+Na]+ 594.47; found [M + H]+ 572.07, [M+Na]+ 594.07. Analysis calcd. for C25H26N5O9P, %: C, 52.54; H, 4.59; N, 12.25; P, 5.42. Found, %: C, 52.51; H, 4.56; N, 12.26; P, 5.44.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl diphenyl phosphate (12b)
A sticky solid, 5% yield. 1H NMR (CDCl3, 400 MHz): δ 9.77 (s, 1H, NH), 7.64 (s, 1H, H-5′′), 7.34-7.10 (m, 10H, H-Ar), 6.08 (d, 1H, J = 1.1 Hz, H-1′), 5.49 (s, 1H, H-5), 4.60-4.51 (m, 3H, H-2′, H-7), 4.46-4.38 (m, 1H, H-3′), 4.38-4.32 (m, 1H, H-4′), 3.80-3.61 (m. 2H, H-5′), 2.72-2.58 (m, 2H, H-10), 2.17 (s, 3H, CH3), 1.71-1.52 (m, 4H, H-8, H-9). 13C NMR (CDCl3, 100 MHz): δ 166.85 (s, C = O, C-4), 152.91 (s, C = O, C-2), 151.43 (s, C-Ar), 148.77 (s, C-6), 143.11 (s, C-4′′), 131.05, 130.24, 126.89, 124,49, 121.18 (s, C-Ar), 122.42 (s, C-5′′), 111.14 (s, C-5), 94.08 (s, C-1′), 84.15 (d, J = 7.7 Hz, C-4′), 76.30 (s, C-3′), 71.64 (s, C-2′), 65.61 (dd, J = 6.0, 2.8 Hz, C-5′), 29.37, 27.21, 25,71, 16.35 (s, C-7, C-8, C-9, C-10), 12.17 (s, C-CH3). 31P NMR (CDCl3, 100 MHz): δ -12.16. MALDI MS m/z: calcd. for C28H32N5O9P [M + H]+ 614.57, [M+Na]+ 636.55; found [M + H]+ 614.40, [M+Na]+ 636.40. Analysis calcd. for C28H32N5O9P, %: C, 54.81; H, 5.26; N, 11.41; P, 5.05. Found, %: C, 54.78; H, 5.29; N, 11.45; P, 5.01.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl diphenyl phosphate (13a)
A sticky solid, 83% yield. 1H NMR (CD3OD, 400 MHz):δ 8.08 (s, 1H, H-5′′), 7.45 (d, 1H, J = 1.2 Hz, H-6), 7.33 (t, 4H, J = 7.3 Hz, H-Ar), 7.24-7.12 (m, 6H, H-Ar), 6.03 (d, 1H, J = 3.1 Hz, H-1′), 4.91 (s, 2H, C-7), 4.55-4.48 (m, 2H, H-2′, H-3′), 4.45–4.38 (m, 2H, H-5′), 4.33-4.28 (m, 1H, H-4′), 1.79 (d, 3H, J = 1.0 Hz, CH3). 13C NMR (CD3OD, 100 MHz): δ 166.74 (s, C = O, C-4), 152.65 (s, C = O, C-2), 151.63 (dd, J = 7.3, 1.9 Hz, C-Ar), 144.09 (s, C-4′′), 142.50 (s, C-6), 131.06, 126.90 (s, C-Ar), 124.36 (s, C-5′′), 121.17 (d, J = 0.5 Hz, C-Ar),111.57 (s, C-5), 94.24 (s, C-1′), 84.13 (d, J = 7.7 Hz, C-4′), 76.24 (s, C-3′), 71.56 (s, C-2′), 69.72 (d, J = 6.2 Hz, C-5′),43.55 (s, C-7), 12.20 (s, CH3). 31P NMR (CD3OD, 100 MHz): δ -12.08. ESI MS m/z: calcd. for C25H26N5O9P [M+Na]+ 594.47; found [M+Na]+ 594.10. Analysis calcd. for C25H26N5O9P, %: C, 52.54; H, 4.59; N, 12.25; P, 5.42. Found, %: C, 52.52; H, 4.61; N, 12.23; P, 5.43.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl diphenyl phosphate (13b)
A sticky solid, 11% yield. 1H NMR (CD3OD, 400 MHz):δ 7.82 (s, 1H, H-5′′), 7.82-7.12 (m, 10H, H-Ar), 6.02 (d, 1H, J = 3.3 Hz, H-1′), 4.59–4.49 (m, 2H, H-2′, H-3′), 4.46-4.38 (m, 2H, H-5′), 4.34-4.29 (m, 1H, H-4′), 3.66 (t, 2H, J = 6.9 Hz, H-7), 2.65 (t, 2H, J = 6.9 Hz, H-10), 1.82 (d, 3H, J = 1.1 Hz, CH3), 1.67-1.55 (m, 4H, H-8, H-9). 13C NMR (CD3OD, 100 MHz): δ 166.87 (s, C = O, C-4), 152.89 (s, C = O, C-2), 151.66 (d, J = 7.3 Hz, C-Ar), 148.82 (s, C-4′′),143.08 (s, C-6), 131.11, 126.94 (s, C-Ar), 122.20 (s, C-5′′), 121.16 (d, J = 4.7 Hz, C-Ar), 111.13 (s, C-5), 94.22 (s, C-1′), 84.00 (d, J = 7.7 Hz, C-4′), 76.33 (s, C-3′), 71.53 (s, C-2′), 69.76 (d, J = 6.2 Hz, C-5′),30.73, 29.31, 27.03, 25.68 (s, C-7, C-8, C-9, C-10), 12.16 (s, CH3). 31P NMR (CD3OD, 100 MHz): δ -12.19. MALDI MS m/z: calcd. for C28H32N5O9P [M + H]+ 614.57, [M+Na]+ 636.55; found [M + H]+ 614.20, [M+Na]+ 636.20. Analysis calcd. for C28H32N5O9P, %: C, 54.81; H, 5.26; N, 11.41; P, 5.05. Found, %: C, 54.80; H, 5.28; N, 11.44; P, 5.04.
General procedure for the synthesis of 5′-(phenyl methoxy-L-alaninyl)phosphates of 1′′,2′′,3′′-triazolyl nucleoside analogues
1 eq of starting nucleoside analogue (8a,b, 9a,b) was heated in vacuo (60 °C, 0.05 Torr) for 30 min, then the flask was filled with argon, and this operation was repeated 2 more times. The dried nucleoside analogue was dissolved in 15 mL of dry DCM, and a flask was equipped with a rubber septum and argon inlet tube and cooled with a water bath (18–20 °C). Then 2.2–2.5 eq of a methyl (chloro(phenoxy)phosphoryl)-L-alaninate 16 solution in DCM was added by the syringe under an argon atmosphere. The flask was tightly closed using a PTFE seal and left to stir for 12-24 h at room temperature. Upon completion of the reaction (under TLC and 31P NMR control), 1 mL of dry MeOH was added to the resulting mixture and left to stir for another 30 min. Then all volatile components were removed using a water jet pump and dried in vacuo (40 °C, 0.05 Torr). The residue was purified by flash chromatography to isolate compounds 17a,b or 18a,b (eluent - a mixture of EtOAc: ethanol 10: 1). To remove the isopropylidene protection group, 4 mL of trifluoroacetic acid (50% v/v) was added and left to stir for 45 minutes at room temperature. Then, all the volatiles were removed in vacuo. The residue was purified by flash chromatography to isolate the target product (19a,b or 20a,b) (eluent - a mixture of CHCl3/MeOH+0.5% Et3N from 30:1 to MeOH+0.5% Et3N).
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-(phenyl methoxy-L-alaninyl)phosphate (17a)
A sticky solid, 57% yield. Mixture of two diastereomers with ratio 1:1. 1H NMR (CDCl3, 400 MHz): δ 9.30 (br s, 2H, NH), 8.11 (s, 1H, H-5′′), 8.09 (s, 1H, H-5′′), 7.33-7.10 (m, 10H, Ar), 6.20 (d, 1H, J = 1.8 Hz, H-1′), 6.15 (d, 1H, J = 2.2 Hz, H-1′), 5.55 (s, 1H, H-5), 5.53 (s, 1H, H-5), 5.20 (dd, 1H, J = 6.0, 1.8 Hz, H-2′), 5.07 (dd, 2H, J = 66.8, 15.4 Hz, H-7), 5.02 (s, 2H, H-7), 4.93 (dd, 1H, J = 5.8, 2.2 Hz, H-2′), 4.75 (dd, 1H, J = 5.8, 2.6 Hz, H-3′), 4.71 (dd, 1H, J = 5.8, 1.5 Hz, H-3′), 4.57-4.51 (m, 4H, H-4′, NH-Ala), 4.27-4.21 (m, 1H, H-8), 4.18-4.04 (m, 5H, H-8, H-5′), 3.72 (s, 3H, H-11), 3.69 (s, 3H, H-11), 2.51 (s, 3H, CH3), 2.50 (s, 3H, CH3), 1.58 (s, 3H, H-7′), 1.55 (s, 3H, H-7′), 1.37-1.30 (m, 9H, H-8′, H-9), 1.28 (s, 3H, H-8′). 13C NMR (CDCl3, 100 MHz): δ 174.60 (d, J = 9.9 Hz, C-10), 174.45 (d, J = 8.8 Hz, C-10), 162.18 (s, C = O, C-4), 162.14 (s, C = O, C-4), 153.59 (s, C = O, C-2), 153.55 (s, C = O, C-2), 152.00 (s, C-6), 150.62 (s, C-Ar), 150.52 (s, C-Ar), 142.95 (s, C-4′′), 142.88 (s, C-4′′), 129.71 (s, C-Ar), 129.63 (s, C-Ar), 125.16 (s, C-Ar), 124.96 (s, C-Ar), 123.50 (s, C-5′′), 122.99 (s, C-5′′), 120.34 (d, J = 4.8 Hz, C-Ar), 120.22 (d, J = 4.8 Hz, C-Ar), 114.19 (s, C-6′), 114.07 (s, C-6′), 102.67 (s, C-5), 102.62 (s, C-5), 95.28 (s, C-1′), 94.33 (s, C-1′),86.08 (d, J = 8.9 Hz, C-4′), 85.80 (d, J = 8.8 Hz, C-4′), 85.16 (s, C-3′), 85.07 (s, C-3′), 81.37 (s, C-2′), 81.09 (s, C-2′), 66.31 (d, J = 4.8 Hz, C-5′), 66.01 (d, J = 5.5 Hz, C-5′), 52.63 (s, C-11), 52.59 (s, C-11), 50.54 (d, J = 2.2 Hz, C-8), 50.19 (d, J = 2.2 Hz, C-8), 39.31 (s, C-7), 39.24 (s, C-7), 26.95 (c, C-7′), 25.15 (s, C-8′), 25.02 (s, C-8′), 21.19 (d, J = 4.0 Hz, C-9), 21.08 (d, J = 3.7 Hz, C-9), 20.43 (s, CH3). 31P NMR (CDCl3, 100 MHz): δ 3.70, 3.37. MALDI MS m/z: calcd. for C26H33N6O10P [M+Na]+ 643.56; found [M+Na]+ 643.50. Analysis calcd. for C26H33N6O10P, %: C, 50.32; H, 5.36; N, 13.54; P, 4.99. Found, %: C, 50.30; H, 5.39; N, 13.57; P, 4.97.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-(phenyl methoxy-L-alaninyl)phosphate (17b)
A sticky solid, 78% yield. Mixture of two diastereomers with ratio approximately 1:1. 1H NMR (CDCl3, 400 MHz): δ 9.58 (br s, 1H, NH), 9.51 (br s, 1H, NH), 7.68 (s, 1H, H-5′′), 7.66 (s, 1H, H-5′′), 7.41–7.17 (m, 10H, Ar), 6.19 (d, 1H, J = 1.1 Hz, H-1′), 6.18 (d, 1H, J = 1.7 Hz, H-1′), 5.59 (s, 2H, H-5), 5.49 (dd, 1H, J = 6.0, 1.1 Hz, H-2′), 5.35 (dd, 1H, J = 6.0, 1.6 Hz, H-2′), 5.00-4.95 (m, 2H, H-3′), 4.63-4.54 (m, 2H, H-4′), 4.21-4.02 (m, 8H, H-5′, H-7, NH-Ala), 3.97-3.80 (m, 4H, H-7, H-11, H-11), 3.77 (s, 3H, H-14), 3.76 (s, 3H, H-14), 2.88-2.79 (m, 4H, H-10), 2.30 (s, 3H, CH3), 2.29 (s, 3H, CH3), 1.86-1.69 (m, 8H, H-8, H-9), 1,65 (s, 6H, H-7′), 1.46-1.37 (m, 12H, H-8′, H-12). 13C NMR (CDCl3, 100 MHz): δ 174.09 (s, C-13), 174.02 (s, C-13), 162.72 (s, C = O, C-4), 162.57 (s, C = O, C-4), 153.48 (s, C = O, C-2), 153.41 (s, C = O, C-2), 151.68 (s, C-6), 151.60 (s, C-6), 150.56 (s, C-Ar), 150.49 (s, C-Ar), 147.72 (s, C-4′′), 147.64 (s, C-4′′), 129.59 (s, C-Ar), 124.94 (s, C-5′′), 124.88 (s, C-5′′), 120.78 (s, C-Ar), 120.64 (s, C-Ar), 120.21 (s, C-Ar), 120.16 (s, C-Ar), 120.11 (s, C-Ar), 120.06 (s, C-Ar), 113.93 (s, C-6′), 113.92 (s, C-6′), 102.22 (s, C-5), 102.15 (s, C-5), 93.80 (s, C-1′), 93.52 (s, C-1′), 86.10 (d, J = 8.1 Hz, C-4′), 85.86 (d, J = 8.0 Hz, C-4′), 84.34 (s, C-3′), 84.29 (s, C-3′), 81.60 (s, C-2′), 81.46 (s, C-2′), 65.78 (d, J = 5.5 Hz, C-5′), 65.73 (d, J = 6.2 Hz, C-5′), 52.63 (s, C-14), 50.20 (d, J = 1.5 Hz, C-11), 50.07 (s, C-11), 43.76 (s, C-7), 43.73 (s, C-7), 27.96 (s, C-10), 27.82 (s, C-10), 26.86 (s, C-7′), 26.81 (s, C-7′), 26.10 (s, C-8), 26.01 (s, C-8), 25.10 (s, C-8′), 24.66 (s, C-9), 24.55 (s, C-9), 20.74 (d, J = 3.3 Hz, C-12), 20.74 (d, J = 3.3 Hz, C-12), 19.83 (s, CH3). 31P NMR (CDCl3, 100 MHz): δ 2.63, 2.35. MALDI MS m/z: calcd. for C29H39N6O10P [M + H]+ 663.64, [M+Na]+ 685.63; found [M + H]+ 663.40, [M+Na]+ 685.40. Analysis calcd. for C29H39N6O10P, %: C, 52.57; H, 5.93; N, 12.68; P, 4.67. Found, %: C, 52.55; H, 5.95; N, 12.67; P, 4.70.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-(phenyl methoxy-L-alaninyl)phosphate (18a)
A sticky solid, 78% yield. Mixture of two diastereomers with ratio approximately 1:1. 1H NMR (CDCl3, 400 MHz): δ 9.58 (br s, 1H, NH), 9.51 (br s, 1H, NH), 7.68 (s, 1H, H-5′′), 7.66 (s, 1H, H-5′′), 7.41–7.17 (m, 10H, Ar), 6.19 (d, 1H, J = 1.1 Hz, H-1′), 6.18 (d, 1H, J = 1.7 Hz, H-1′), 5.59 (s, 2H, H-5), 5.49 (dd, 1H, J = 6.0, 1.1 Hz, H-2′), 5.35 (dd, 1H, J = 6.0, 1.6 Hz, H-2′), 5.00-4.95 (m, 2H, H-3′), 4.63-4.54 (m, 2H, H-4′), 4.21-4.02 (m, 8H, H-5′, H-7, NH-Ala), 3.97-3.80 (m, 4H, H-7, H-11, H-11), 3.77 (s, 3H, H-14), 3.76 (s, 3H, H-14), 2.88-2.79 (m, 4H, H-10), 2.30 (s, 3H, CH3), 2.29 (s, 3H, CH3), 1.86-1.69 (m, 8H, H-8, H-9), 1,65 (s, 6H, H-7′), 1.46-1.37 (m, 12H, H-8′, H-12). 13C NMR (CDCl3, 100 MHz): δ 174.09 (s, C-13), 174.02 (s, C-13), 162.72 (s, C = O, C-4), 162.57 (s, C = O, C-4), 153.48 (s, C = O, C-2), 153.41 (s, C = O, C-2), 151.68 (s, C-6), 151.60 (s, C-6), 150.56 (s, C-Ar), 150.49 (s, C-Ar), 147.72 (s, C-4′′), 147.64 (s, C-4′′), 129.59 (s, C-Ar), 124.94 (s, C-5′′), 124.88 (s, C-5′′), 120.78 (s, C-Ar), 120.64 (s, C-Ar), 120.21 (s, C-Ar), 120.16 (s, C-Ar), 120.11 (s, C-Ar), 120.06 (s, C-Ar), 113.93 (s, C-6′), 113.92 (s, C-6′), 102.22 (s, C-5), 102.15 (s, C-5), 93.80 (s, C-1′), 93.52 (s, C-1′), 86.10 (d, J = 8.1 Hz, C-4′), 85.86 (d, J = 8.0 Hz, C-4′), 84.34 (s, C-3′), 84.29 (s, C-3′), 81.60 (s, C-2′), 81.46 (s, C-2′), 65.78 (d, J = 5.5 Hz, C-5′), 65.73 (d, J = 6.2 Hz, C-5′), 52.63 (s, C-14), 50.20 (d, J = 1.5 Hz, C-11), 50.07 (s, C-11), 43.76 (s, C-7), 43.73 (s, C-7), 27.96 (s, C-10), 27.82 (s, C-10), 26.86 (s, C-7′), 26.81 (s, C-7′), 26.10 (s, C-8), 26.01 (s, C-8), 25.10 (s, C-8′), 24.66 (s, C-9), 24.55 (s, C-9), 20.74 (d, J = 3.3 Hz, C-12), 20.74 (d, J = 3.3 Hz, C-12), 19.83 (s, CH3). 31P NMR (CDCl3, 100 MHz): δ 2.63, 2.35. MALDI MS m/z: calcd. for C29H39N6O10P [M + H]+ 663.64, [M+Na]+ 685.63; found [M + H]+ 663.40, [M+Na]+ 685.40. Analysis calcd. for C29H39N6O10P, %: C, 52.57; H, 5.93; N, 12.68; P, 4.67. Found, %: C, 52.55; H, 5.95; N, 12.67; P, 4.70.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-(phenyl methoxy-L-alaninyl)phosphate (18b)
A sticky solid, 52% yield. Mixture of two diastereomers with ratio approximately 1:1. 1H NMR (CDCl3, 400 MHz): δ 8.99 (br s, 1H, NH), 8.84 (br s, 1H, NH), 7.56 (s, 1H, H-5′′), 7.55 (s, 1H, H-5′′), 7.34-7.27 (m, 4H, Ar), 7.20-7.11 (m, 6H, Ar), 6.96 (d, 2H, J = 0.5 Hz, H-6), 6.10 (s, 1H, H-1′), 6.09 (s, 1H, H-1′), 5.45-5.42 (m, 1H, H-2′), 5.26 (dd, 1H, J = 6.0, 1.9 Hz, H-2′), 4.91-4.86 (m, 2H, H-3′), 4.54-4.48 (m, 2H, H-4′), 4.08-3.95 (m, 8H, H-5′, H-7), 3.80-3.59 (m, 10H, H-11, H-14, NH-Ala), 2.78-2.72 (m, 4H, H-10), 1.89 (s, 6H, CH3), 1.73-1.67 (m, 8H, H-8, H-9), 1,57 (s, 6H, H-7′), 1.40-1.29 (m, 12H, H-8′, H-12). 13C NMR (CDCl3, 100 MHz): δ 174.14 (d, J = 7.5 Hz, C-13), 174.02 (d, J = 6.8 Hz, C-13), 164.07 (s, C = O, C-4), 163.95 (s, C = O, C-4), 150.99 (s, C = O, C-2), 150.88 (s, C = O, C-2), 150.60 (s, C-6), 150.55 (s, C-6), 147.81 (s, C-Ar), 147.73 (s, C-Ar), 140.29 (s, C-4′′), 140.18 (s, C-4′′), 129.66 (s, C-Ar), 125.01 (s, C-5′′), 124.94 (s, C-5′′), 120.75 (s, C-Ar), 120.55 (s, C-Ar), 120.25 (s, C-Ar), 120.21 (s, C-Ar), 120.14 (s, C-Ar), 120.10 (s, C-Ar), 114.00 (s, C-6′), 113.98 (s, C-6′), 110.82 (s, C-5), 110.70 (s, C-5), 93.92 (s, C-1′), 93.55 (s, C-1′), 86.24 (d, J = 8.2 Hz, C-4′), 85.91 (d, J = 8.2 Hz, C-4′), 84.40 (s, C-3′), 84.30 (s, C-3′), 81.59 (s, C-2′), 81.46 (s, C-2′), 65.85 (s, C-5′), 65.81 (s, C-5′), 52.49 (s, C-14), 50.27 (s, C-11), 50.14 (s, C-11), 47.96 (s, C-7), 47.89 (s, C-7), 28.24 (s, C-10), 28.17 (s, C-10), 26.90 (s, C-7′), 26.85 (s, C-7′), 25.91 (s, C-8), 25.85 (s, C-8), 25.14 (s, C-8′), 24.71 (s, C-9), 24.66 (s, C-9), 20.86 (s, C-12), 20.82 (s, C-12), 12.26 (s, CH3). 31P NMR (CDCl3, 100 MHz): δ 2.61, 2.31. MALDI MS m/z: calcd. for C29H39N6O10P [M + H]+ 663.64, [M+Na]+ 685.63; found [M + H]+ 663.40, [M+Na]+ 685.40. Analysis calcd. for C29H39N6O10P, %: C, 52.57; H, 5.93; N, 12.68; P, 4.67. Found, %: C, 52.54; H, 5.95; N, 12.69; P, 4.69.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-(phenyl methoxy-L-alaninyl)phosphate (19a)
A sticky solid, 82% yield. Mixture of two diastereomers with ratio 1:1. 1H NMR (CD3OD, 400 MHz): δ 8.15 (s, 1H, H-5′′), 8.13 (s, 1H, H-5′′), 7.36-7.12 (m, 10H, Ar), 6.03 (t, 2H, J = 3.5 Hz, H-1′), 5.55 (d, 2H, J = 1.5 Hz, H-5), 5.12 (s, 2H, H-7), 5.09 (s, 2H, H-7), 4.53-4.48 (m, 2H, H-2′), 4.41-4.20 (m, 8H, H-3′, H-4′, H-5′), 4.00-3.86 (m, 2H, H-8), 3.63 (s, 3H, H-11), 3.62 (s, 3H, H-11), 2.43 (s, 3H, CH3), 2.42 (s, 3H, CH3), 1.33-1.23 (m, 6H, H-9). 13C NMR (CD3OD, 100 MHz): δ 175.48 (d, J = 4.8 Hz, C-10), 175.37 (d, J = 5.6 Hz, C-10), 165.50 (br s, C = O, C-4), 156.79 (s, C-Ar), 156.75 (br s, C = O, C-2), 153.24 (s, C-Ar), 152.04 (s, C-6), 151.97 (s, C-6), 144.32 (s, C-4′′), 144.29 (s, C-4′′), 130.75 (s, C-Ar), 130.73 (s, C-Ar), 126.17 (s, C-Ar), 126.12 (s, C-Ar), 124.23 (br s, C-5′′), 121.43 (t, J = 4.8 Hz, C-Ar), 102.50 (br s, C-5), 94.11 (s, C-1′), 94.07 (s, C-1′), 84.71 (d, J = 8.4 Hz, C-4′), 84.50 (d, J = 8.1 Hz, C-4′), 76.42 (s, C-3′), 76.39 (s, C-3′), 71.70 (s, C-2′), 71.65 (s, C-2′), 67.56 (d, J = 5.5 Hz, C-5′), 67.17 (d, J = 5.5 Hz, C-5′), 52.82 (s, C-11), 52.80 (s, C-11), 51.50 (s, C-8), 51.40 (s, C-8), 40.07 (br s, C-7), 20.49 (d, J = 6.3 Hz, C-9), 20.39-20.27 (m, C-9, CH3). 31P NMR (CD3OD, 100 MHz): δ 3.79, 3.61. MALDI MS m/z: calcd. for C23H29N6O10P [M + H]+ 581.50, [M+Na]+ 603.48; found [M + H]+ 581.23, [M+Na]+ 603.25. Analysis calcd. for C23H29N6O10P, %: C, 47.59; H, 5.04; N, 14.48; P, 5.34. Found, %: C, 47.57; H, 5.06; N, 14.52; P, 5.30.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-(phenyl methoxy-L-alaninyl)phosphate (19b)
A sticky solid, 82% yield. Mixture of two diastereomers with ratio 1:1. 1H NMR (CD3OD, 400 MHz): δ 8.15 (s, 1H, H-5′′), 8.13 (s, 1H, H-5′′), 7.36-7.12 (m, 10H, Ar), 6.03 (t, 2H, J = 3.5 Hz, H-1′), 5.55 (d, 2H, J = 1.5 Hz, H-5), 5.12 (s, 2H, H-7), 5.09 (s, 2H, H-7), 4.53-4.48 (m, 2H, H-2′), 4.41-4.20 (m, 8H, H-3′, H-4′, H-5′), 4.00-3.86 (m, 2H, H-8), 3.63 (s, 3H, H-11), 3.62 (s, 3H, H-11), 2.43 (s, 3H, CH3), 2.42 (s, 3H, CH3), 1.33-1.23 (m, 6H, H-9). 13C NMR (CD3OD, 100 MHz): δ 175.48 (d, J = 4.8 Hz, C-10), 175.37 (d, J = 5.6 Hz, C-10), 165.50 (br s, C = O, C-4), 156.79 (s, C-Ar), 156.75 (br s, C = O, C-2), 153.24 (s, C-Ar), 152.04 (s, C-6), 151.97 (s, C-6), 144.32 (s, C-4′′), 144.29 (s, C-4′′), 130.75 (s, C-Ar), 130.73 (s, C-Ar), 126.17 (s, C-Ar), 126.12 (s, C-Ar), 124.23 (br s, C-5′′), 121.43 (t, J = 4.8 Hz, C-Ar), 102.50 (br s, C-5), 94.11 (s, C-1′), 94.07 (s, C-1′), 84.71 (d, J = 8.4 Hz, C-4′), 84.50 (d, J = 8.1 Hz, C-4′), 76.42 (s, C-3′), 76.39 (s, C-3′), 71.70 (s, C-2′), 71.65 (s, C-2′), 67.56 (d, J = 5.5 Hz, C-5′), 67.17 (d, J = 5.5 Hz, C-5′), 52.82 (s, C-11), 52.80 (s, C-11), 51.50 (s, C-8), 51.40 (s, C-8), 40.07 (br s, C-7), 20.49 (d, J = 6.3 Hz, C-9), 20.39-20.27 (m, C-9, CH3). 31P NMR (CD3OD, 100 MHz): δ 3.79, 3.61. MALDI MS m/z: calcd. for C23H29N6O10P [M + H]+ 581.50, [M+Na]+ 603.48; found [M + H]+ 581.23, [M+Na]+ 603.25. Analysis calcd. for C23H29N6O10P, %: C, 47.59; H, 5.04; N, 14.48; P, 5.34. Found, %: C, 47.57; H, 5.06; N, 14.52; P, 5.30.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-(phenyl methoxy-L-alaninyl)phosphate (20a)
A sticky solid, 65% yield. Mixture of two diastereomers with ratio 1:1. 1H NMR (CD3OD, 400 MHz): δ 8.17 (s, 1H, H-5′′), 8.14 (s, 1H, H-5′′), 7.52 (d, 1H, J = 1.5 Hz, H-6), 7.51 (d, 1H, J = 1.1 Hz, H-6), 7.35-7.28 (m, 4H, Ar), 7.20-7.14 (m, 6H, Ar), 6.03 (d, 1H, J = 1.9 Hz, H-1′), 6.02 (d, 1H, J = 2.2 Hz, H-1′), 4.98 (d, 2H, J = 3.7 Hz, H-7), 4.95 (s, 2H, H-7), 4.51-4.47 (m, 2H, H-2′), 4.40-4.19 (m, 8H, H-3′, H-4′, H-5′), 3.98-3.85 (m, 2H, H-8), 3.64 (s, 3H, H-11), 3.63 (s, 3H, H-11), 1.83 (t, 6H, J = 1.1 Hz, CH3), 1.32-1.25 (m, 6H, H-9). 13C NMR (CD3OD, 100 MHz): δ 175.52 (d, J = 4.7 Hz, C-10), 174.45 (d, J = 5.2 Hz, C-10), 166.74 (s, C = O, C-4), 166.72 (s, C = O, C-4), 152.70 (s, C-Ar), 152.10 (s, C = O, C-2), 152.03 (s, C = O, C-2), 144.10 (s, C-4′′), 144.08 (s, C-4′′), 142.60 (s, C-6), 142.57 (s, C-6), 130.77 (s, C-Ar), 126.20 (s, C-Ar), 126.15 (s, C-Ar), 124.19 (br s, C-4′′), 121.46 (t, J = 4.8 Hz, C-Ar), 111.58 (s, C-5), 94.24 (s, C-1′), 94.23 (s, C-1′), 84.76 (d, J = 8.4 Hz, C-4′), 84.57 (d, J = 8.2 Hz, C-4′), 76.52 (s, C-3′), 76.47 (s, C-3′), 71.70 (s, C-2′), 71.63 (s, C-2′), 67.54 (d, J = 5.5 Hz, C-5′), 67.13 (d, J = 5.1 Hz, C-5′), 52.80 (s, C-11), 52.78 (s, C-11), 51.55 (s, C-8), 51.45 (s, C-8), 43.66 (s, C-7), 20.48 (d, J = 6.2 Hz, C-9), 20.29 (d, J = 7.0 Hz, C-9), 12.21 (s, CH3). 31P NMR (CD3OD, 100 MHz): δ 3.83, 3.63. MALDI MS m/z: calcd. for C23H29N6O10P [M + H]+ 581.50, [M+Na]+ 603.48; found [M + H]+ 581.23, [M+Na]+ 603.24. Analysis calcd. for C23H29N6O10P, %: C, 47.59; H, 5.04; N, 14.48; P, 5.34. Found, %: C, 47.55; H, 5.07; N, 14.52; P, 5.32.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-(phenyl methoxy-L-alaninyl)phosphate (20b)
A sticky solid, 76% yield. Mixture of two diastereomers with ratio approximately 1:1. 1H NMR (CD3OD, 400 MHz): δ 7.92 (s, 1H, H-5′′), 7.90 (s, 1H, H-5′′), 7.38-7.28 (m, 6H, H-6, Ar), 7.22-7.12 (m, 6H, Ar), 6.04-6.00 (m, 2H, H-1′), 4.52-4.45 (m, 2H, H-2′), 4.42-4.20 (m, 8H, H-3′, H-4′, H-5′), 3.97-3.85 (m, 2H, H-11), 3.72-3.66 (m, 4H, H-7), 3.64 (s, 3H, H-14), 3.63 (s, 3H, H-14), 2.75-2.66 (m, 4H, H-10), 1.84 (br s, 6H, CH3), 1.70-1.62 (m, 8H, H-8, H-9), 1.32-1.24 (m, 6H, H-12). 13C NMR (CD3OD, 100 MHz): δ 174.45 (d, J = 4.4 Hz, C-13), 175.27 (d, J = 5.1 Hz, C-13), 166.78 (br s, C = O, C-4), 152.83 (br s, C = O, C-2), 152.01 (s, C-6), 151.94 (s, C-6), 148.83 (br s, C-4′′), 143.05 (s, C-Ar), 130.77 (s, C-Ar), 126.18 (s, C-Ar), 122.08 (s, C-5′′), 122.05 (s, C-5′′), 121.40 (t, J = 4.2 Hz, C-Ar), 111.10 (s, C-5), 94.06 (s, C-1′), 84.54 (d, J = 8.5 Hz, C-4′), 84.40 (d, J = 8.1 Hz, C-4′), 76.49 (s, C-3′), 76.45 (s, C-3′), 71.72 (s, C-2′), 71.67 (s, C-2′), 67.56 (d, J = 5.1 Hz, C-5′), 67.25 (d, J = 5.1 Hz, C-5′), 52.80 (s, C-14), 52.78 (s, C-14), 51.49 (s, C-11), 51.39 (s, C-11), 48.91 (s, C-7), 29.34 (s, C-10), 29.32 (s, C-10), 27.13 (s, C-8), 27.11 (s, C-8), 25.71 (s, C-9), 25.69 (s, C-9), 20.44 (d, J = 6.6 Hz, C-12), 20.32 (d, J = 7.0 Hz, C-12), 12.20 (s, CH3). 31P NMR (CD3OD, 100 MHz): δ 3.75, 3.62. MALDI MS m/z: calcd. for C26H35N6O10P [M + H]+ 623.58; found [M+Na]+ 623.30. Analysis calcd. for C26H35N6O10P, %: C, 50.16; H, 5.67; N, 13.50; P, 4.98. Found, %: C, 50.18; H, 5.65; N, 13.52; P, 5.00.
General procedure for the synthesis of 5′-H-phosponates of 1′′,2′′,3′′-triazolyl nucleoside analogues
1 eq of nucleoside analogue 8a or 8b or 9a,b was heated in vacuo (60 °C, 0.05 Torr) for 30 min, then the flask was filled with argon, and this operation was repeated 2 more times. The dried nucleoside analogue was dissolved in dry DCM (10 mL per 1 mmoL of the nucleoside analogue) and 10 eq of pyridine was added. A flask was equipped with a rubber septum, and argon inlet tube and cooled with a water bath (18–20 °C). Then 2.2–2.5 eq of the 20% solution of salicyl phosphorochloridate 22 in MeCN was added by the syringe under argon atmosphere. The flask was stirred for 30 min at room temperature and then 0.5 ml of distilled water and 0.5 ml of Et3N were added in one portion and stirred for an additional 30 min. Then all volatile components were removed on a rotary evaporator and dried in vacuo (40 °C, 0.05 Torr). The residue was purified by flash chromatography to isolate 5ʹ-H-phosphonate derivatives of 2ʹ,3ʹ-O-isopropylidene-protected nucleoside analogues 25a,b, 26a,b (gradient elution from СH2Cl2:MeOH+0.5% Et3N 25:1 to MeOH+0.5% Et3N).
To remove the isopropylidene protection group, compounds 25a,b, 26a,b were dissolved in aqueous trifluoroacetic acid (50% v/v; 4 mL per 1 mmoL of the compound) and left to stir for 45 min at room temperature. The solution was concentrated on rotary evaporator and dried in vacuo (40 °C, 0.05 Torr). The residue was purified by flash chromatography to give compounds 27a,b, 28a,b (eluent - a mixture of CHCl3/MeOH+0.5% Et3N from 30:1 to MeOH+0.5% Et3N).
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-yl H-phosphonate triethylammonium salt (25a)
A sticky solid, 11% yield. 1H NMR (CD3OD, 400 MHz): δ 8.19 (s, 1H, H-5′′), 6.65 (d, 1H, J = 622.0 Hz, PH), 6.21 (d, 1H, J = 1.9 Hz, H-1′), 5.58 (d, 1H, J = 0.6 Hz, H-5), 5.36 (dd, 1H, J = 5.9, 1.9 Hz, H-2′),5.14 (s, 2H, H-7), 5.01 (dd, 1H, J = 5.9, 1.7 Hz, H-3′), 4.50-4.45 (m, 1H, H-4′), 3.83 (dd, 2H, J = 7.2, 5.6 Hz, H-5′), 3.16 (q, 6H, J = 7.3 Hz, (CH3CH2)3N), 2.43 (s, 3H, CH3), 1.55 (s, 3H, H-7′), 1.37 (s, 3H, H-8′), 1.28 (t, 9H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 165.63 (s, C = O, C-4), 156.83 (s, C = O, C-2), 153.25 (s, C-6), 144.66 (s, C-4′′), 124.27 (s, C-5′′), 114.94 (s, C-6′), 102.42 (s, C-5), 95.82 (s, C-1′), 88.11 (d, 1H, J = 8.0 Hz, C-4′), 85.98 (s, C-3′), 83.45 (s, C-2′), 64.38 (d, 1H, J = 4.4 Hz, C-5′), 47.48 (s, (CH3CH2)3N), 40.07 (s, C-7), 27.27, 25.38 (s, C-7′, C-8′), 20.31 (s, CH3), 9.72 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 4.08. ESI MS m/z: calcd. for C16H21N5O8Pˉ [M]ˉ 442.35, found [M]ˉ 441.97. Analysis calcd. for C22H37N6O8P, %: C, 48.53; H, 6.85; N, 15.43; P, 5.69. Found, %: C, 48.58; H, 6.90; N, 15.41; P, 5.67.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-yl H-phosphonate triethylammonium salt (25b)
A sticky solid, 72% yield. 1H NMR (CD3OD, 400 MHz): δ 8.04 (s, 1H, H-5′′), 6.66 (d, 1H, J = 620.7 Hz, PH), 6.19 (d, 1H, J = 2.3 Hz, H-1′), 5.54 (d, 1H, J = 0.7 Hz, H-5), 5.33 (dd, 1H, J = 6.0, 2.3 Hz, H-2′), 5.02 (dd, 1H, J = 6.1, 1.9 Hz, H-3′), 4.50-4.46 (m, 1H, H-4′), 3.88-3.81 (m, 4H, H-7, H-5′), 3.14 (q, 6H, J = 7.3 Hz, (CH3CH2)3N), 2.8–2.74 (m, 2H, H-10), 2.30 (s, 3H, CH3), 1.79-1.65 (m, 4H, H-8, H-9), 1.56 (s, 3H, H-7′), 1.38 (s, 3H, H-8′), 1.28 (t, 9H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 165.69 (s, C = O, C-4), 156.95 (s, C = O, C-2), 153.31 (s, C-6), 149.23 (s, C-4′′), 122.47 (s, C-5′′), 114.96 (s, C-6′), 102.18 (s, C-5), 95.99 (s, C-1′), 87.72 (d, 1H, J = 7.9 Hz, C-4′), 86.03 (s, C-3′), 83.42 (s, C-2′), 64.48 (d, 1H, J = 4.3 Hz, C-5′), 47.70 (s, (CH3CH2)3N), 44.99 (s, C-7), 29.06, 27.42, 27.33, 25.73, 24.41 (s, C-8, C-9, C-10, C-7′, C-8′), 19.91 (s, CH3), 9.38 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 4.14. ESI MS m/z: calcd. for C19H27N5O8Pˉ [M]ˉ 484.43, found [M]ˉ 484.09. Analysis calcd. for C25H43N6O8P, %: C, 51.19; H, 7.39; N, 14.33; P, 5.28. Found, %: C, 51.24; H, 7.44; N, 14.31; P, 5.26.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-yl H-phosphonate triethylammonium salt (26a)
A sticky solid, 62% yield. 1H NMR (CD3OD, 400 MHz): δ 8.25 (s, 1H, H-5′′), 7.56 (d, 1H, J = 1.0 Hz, H-6), 6.65 (d, 1H, J = 621.5 Hz, PH), 6.21 (d, 1H, J = 2.1 Hz, H-1′), 5.34 (dd, 1H, J = 6.1, 2.0 Hz, H-2′), 5.04-4.99 (m, 3H, H-7, H-3′), 4.52-4.46 (m, 1H, H-4′), 3.84 (dd, 2H, J = 7.2, 5.3 Hz, H-5′), 3.12 (q, 6H, J = 7.3 Hz, (CH3CH2)3N), 1.86 (s, 3H, CH3), 1.55 (s, 3H, H-7′), 1.37 (s, 3H, H-8′), 1.26 (t, 9H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 166.81 (s, C = O, C-4), 152.69 (s, C = O, C-2), 144.41 (s, C-4′′), 142.66 (s, C-6), 124.29 (s, C-5′′), 114.96 (s, C-6′), 111.55 (s, C-5), 96.15 (s, C-1′), 88.08 (d, 1H, J = 8.0 Hz, C-4′), 86.14 (s, C-3′), 83.43 (s, C-2′), 64.45 (d, 1H, J = 4.4 Hz, C-5′), 47.80 (s, (CH3CH2)3N), 43.49 (s, C-7), 27.28, 25.37 (s, C-7′, C-8′), 12.22 (s, CH3), 9.20 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 4.02. ESI MS m/z: calcd. for C16H21N5O8Pˉ [M]ˉ 442.34, found [M]ˉ 442.03. Analysis calcd. for C22H37N6O8P, %: C, 48.53; H, 6.85; N, 15.43; P, 5.69. Found, %: C, 48.59; H, 6.89; N, 15.47; P, 5.70.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-yl H-phosphonate triethylammonium salt (26b)
A sticky solid, 70% yield. 1H NMR (CD3OD, 400 MHz): δ 8.03 (s, 1H, H-5′′), 7.44 (s, 1H, H-6), 6.66 (d, 1H, J = 619.6 Hz, PH), 6.18 (d, 1H, J = 2.2 Hz, H-1′), 5.32 (dd, 1H, J = 6.0, 2.2 Hz, H-2′), 5.02 (dd, 1H, J = 6.0, 1.8 Hz, H-3′), 4.50-4.46 (m, 1H, H-4′), 3.86-3.80 (m, 2H, H-5′), 3.78-3.72 (m, 2H, H-7), 3.09 (q, 6H, J = 7.3 Hz, (CH3CH2)3N), 2.79-2.72 (m, 2H, H-10), 1.86 (s, 3H, CH3), 1.75-1.67 (m, 4H, H-8, H-9), 1.56 (s, 3H, H-7′), 1.37 (s, 3H, H-8′), 1.26 (t, 9H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 166.84 (s, C = O, C-4), 152.87 (s, C = O, C-2), 149.20 (s, C-6), 143.19 (s, C-4′′), 122.51 (s, C-5′′), 114.94 (s, C-6′), 111.05 (s, C-5), 95.84 (s, C-1′), 87.74 (d, 1H, J = 8.1 Hz, C-4′), 85.96 (s, C-3′), 83.38 (s, C-2′), 64.48 (d, 1H, J = 4.0 Hz, C-5′), 48.95 (s, C-7), 47.71 (s, (CH3CH2)3N), 29.38, 27.32, 27.19, 25.72, 25.41 (s, C-8, C-9, C-10, C-7′, C-8′), 12.19 (s, CH3), 9.18 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 4.13. ESI MS m/z: calcd. for C19H27N5O8Pˉ [M]ˉ 484.43, found [M]ˉ 484.09. Analysis calcd. for C25H43N6O8P, %: C, 51.19; H, 7.39; N, 14.33; P, 5.28. Found, %: C, 51.23; H, 7.42; N, 14.37; P, 5.25.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl H-phosphonate triethylammonium salt (27a)
A sticky solid, 60% yield. 1H NMR (CD3OD, 500 MHz): δ 8.24 (s, 1H, H-5′′), 6.74 (d, 1H, J = 622.1 Hz, PH), 6.02 (d, 1H, J = 4.7 Hz, H-1′), 5.58 (d, 1H, J = 0.6 Hz, H-5), 5.16 (s, 2H, H-7), 4.53 (7, 1H, J = 4.9 Hz, H-2′), 4.33 (t, 1H, J = 4.9 Hz, H-3′), 4.21 (q, 1H, J = 3.8 Hz, H-4′), 4.08-3.96 (m, 2H, H-5′), 3.04 (q, 6H, J = 7.3 Hz, (CH3CH2)3N), 2.41 (s, 3H, CH3), 1.23 (t, 9H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 165.69 (s, C = O, C-4), 158.92 (s, C = O, C-2), 153.27 (s, C-6), 144.54 (s, C-4′′), 123.33 (s, C-5′′), 102.39 (s, C-5), 94.29 (s, C-1′), 88.11 (d, 1H, J = 7.5 Hz, C-4′), 76.77 (s, C-3′), 72.24 (s, C-2′), 64.41 (d, 1H, J = 4.3 Hz, C-5′), 47.57 (s, (CH3CH2)3N), 40.11 (s, C-7), 9.57 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 4.53. ESI MS m/z: calcd. for C13H17N5O8Pˉ [M]ˉ 402.08, found [M]ˉ 402.10. Analysis calcd. for C19H33N6O8P, %: C, 45.24; H, 6.59; N, 16.66; P, 6.14. Found, %: C, 45.27; H, 6.64; N, 16.69; P, 6.10.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl H-phosphonate triethylammonium salt (27b)
A sticky solid, 75% yield. 1H NMR (CD3OD, 400 MHz): δ 8.10 (s, 1H, H-5′′), 6.75 (d, 1H, J = 620.1 Hz, PH), 6.01 (d, 1H, J = 4.6 Hz, H-1′), 5.53 (d, 1H, J = 0.8 Hz, H-5), 4.5 (t, 1H, J = 4.7 Hz, H-2′), 4.32 (t, 1H, J = 4.7 Hz, H-3′), 4.23-4.19 (m, 1H, H-4′), 4.10-3.96 (m, 2H, H-5′), 3.89-3.82 (m, 2H, H-7), 3.14 (q, 6H, J = 7.3 Hz, (CH3CH2)3N), 2.77 (t, 2H, J = 7.2 Hz, H-10), 2.29 (d, 1H, J = 0.8 Hz, CH3), 1.80-1.64 (m, 4H, H-8, H-9), 1.27 (t, 9H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 165.68 (s, C = O, C-4), 156.97 (s, C = O, C-2), 153.29 (s, C-6), 149.03 (s, C-4′′), 121.71 (s, C-5′′), 102.16 (s, C-5), 94.26 (s, C-1′), 88.77 (d, 1H, J = 7.7 Hz, C-4′), 76.88 (s, C-3′), 72.26 (s, C-2′), 64.45 (d, 1H, J = 4.4 Hz, C-5′), 47.69 (s, (CH3CH2)3N), 44.99 (s, C-7), 29.03, 27.45, 25.74, (s, C-8, C-9, C-10), 19.91 (s, CH3), 9.30 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 4.56. ESI MS m/z: calcd. for C16H23N5O8Pˉ [M]ˉ 444.36, found [M]ˉ 444.18. Analysis calcd. for C22H39N6O8P, %: C, 48.35; H, 7.19; N, 15.38; P, 5.67. Found, %: C, 48.33; H, 7.23; N, 15.35; P, 5.64.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl H-phosphonate triethylammonium salt (28a)
A sticky solid, 94% yield. 1H NMR (CD3OD, 400 MHz): δ 8.31 (s, 1H, H-5′′), 7.55 (d, 1H, J = 1.2 Hz, H-6), 6.75 (d, 1H, J = 621.3 Hz, PH), 6.03 (d, 1H, J = 4.6 Hz, H-1′), 5.02 (s, 2H, H-7), 4.52 (t, 1H, J = 4.8 Hz, H-2′), 4.33 (t, 1H, J = 4.7 Hz, H-3′), 4.24-4.20 (m, 1H, H-4′), 4.10-3.96 (m, 2H, H-5′), 3.18 (q, 6H, J = 7.3 Hz, (CH3CH2)3N), 1.86 (d, 1H, J = 1.1 Hz, CH3), 1.29 (t, 9H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 165.69 (s, C = O, C-4), 156.92 (s, C = O, C-2), 153.27 (s, C-6), 144.57 (s, C-4′′), 123.33 (s, C-5′′), 102.39 (s, C-5), 94.29 (s, C-1′), 85.88 (d, 1H, J = 7.5 Hz, C-4′), 76.77 (s, C-3′), 72.24 (s, C-2′), 64.41 (d, 1H, J = 4.3 Hz, C-5′), 47.57 (s, (CH3CH2)3N), 40.11 (s, C-7), 20.28 (s, CH3), 9.57 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 4.54. ESI MS m/z: calcd. for C13H17N5O8Pˉ [M]ˉ 402.28, found [M]ˉ 402.10. Analysis calcd. for C19H33N6O8P, %: C, 45.24; H, 6.59; N, 16.66; P, 6.14. Found, %: C, 45.28; H, 6.63; N, 16.68; P, 6.16.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl H-phosphonate triethylammonium salt (28b)
A sticky solid, 95% yield. 1H NMR (CD3OD, 400 MHz): δ 8.09 (s, 1H, H-5′′), 7.44 (d, 1H, J = 1.0 Hz, H-6), 6.75 (d, 1H, J = 620.2 Hz, PH), 6.01 (d, 1H, J = 4.7 Hz, H-1′), 4.50 (t, 1H, J = 4.9 Hz, H-2′), 4.33 (t, 1H, J = 4.4 Hz, H-3′), 4.24-4.19 (m, 1H, H-4′), 4.10-3.96 (m, 2H, H-5′), 3.79-3.71 (m, 2H, H-7), 3.18 (q, 6H, J = 7.3 Hz, (CH3CH2)3N), 2.79-2.72 (m, 2H, H-10), 1.86 (s, 3H, CH3), 1.76-1.67 (m, 4H, H-8, H-9), 1.29 (t, 9H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 166.90 (s, C = O, C-4), 152.89 (s, C = O, C-2), 149.06 (s, C-6), 143.24 (s, C-4′′), 121.65 (s, C-5′′), 111.06 (s, C-5), 95.29 (s, C-1′), 87.78 (d, 1H, J = 7.7 Hz, C-4′), 76.91 (s, C-3′), 72.25 (s, C-2′), 64.40 (d, 1H, J = 4.4 Hz, C-5′), 47.81 (s, (CH3CH2)3N), 29.40, 27.27, 25.77 (s, C-8, C-9, C-10), 12.16 (s, CH3), 9.19 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 4.57. ESI MS m/z: calcd. for C16H23N5O8Pˉ [M]ˉ 444.36, found [M]ˉ 444.18. Analysis calcd. for C22H39N6O8P, %: C, 48.35; H, 7.19; N, 15.38; P, 5.67. Found, %: C, 48.39; H, 7.22; N, 15.39; P, 5.65.
General procedure for the synthesis of 5′-phosphates of 1′′,2′′,3′′-triazolyl nucleoside analogues
1 eq of 5ʹ-H-phosphonate (25a,b, 26a,b) was heated in vacuo (60 °C, 0.05 Torr) for 30 min, then the flask was filled with argon, and this operation was repeated 2 more times. The dried nucleotide analogue was dissolved in dry DCM (10 mL per 1 mmoL of the nucleotide analogue) and 10 eq of triethylamine was added. A flask was equipped with a rubber septum, and argon inlet tube and cooled with a water bath (18–20 °C). Then 5 eq of the TMSCl was added by the syringe under an argon atmosphere. The flask was stirred for 30 min at room temperature and then 1.05 eq of crystalline iodine was added in one portion and the mixture was stirred for an additional 30 min. Then all volatile components were removed on a rotary evaporator and dried in vacuo (40 °C, 0.05 Torr). The residue was purified by flash chromatography to isolate compounds 33a,b or 34a,b (gradient elution СH2Cl2:MeOH+0.5% Et3N 25:1 to MeOH+0.5% Et3N).
To remove the isopropylidene protection group, compounds 33a,b, 34a,b were dissolved in aqueous trifluoroacetic acid (50% v/v; 4 ml per 1 mmoL of the nucleotide analogue) and left to stir for 45 min at room temperature. The solution was concentrated on rotary evaporator and dried in vacuo (40 °C, 0.05 Torr). The residue was purified by flash chromatography to give compounds 35a,b, 36a,b (eluent - a mixture of CHCl3/MeOH+0.5% Et3N from 30:1 to MeOH+0.5% Et3N).
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-yl phosphate bis(triethylammonium) salt (33a)
A sticky solid, 19% yield. 1H NMR (CD3OD, 400 MHz): δ 8.27 (s, 1H, H-5′′), 6.19 (d, 1H, J = 2.3 Hz, H-1′), 5.58 (d, 1H, J = 0.8 Hz, H-5), 5.29 (dd, 1H, J = 5.9, 2.4 Hz, H-2′), 5.15 (s, 2H, H-7), 5.06 (dd, 1H, J = 5.9, 1.6 Hz, H-3′), 4.53-4.49 (m, 1H, H-4′), 3.89-3.85 (m, 2H, H-5′), 3.10 (q, 12H, J = 7.4 Hz, (CH3CH2)3N), 2.43 (s, 3H, CH3), 1.56 (s, 3H, H-7′), 1.37 (s, 3H, H-8′), 1.26 (t, 18H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 165.66 (s, C = O, C-4), 156.97 (s, C = O, C-2), 153.36 (s, C-6), 144.61 (s, C-4′′), 123.99 (s, C-5′′), 114.84 (s, C-6′), 102.49 (s, C-5), 96.46 (s, C-1′), 88.03 (d, 1H, J = 9.2 Hz, C-4′), 86.24 (s, C-3′), 83.58 (s, C-2′), 65.89 (d, 1H, J = 5.1 Hz, C-5′), 47.53 (s, (CH3CH2)3N), 40.13 (s, C-7), 27.33, 25.40 (s, C-7′, C-8′), 20.33 (s, CH3), 9.32 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 0.92. ESI MS m/z: calcd. for C16H20N5O9P2ˉ [M + 3H] + 460.36, found [M + 3H] + 460.18. Analysis calcd. for C28H52N7O9P, %: C, 50.82; H, 7.92; N, 14.82; P, 4.68. Found, %: C, 50.88; H, 7.94; N, 14.85; P, 4.66.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-yl phosphate bis(triethylammonium) salt (33b)
A sticky solid, 24% yield. 1H NMR (CD3OD, 400 MHz): δ 8.12 (s, 1H, H-5′′), 6.17 (d, 1H, J = 2.3 Hz, H-1′), 5.54 (d, 1H, J = 0.8 Hz, H-5), 5.24 (dd, 1H, J = 5.8, 2.7 Hz, H-2′), 5.05 (dd, 1H, J = 5.8, 1.5 Hz, H-3′), 4.53-4.48 (m, 1H, H-4′), 3.94-3.83 (m, 4H, H-7, H-5′), 3.18 (q, 12H, J = 7.3 Hz, (CH3CH2)3N), 2.80-2.74 (m, 2H, H-10), 2.29 (d, 3H, J = 0.8 Hz, CH3), 1.80-1.66 (m, 4H, H-8, H-9), 1.57 (s, 3H, H-7′), 1.37 (s, 3H, H-8′), 1.30 (t, 18H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 165.73 (s, C = O, C-4), 157.04 (s, C = O, C-2), 153.41 (s, C-6), 149.33 (s, C-4′′), 122.28 (s, C-5′′), 114.86 (s, C-6′), 102.23 (s, C-5), 96.52 (s, C-1′), 87.57 (d, 1H, J = 8.8 Hz, C-4′), 86.22 (s, C-3′), 83.51 (s, C-2′), 66.00 (d, 1H, J = 5.1 Hz, C-5′), 47.74 (s, (CH3CH2)3N), 45.01 (s, C-7), 29.04, 27.40, 27.37, 25.72, 25.43 (s, C-8, C-9, C-10, C-7′, C-8′), 19.92 (s, CH3), 9.17 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 1.22. ESI MS m/z: calcd. for C19H26N5O8P2ˉ [M + 3H]+ 502.42, found [M + 3H]+ 502.23. Analysis calcd. for C31H58N7O9P, %: C, 52.90; H, 8.31; N, 13.93; P, 4.40. Found, %: C, 52.94; H, 8.34; N, 13.95; P, 4.44.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-yl phosphate bis(triethylammonium) salt (34a)
A sticky solid, 48% yield. 1H NMR (CD3OD, 400 MHz): δ 8.32 (s, 1H, H-5′′), 7.56 (d, 1H, J = 1.1 Hz, H-6), 6.21 (d, 1H, J = 2.4 Hz, H-1′), 5.28 (dd, 1H, J = 5.8, 2.3 Hz, H-2′), 5.07-5.00 (m, 3H, H-7, H-3′), 4.54-4.50 (m, 1H, H-4′), 3.89 (t, 2H, J = 5.1 Hz, H-5′), 3.16 (q, 12H, J = 7.3 Hz, (CH3CH2)3N), 1.86 (d, 3H, J = 1.0 Hz, CH3), 1.55 (s, 3H, H-7′), 1.36 (s, 3H, H-8′), 1.29 (t, 18H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 166.80 (s, C = O, C-4), 152.77 (s, C = O, C-2), 144.31 (s, C-4′′), 142.73 (s, C-6), 124.28 (s, C-5′′), 114.86 (s, C-6′), 111.62 (s, C-5), 96.48 (s, C-1′), 88.01 (d, 1H, J = 8.8 Hz, C-4′), 86.30 (s, C-3′), 83.49 (s, C-2′), 64.45 (d, 1H, J = 4.4 Hz, C-5′), 47.60 (s, (CH3CH2)3N), 43.48 (s, C-7), 27.32, 25.39 (s, C-7′, C-8′), 12.22 (s, CH3), 9.14 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 0.53. ESI MS m/z: calcd. for C16H20N5O9P2ˉ [M + 3H] + 460.36, found [M + 3H] + 460.17. Analysis calcd. for C28H52N7O9P, %: C, 50.82; H, 7.92; N, 14.82; P, 4.68. Found, %: C, 50.85; H, 7.95; N, 14.86; P, 4.65.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-2′,3′-O-isopropylidene-β-D-ribofuranose-5′-yl phosphate bis(triethylammonium) salt (34b)
A sticky solid, 27% yield. 1H NMR (CD3OD, 400 MHz): δ 8.10 (s, 1H, H-5′′), 7.45 (d, 1H, J = 1.5 Hz, H-6), 6.17 (d, 1H, J = 2.4 Hz, H-1′), 5.26 (dd, 1H, J = 6.0, 2.5 Hz, H-2′), 5.05 (dd, 1H, J = 5.9, 1.2 Hz, H-3′), 4.52-4.48 (m, 1H, H-4′), 3.94-3.82 (m, 2H, H-5′), 3.78-3.71 (m, 2H, H-7), 3.17 (q, 12H, J = 7.3 Hz, (CH3CH2)3N), 2.78-2.72 (m, 2H, H-10), 1.85 (s, 3H, CH3), 1.76-1.66 (m, 4H, H-8, H-9), 1.56 (s, 3H, H-7′), 1.37 (s, 3H, H-8′), 1.30 (t, 18H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 166.86 (s, C = O, C-4), 152.97 (s, C = O, C-2), 149.28 (s, C-6), 143.23 (s, C-4′′), 122.35 (s, C-5′′), 114.84 (s, C-6′), 111.11 (s, C-5), 96.33 (s, C-1′), 87.70 (d, 1H, J = 8.8 Hz, C-4′), 86.16 (s, C-3′), 83.50 (s, C-2′), 66.00 (d, 1H, J = 4.8 Hz, C-5′), 53.66 (s, C-7), 47.63 (s, (CH3CH2)3N), 29.39, 27.39, 27.17, 25.75, 25.44 (s, C-8, C-9, C-10, C-7′, C-8′), 12.18 (s, CH3), 9.16 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 0.63. ESI MS m/z: calcd. for C19H26N5O8P2ˉ [M + 3H]+ 502.42, found [M + 3H]+ 502.24. Analysis calcd. for C31H58N7O9P, %: C, 52.90; H, 8.31; N, 13.93; P, 4.40. Found, %: C, 52.92; H, 8.30; N, 13.96; P, 4.38.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl phosphate bis(triethylammonium) salt (35a)
A sticky solid, 68% yield. 1H NMR (CD3OD, 500 MHz): δ 8.34 (s, 1H, H-5′′), 6.02 (d, 1H, J = 5.0 Hz, H-1′), 5.58 (d, 1H, J = 0.6 Hz, H-5), 5.16 (s, 2H, H-7), 4.55 (t, 1H, J = 4.9 Hz, H-2′), 4.38-4.35 (m, 1H, H-3′), 4.24-4.21 (m, 1H, H-4′), 4.07-3.98 (m, 2H, H-5′), 2.96 (q, 12H, J = 7.3 Hz, (CH3CH2)3N), 2.42 (s, 3H, CH3), 1.20 (t, 18H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 165.87 (s, C = O, C-4), 157.17 (s, C = O, C-2), 153.54 (s, C-6), 144.62 (s, C-4′′), 123.50 (s, C-5′′), 102.62 (s, C-5), 94.61 (s, C-1′), 86.62 (d, 1H, J = 8.6 Hz, C-4′), 77.24 (s, C-3′), 72.62 (s, C-2′), 65.80 (d, 1H, J = 5.0 Hz, C-5′), 47.43 (s, (CH3CH2)3N), 40.33 (s, C-7), 9.91 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 1.81. ESI MS m/z: calcd. for C13H16N5O9P2ˉ [M + H]ˉ 418.27, found [M + H]ˉ 418.11. Analysis calcd. for C25H48N7O9P, %: C, 48.30; H, 7.78; N, 15.77; P, 4.98. Found, %: C, 48.27; H, 7.81; N, 15.75; P, 4.96.
1′-[4′′-(6-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl phosphate bis(triethylammonium) salt (35b)
A sticky solid, 55% yield. 1H NMR (CD3OD, 400 MHz): δ 8.17 (s, 1H, H-5′′), 6.02 (d, 1H, J = 5.2 Hz, H-1′), 5.53 (s, 1H, H-5), 4.53 (t, 1H, J = 5.2 Hz, H-2′), 4.36 (t, 1H, J = 4.2 Hz, H-3′), 4.25–4.20 (m, 1H, H-4′), 4.09-3.99 (m, 2H, H-5′), 3.89-3.82 (m, 2H, H-7), 3.06 (q, 12H, J = 7.3 Hz, (CH3CH2)3N), 2.77 (t, 2H, J = 7.3 Hz, H-10), 2.29 (s, 1H, CH3), 1.80-1.64 (m, 4H, H-8, H-9), 1.25 (t, 9H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 165.71 (s, C = O, C-4), 156.99 (s, C = O, C-2), 153.39 (s, C-6), 149.00 (s, C-4′′), 121.79 (s, C-5′′), 102.22 (s, C-5), 94.38 (s, C-1′), 86.31 (d, 1H, J = 8.4 Hz, C-4′), 77.14 (s, C-3′), 72.50 (s, C-2′), 65.73 (d, 1H, J = 5.1 Hz, C-5′), 47.27 (s, (CH3CH2)3N), 44.97 (s, C-7), 29.03, 27.45, 25.73, (s, C-8, C-9, C-10), 19.92 (s, CH3), 9.37 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 2.19. ESI MS m/z: calcd. for C16H22N5O8P2ˉ [M + H]ˉ 460.35, found [M + H]ˉ 460.18. Analysis calcd. for C28H54N7O9P, %: C, 50.67; H, 8.20; N, 14.77; P, 4.67. Found, %: C, 50.64; H, 8.22; N, 14.75; P, 4.65.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-methyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl phosphate bis(triethylammonium) salt (36a)
A sticky solid, 76% yield. 1H NMR (CD3OD, 400 MHz): δ 8.41 (s, 1H, H-5′′), 7.55 (d, 1H, J = 1.2 Hz, H-6), 6.04 (d, 1H, J = 4.9 Hz, H-1′), 5.02 (s, 2H, H-7), 4.54 (t, 1H, J = 4.9 Hz, H-2′), 4.36 (m, 1H, H-3′), 4.26-4.21 (m, 1H, H-4′), 4.11–3.99 (m, 2H, H-5′), 3.12 (q, 12H, J = 7.3 Hz, (CH3CH2)3N), 1.86 (d, 3H, J = 1.1 Hz, CH3), 1.27 (t, 18H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 166.81 (s, C = O, C-4), 152.69 (s, C = O, C-2), 144.27 (s, C-4′′), 142.65 (s, C-6), 123.56 (s, C-5′′), 111.53 (s, C-5), 94.41 (s, C-1′), 85.88 (d, 1H, J = 7.7 Hz, C-4′), 76.89 (s, C-3′), 72.16 (s, C-2′), 64.29 (d, 1H, J = 4.4 Hz, C-5′), 47.80 (s, (CH3CH2)3N), 43.38 (s, C-7), 12.21 (s, CH3), 9.18 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 0.95. ESI MS m/z: calcd. for C13H16N5O9P2ˉ [M + H]ˉ 418.27, found [M + H]ˉ 418.11. Analysis calcd. for C25H48N7O9P, %: C, 48.30; H, 7.78; N, 15.77; P, 4.98. Found, %: C, 48.28; H, 7.75; N, 15.80; P, 4.95.
1′-[4′′-(5-Methyl-2,4-dioxo-pyrimidine-1-yl-butyl)-1′′,2′′,3′′-triazol-1′′-yl]-β-D-ribofuranose-5′-yl phosphate bis(triethylammonium) salt (36b)
A sticky solid, 70% yield. 1H NMR (CD3OD, 400 MHz): δ 8.14 (s, 1H, H-5′′), 7.44 (d, 1H, J = 1.1 Hz, H-6), 6.02 (d, 1H, J = 5.1 Hz, H-1′), 4.52 (t, 1H, J = 5.1 Hz, H-2′), 4.36 (t, 1H, J = 4.3 Hz, H-3′), 4.25-4.21 (m, 1H, H-4′), 4.12–3.99 (m, 2H, H-5′), 3.78-3.72 (m, 2H, H-7), 3.16 (q, 6H, J = 7.3 Hz, (CH3CH2)3N), 2.78-2.72 (m, 2H, H-10), 1.85 (d, 3H, J = 1.0 Hz, CH3), 1.74-1.67 (m, 4H, H-8, H-9), 1.29 (t, 9H, J = 7.3 Hz, (CH3CH2)3N). 13C NMR (CD3OD, 100 MHz): δ 166.88 (s, C = O, C-4), 152.96 (s, C = O, C-2), 149.05 (s, C-6), 143.24 (s, C-4′′), 121.69 (s, C-5′′), 111.13 (s, C-5), 94.35 (s, C-1′), 86.12 (d, 1H, J = 8.8 Hz, C-4′), 77.11 (s, C-3′), 72.41 (s, C-2′), 65.95 (d, 1H, J = 4.8 Hz, C-5′), 47.62 (s, (CH3CH2)3N), 29.39, 27.22, 25.76 (s, C-8, C-9, C-10), 12.18 (s, CH3), 9.15 (s, (CH3CH2)3N). 31P NMR (CD3OD, 100 MHz): δ 1.31. ESI MS m/z: calcd. for C16H22N5O8P2ˉ [M + H]ˉ 460.35, found [M + H]ˉ 460.18. Analysis calcd. for C28H54N7O9P, %: C, 50.67; H, 8.20; N, 14.77; P, 4.67. Found, %: C, 50.66; H, 8.24; N, 14.74; P, 4.69.
Biology
Antiviral assay
Virus and cells
Influenza virus A/Puerto Rico/8/34 (H1N1) was obtained from the collection of viruses of St. Petersburg Pasteur Institute. Before the experiment, virus was propagated in the allantoic cavity of 10- to 12-day-old chicken embryos for 48 h at 36 °C. The infectious titre of the virus was determined in Madin-Darby Canine Kidney (MDCK) cells (ATCC-CCL-34) grown in 96-well plates in alpha-MEM medium with 10% foetal bovine serum.
Cytotoxicity assay
MDCK cells were seeded onto 96-well culture plates (104 cells per well) and incubated at 36 °C in 5% CO2 until continuous monolayer formation. To assess the toxicity of compounds, a series of their threefold dilutions at concentrations of 300 to 3.7 μg/mL in Eagle’s Minimal Essential Medium (MEM) were prepared. The dilutions were added to the wells of the plates. Cells were incubated for 72 h at 36 °C in a CO2 incubator under 5% CO2. Further, a microtetrazolium (MTT) assay was performed on 96-well plates. The cells were washed 2 times with saline (0.9% NaCl), and 100 μL/well of MTT solution [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] at a concentration of 0.5 μg/mL in MEM was added. The plates were incubated for 1 h at 36 °C, the liquid was removed, and dimethylsulfoxide (DMSO) (0.1 mL per well) was added. The optical density (OD) of the cells was measured on a Thermo Multiskan FC spectrophotometer (Thermo Fisher Scientific, USA) at a wavelength of 540 nm. Based on the obtained data, the CC50, the concentration of the compound that destroys 50% of the cells in the culture, was calculated for each specimen.
CPE reduction assay
The compounds in appropriate concentrations were added to MDCK cells (0.1 ml per well). MDCK cells were further infected with A/Puerto Rico/8/34 (H1N1) influenza virus (m.o.i 0.01 TCID50 per cell). Plates were incubated for 72 h at 36 °C at 5% CO2. After that, cell viability was assessed by the MTT test, as described above. The cytoprotective activity of compounds was considered as their ability to increase the values of the OD compared to the control wells (with virus only; no drugs). Based on the obtained results, the IC50 values, i.e., the concentration of compounds that results in 50% cell protection, were calculated using GraphPad Prism 6.01 software. IC50 values in μg/ml were then calculated into micromoles. For each compound, the value of the selectivity index (SI) was calculated as a ratio of CC50 to IC50.
Cytotoxicity against human cancer cell lines
Cells and Materials
M-HeLa clone 11 (epithelioid carcinoma of the cervix, subline HeLa., clone M-HeLa); HuTu 80, human duodenal adenocarcinoma; MCF7, human breast adenocarcinoma (pleural fluid); HepG2, a human liver cancer cell line; PANC-1, a human pancreatic carcinoma; A549, a human lung carcinoma; PC3, prostate adenocarcinoma cell line from ATCC (American Type Cell Collection, USA; CRL 1435); А-375, a human melanoma cell line from the CLS Cell Lines Service cell repository Eppelheim Germany and WI38, a diploid human embryo lung from the collection of the Institute of Cytology, Russian Academy of Sciences (St. Petersburg) were used in the experiments.
MTT assay
The cytotoxic effect on cells was determined using the colorimetric method of cell proliferation - the MTT test. NADP-H-dependent cellular oxidoreductase enzymes can, under certain conditions, reflect the number of viable cells. These enzymes are able to reduce the tetrazolium dye (MTT) - 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide to insoluble blue-violet formazan, which crystallizes inside the cell. The amount of formazan formed is proportional to the number of cells with active metabolism. Cells were seeded on a 96-well Nunc plate at a concentration of 5 × 103 cells per well in a volume of 100 μl of medium and cultured in a CO2 incubator at 37 °C until a monolayer was formed. Then the nutrient medium was removed and 100 µl of solutions of the test drug in the given dilutions were added to the wells, which were prepared directly in the nutrient medium with the addition of 5% DMSO to improve solubility. After 48 h of incubation of the cells with the tested compounds, the nutrient medium was removed from the plates and 100 µl of the nutrient medium without serum with MTT at a concentration of 0.5 mg/mL was added and incubated for 4 h at 37 °C. Formazan crystals were added 100 µl of DMSO to each well. Optical density was recorded at 540 nm on an Invitrologic microplate reader (Russia). The experiments for all compounds were repeated three times. IC50 values were estimated using the Quest GraphTM IC50 Calculator (AAT Bioquest, Inc., Sunnyvale, CA, USA).
Molecular docking
Molecular docking was carried out using the Autodock Vina 1.1.2 software and AutoDock Tools (ADT 1.5.6) [47]. The standard 3D structures of the compounds tested were constructed using the HyperChem 8.0 [48] and converted into a pdb file by Open Babel [49]. The dimensions of the Grid box were chosen for PA N-ter (PDB code 4AWK) 22 × 20 × 18 (x, y, z), for neuraminidase N1 (PDB code 4B7Q) 60 × 60 × 62 (x, y, z) and for hemagglutinin H1 (PDB code 1RU7) 30 × 30 × 40 (x, y, z) with a spacing of 1.000 Å and grid maps were generated. The docking parameters were used as the default settings.
References
Balzarini J. Metabolism and mechanism of antiretroviral action of purine and pyrimidine derivatives. Pharm World Sci. 1994;16:113–26. https://doi.org/10.1007/BF01880662
Pastuch-Gawołek G, Gillner D, Krol E, Walczak K, Wandzik I. Selected nucleos(t)ide-based prescribed drugs and their multi-target activity. Eur J Pharm. 2019;865:172747. https://doi.org/10.1016/j.ejphar.2019.172747
Eyer L, Nencka R, De Clercq E, Seley-Radtke K, Ruzek D. Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antiviral Chem Chemother. 2018;26:1–28. https://doi.org/10.1177/2040206618761299
Jordheim LP, Durantel D, Zoulim F, Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discovery. 2013;12:447–64. https://doi.org/10.1038/nrd4010
Martin JC, Hitchcock MJM, Kaul S, Dunkle LM, Sterzycki RZ, Mansuri MM, et al. Comparative studies of 2′,3′-didehydro-2′3′-dideoxythymidine (d4T) with other pyrimidine nucleoside analogues. Ann NY Acad Sci. 1990;616:22–28. https://doi.org/10.1111/j.1749-6632.1990.tb17824.x
Gao WY, Agbaria R, Driscoll JS, Mitsuya H. Divergent anti-human immunodeficiency virus activity and anabolic phosphorylation of 2′,3′-dideoxynucleoside analogs in resting and activated human cells. J Biol Chem. 1994;269:12633–8. https://doi.org/10.1016/S0021-9258(18)99923-0
Stein DS, Moore KH. Phosphorylation of nucleoside analog antiretrovirals: a review for clinicians. Pharmacotherapy. 2001;21:11–34. https://doi.org/10.1592/phco.21.1.11.34439
Roy R, Depaix A, Périgaud C, Peyrottes S. Recent trends in nucleotide synthesis. Chem Rev. 2016;116:7854–97. https://doi.org/10.1021/acs.chemrev.6b00174
Kataev VE, Garifullin BF. Antiviral nucleoside analogs. Chem Heterocycl Comp. 2021;57:326–41. https://doi.org/10.1007/s10593-021-02912-8
Li Y, Mao S, Hager MW, Becnel KD, Schinazi RF, Liotta DC. Synthesis and evaluation of 2ʹ-substituted cyclobutyl nucleosides and nucleotides as potential anti-HIV agents. Bioorg Med Chem Lett. 2007;17:3398–401. https://doi.org/10.1016/j.bmcl.2007.03.094
Toti KS, Derudas M, Pertusati F, Sinnaeve D, Van den Broeck F, Margamuljana L, et al. Synthesis of an apionucleoside family and discovery of a prodrug with anti-HIV activity. J Org Chem. 2014;79:5097–112. https://doi.org/10.1021/jo500659e
Chien M, Anderson TK, Jockusch D, Tao C, Li X, Kumar S, et al. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase, a Key Drug Target for COVID-19. J Proteome Res. 2020;19:4690–7. https://doi.org/10.1021/acs.jproteome.0c00392
Montgomery JA, Thomas HJ, Schaeffer HJ. Synthesis of potential anticancer agents. XXVIII. Simple esters of 6-mercaptopurine ribonucleotide. J Org Chem. 1961;26:1929–33. https://doi.org/10.1021/jo01065a058
Wagner CR, Iyer VV, McIntee EJ. Pronucleotides:toward the in vivo delivery of antiviral and anticancer nucleotides. Med Res Rev. 2000;20:417–51. https://doi.org/10.1002/1098-1128(200011)20:6<417::aid-med1>3.0.co;2-z
Cahard D, McGuigan C, Balzarini J. Aryloxy phosphoramidate triesters as Pro-Tides. Mini Rev Med Chem. 2004;4:371–81. https://doi.org/10.2174/1389557043403936.
Mehellou Y, Balzarini J, McGuigan C. Aryloxy phosphoramidate triesters: a technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem. 2009;4:1779–91. https://doi.org/10.1002/cmdc.200900289
Singh US, Mulamoottil VA, Chu CK. 2′-Fluoro-6′-methylene carbocyclic adenosine and its phosphoramidate prodrug: A novel anti-HBV agent, active against drug-resistant HBVmutants. Med Res Rev. 2018;38:977–1002. https://doi.org/10.1002/med.21490
McGuigan C, Madela K, Aljarah M, Gilles A, Brancale A, Zonta N, et al. Design, synthesis and evaluation of a novel double pro-drug: INX-08189. A new clinical candidate for hepatitis C virus. Bioorg Med Chem Lett. 2010;20:4850–4. https://doi.org/10.1016/j.bmcl.2010.06.094
Mehellou Y, Rattan HS, Balzarini J. The ProTide prodrug technology: from the concept to the clinic. J Med Chem. 2018;61:2211–26. https://doi.org/10.1021/acs.jmedchem.7b00734
Wang G, Lim SP, Chen YL, Hunziker J, Rao R, Gu F, et al. Structure-activity relationship of uridine-based nucleoside phosphoramidate prodrugs for inhibition of dengue virus RNA-dependent RNA polymerase. Bioorg Med Chem Lett. 2018;28:2324–7. https://doi.org/10.1016/j.bmcl.2018.04.069
Schooley RT, Carlin AF, Beadle JR, Valiaeva N, Zhang XQ, Clark AE, et al. Rethinking Remdesivir: synthesis, antiviral activity, and pharmacokinetics of oral lipid prodrugs. Antimicrob Agents Chemother. 2021;65:e01155–21. https://doi.org/10.1128/AAC.01155-21
Seley-Radtke KL, Yates MK. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir Res. 2018;154:66–86. https://doi.org/10.1016/j.antiviral.2018.04.004
Slusarczyk M, Serpi M, Pertusati F. Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antivir Chem Chemother. 2018;26:1–31. https://doi.org/10.1177/2040206618775243
Pruijssers AJ, Denison MR. Nucleoside analogues for the treatment of coronavirus infections. Curr Opinion Virology. 2019;35:57–62. https://doi.org/10.1016/j.coviro.2019.04.002
Tatarinov DA, Garifullin BF, Belenok MG, Andreeva OV, Strobykina IYU, Shepelina AV, et al. The first 5′-phosphorylated 1,2,3-triazolyl nucleoside analogues with uracil and quinazoline-2,4-dione moieties. Synthesis and antiviral evaluation. Molecules. 2022;27:6214. https://doi.org/10.3390/molecules27196214
Andreeva OV, Garifullin BF, Zarybaev VV, Slita AV, Yesaulkova IL, Saifina LF, et al. Synthesis and biological evaluation of 1,2,3-triazolyl nucleoside analogues as potential antiviral agents against influenza virus A (H1N1) and coxsackievirus B4. Mol Diversity. 2021;25:473–90. https://doi.org/10.1007/s11030-020-10141-y
Fan WQ, Katritzky AR 1,2,3-Triazoles. In: Katritzky AR, Rees CW, Scriven EFV, editors. Comprehensive Heterocyclic Chemistry. Oxford, UK; Pergamon; 1997. Vol. 4. p. 2-129.
Alexandrova LA, Efremenkova OV, Andronova VL, Galegov GA, Solyev PN, Karpenko IL, et al. 5-(4-Alkyl-1,2,3-triazol-1-yl)methyl derivatives of 2’-deoxyuridine as inhibitors of viral and bacterial growth. Russ J Bioorg Chem. 2016;42:677–84. https://doi.org/10.1134/S1068162016050022
Chatzileontiadou DSM, Parmenopoulou V, Manta S, Kantsadi AL, Kylindri P, Griniezaki M, et al. Triazole double-headed ribonucleosides as inhibitors of eosinophil derived neurotoxin. Bioorg Chem. 2015;63:152–65. https://doi.org/10.1016/j.bioorg.2015.10.007
Malin AA, Ostrovskii VA. Synthesis of thymidine derivatives as potential pharmaceuticals against HIV/AIDS infection. Russ J Org Chem. 2001;37:759–80. https://doi.org/10.1023/A:1012441026853
Kowalinski E, Zubieta C, Wolkerstorfer A, Szolar OHJ, Ruigrok RWH, Cusack S. Structural analysis of specific metal chelating inhibitor binding to the endonuclease domain of influenza pH1N1 (2009) polymerase. PloS Pathog. 2012;8:e1002831. https://doi.org/10.1371/journal.ppat.1002831
Jockusch S, Tao C, Li X, Anderson TK, Chien M, Kumar S, et al. A library of nucleotide analogues terminate RNA synthesis catalyzed by polymerases of coronaviruses that cause SARS and COVID-19. Antiviral Res. 2020;180:104857. https://doi.org/10.1016/j.antiviral.2020.104857
Jácome R, Campillo-Balderas JA, Ponce de León S, Becerra A, Lazcano A. Sofosbuvir as a potential alternative to treat the SARS-CoV-2 epidemic. Sci. Rep. 2020;10:9294. https://doi.org/10.1038/s41598-020-66440-9
Das K, Aramini JM, Ma LC, Krug RM, Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nature SMB. 2010;17:530–8. https://doi.org/10.1038/nsmb.1779
Fudo S, Yamamoto N, Nukaga M, Odagiri T, Tashiro M, Hoshino T. Two distinctive binding modes of endonuclease inhibitors to the N‑terminal region of influenza virus polymerase acidic subunit. Biochemistry. 2016;55:2646–60. https://doi.org/10.1021/acs.biochem.5b01087
Lao J, Vanet A. A new strategy to reduce influenza escape: detecting therapeutic targets constituted of invariance groups. Viruses. 2017;9:38. https://doi.org/10.3390/v9030038
Luganini A, Terlizzi ME, Catucci G, Gilardi G, Maffei ME, Gribaudo G. The cranberry extract Oximacro® exerts in vitro virucidal activity against influenza virus by interfering with hemagglutinin. Front Microbiol. 2018;9:1826. https://doi.org/10.3389/fmicb.2018.01826.
Zhang W, Chen ST, He QY, Huang LQ, Li X, Lai XP, et al. Asprellcosides B of Ilex asprella Inhibits Influenza A virus infection by blocking the hemagglutinin- mediated membrane fusion. Front Microbiol. 2019;9:3325. https://doi.org/10.3389/fmicb.2018.03325
Gamblin SJ, Haire LF, Russell RJ, Stevens DJ, Xiao B, Ha Y, et al. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science. 2004;303:1838–43. https://doi.org/10.1126/science.1093155
Berman H, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–42. https://doi.org/10.1093/nar/28.1.235
Benton DJ, Wharton SA, Martin SR, McCauley JW. Role of neuraminidase in influenza A(H7N9) virus receptor binding. J Virol. 2017;91:e02293–16. https://doi.org/10.1128/JVI.02293-16
Dou D, Revol R, Östbye H, Wang H, Daniels R. Influenza A virus cell entry, replication, virion assembly and movement. Front Immunol. 2018;9:1581. https://doi.org/10.3389/fimmu.2018.01581
D’Souza C, Kanyalkar M, Joshi M, Coutinho E, Srivastava S. Search for novel neuraminidase inhibitors: design, synthesis and interaction of oseltamivir derivatives with model membrane using docking, NMR and DSC methods. Biochim Biophys Acta Biomembr. 2009;1740:1740–51. https://doi.org/10.1016/j.bbamem.2009.04.014
Herlambang SJ, Saleh R. Molecular docking investigation for Indonesian H274Y mutant neuraminidase type 1 with neuraminidase inhibitors. Am J Mol Biol. 2012;2:49–59. https://doi.org/10.4236/ajmb.2012.21006
Le K, Tran D, Nguyen A, Le L. A screening of neuraminidase inhibition activities of isoquinolone alkaloids in Coptis chinensis using molecular docking and pharmacophore analysis. ACS Omega. 2020;5:30315–22. https://doi.org/10.1021/acsomega.0c04847
van der Vries E, Collins PJ, Vachieri SG, Xiong X, Liu J, Walker PA, et al. H1N1 2009 pandemic influenza virus: resistance of the I223R neuraminidase mutant explained by kinetic and structural analysis. PLoS Pathog. 2012;8:e1002914. https://doi.org/10.1371/journal.ppat.1002914
Trott O, Olson FJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–61. https://doi.org/10.1002/jcc.21334
HyperChem Professional 8.0 (2007). Hypercube, Inc. http://www.hyper .com/?tabid=360. Accessed 26 November 2022.
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: an open chemical toolbox. J Cheminform. 2011;3:33. https://doi.org/10.1186/1758-2946-3-33
Acknowledgements
We are grateful to the Assigned Spectral-Analytical Centre of FRC Kazan Scientific Centre of RAS for technical assistance in research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Garifullin, B.F., Tatarinov, D.A., Andreeva, O.V. et al. Synthesis, antiviral evaluation, molecular docking study and cytotoxicity of 5′-phosphorylated 1,2,3-triazolyl nucleoside analogues with thymine and 6-methyl uracil moieties. Med Chem Res 32, 1770–1803 (2023). https://doi.org/10.1007/s00044-023-03112-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-023-03112-z