
Abstract
Based on the cholinergic hypothesis of the reported compound, N-(4-(4-methoxy-phenyl)thiazol-2-yl)-3-(pyrrolidin-1-yl)propionamide, which had a good inhibitory activity to acetylcholinesterase (AChE), the new 4-methoxy-phenylthiazole-2-amine derivatives as AChE inhibitors (AChEIs) have been designed and synthesized in this study. Their chemical structures were confirmed by proton nuclear magnetic resonance, carbon-13 nuclear magnetic resonance, mass spectrometry, and infrared. Furthermore, their inhibitory activities against AChE in vitro were also tested by Ellman spectrophotometry, and the inhibitory activity test results showed that most of the compounds of 4-methoxy-phenylthiazole-2-amine derivatives had a certain AChE inhibitory activity in vitro, and the IC50 (half-maximal inhibitory concentration) value of compound 5g was 5.84 μmol/L, which was higher than that of the reference compound, rivastigmine. Moreover, it had almost no inhibitory effect on butyrylcholinesterase. In addition, compound 5g was subjected to enzyme inhibition kinetics experiments, and the result of Lineweaver–Burk’s V−1–[S]−1 double-reciprocal plot showed that the acting type of compound 5g was mixed inhibition type. Furthermore, the AChE inhibitory activity mechanism of compound 5g was explored by the conformational analysis and molecular docking, which was based on the principle of the four-point pharmacophore model necessary for AChE inhibition. Finally, in silico molecular property and ADMET (absorption, distribution, metabolism, excretion, and toxicity) of the synthesized compounds were predicted by using Molinspiration and PreADMET online servers, respectively. It can be concluded that the lead AChEI compound 5g presented satisfactory drug-like characteristics and ADME properties.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD), the most common cause of dementia, is a neurodegenerative disorder characterized by progressive deterioration of memory and cognition (Hiremathad et al. 2018). AD is thought to be the leading cause of senile dementia affecting nearly 50 million people worldwide (Schelke et al. 2018). In the past two decades, considerable research efforts devoted to unraveling the molecular, biochemical, and cellular mechanisms of cognitive deficit have been witnessed. Several factors such as, both genetic and environmental, reduced levels of acetylcholine, amyloid β-protein aggregation, tau-hyperphosphorylation, hypoxia, and so on have been considered to play a crucial role in cognition decline (Zhang et al. 2016; Grozeva et al. 2019). Despite several attempts made on the development of new AD therapies, efficacious therapeutic interventions for the suppression of AD have not yet been established. AD is characterized by the disturbance of forebrain cholinergic system and the dramatic decrease of acetylcholine (ACh), choline acetyltransferase, and a severe loss of cholinergic neurons (Pepeu et al. 2017).
Acetylcholinesterase (AChE) belongs to the class of serine hydrolase family of enzymes and is responsible for catalyzing the hydrolysis of neurotransmitter ACh into choline and acetic acid, thereby causing the termination of cholinergic neurotransmission (Lopes et al. 2014; Kaushik et al. 2018). The inhibition of AChE would prevent the hydrolysis of ACh, so that the level of ACh in cholinergic synapses is increased.
Nowadays, many AChE inhibitors have been approved for the therapeutic treatment of AD (Fig. 1), which include tacrine, huperzine-A, donepezil, rivastigmine, physostigmine, and galantamine, and so on (Atri et al. 2018). However, none of these drugs is able to combat or reverse the progression of the disease. Because of the limited number of available clinical AChEIs, the search for new AChEI is gaining wide attention.

Acetylcholinesterase (AChE) inhibitors clinically used for Alzheimer’s disease (AD) treatment
In recent years, substantial attention has been given to the chemistry of thiazole and its fused heterocyclic derivatives due to biological importance. Cholinesterase inhibition activity of compounds bearing thiazole scaffold have been reported (Ibrar et al. 2018; Tripathi et al. 2018). Based on the structural characteristics of donepezil, Yurttaş et al. (2013) designed a series of new thiazole-piperazine derivatives, and IC50 (half maximal inhibitory concentration) value of their AChE inhibitory activity was up to 0.011 μmol/L, which is higher than donepezil (IC50 = 0.054 μmol/L) as a reference compound. A series of new N-acyl-4-phenylthiazole-2-amine derivatives were designed by Ma et al. (2014) and Tian et al. (2015), and the IC50 value of their AChE inhibitory activity was up to 0.51 μmol/L, better than that of rivastigmine and huperzine-A as reference compounds.
Based on these results (Ma et al. 2014), 4-methoxy-phenylthiazole-2-amine derivatives as AChE inhibitors (AChEI) were designed and synthesized in this paper. Interestingly, three pharmacophoric features out of four-point pharmacophore model for AChEIs, which was reported by Bag et al. (2013), namely aromatic ring(s), H-bond acceptor, and H-bond donor, are identified in 4-methoxy-phenylthiazole-2-amine derivatives (Fig. 2). Based on these evidences, the structure–activity relationships (SAR) for AChE inhibition by 4-methoxy-phenylthiazole-2-amine derivatives in vitro was explored.

Structural hypothesis of 4-methoxythiazole-2-amine derivatives showing the shared pharmacophoric features essential for acetylcholinesterase (AChE) inhibition
Further, in order to better understand the action mechanism of AChE inhibitory activity, the molecular docking was conducted to investigate the potential binding mode of compounds with the catalytic domain of AChE (PDB: 4EY7) (Cheung et al. 2012), which was performed using Autodock 4.2.
Moreover, in order to predict whether these compounds have the potential to become clinical drugs, and get insights into their pharmacokinetics profile, the compounds were subjected to in silico molecular property and ADMET (absorption, distribution, metabolism, excretion, and toxicity) predictions by using Molinspiration and PreADMET online servers, respectively.
Materials and methods
Instrumentation and chemicals
All experiments were carried out under air atmosphere; the reagent materials were commercially available analytically pure or chemically pure, unless stated otherwise. High-resolution mass spectrometry was determined by Bruker Maxis 4G Mass Spectrometer, and the nuclear magnetic resonance spectrum was measured by Bruker Avance III 600 MHz nuclear magnetic resonance spectrometer. The melting point was determined by SGW X-4 micro melting point apparatus (Shanghai Precision Scientific Instrument Co., Ltd). The infrared (IR) spectra was obtained by FTIR-8400S Fourier transform infrared splitting Photometer (KBr disks). The 96-well plate was read by 1420 Victor Microplate Reader. AChE was purchased from Sigma, and huperzine-A and rivastigmine were purchased from Shanghai Yuanye Biotechnology Co., Ltd.
Syntheses
General procedure for the synthesis of (2-amino-4-phenylthiazole) amide derivatives (4a–4e)
Compounds 1, 2, and 3 (Ma et al. 2014) were obtained according to the previously described method. Chloroacetyl chloride (1.45 mmol) in CH2Cl2 (15 mL) was added dropwise to a cooled solution (0 °C) of 4-(4-methoxyphenyl)thiazol-2-amine 3 (1.21 mmol) and diisopropylethylamine (DIEA) (1.24 mmol) in CH2Cl2 (25 mL). The completion of the reaction was assessed by thin-layer chromatography (TLC). The organic layer was separated, washed with saturated NaHCO3 solution (100 mL), water, and brine, the aqueous phase was extracted with CH2Cl2 (60 mL), and the organic phases were collected, dried over anhydrous MgSO4, and evaporated in vacuo. The product was separated and purified by column chromatography on silica gel using an ethyl acetate/petroleum ether mixture (1/4) as the eluent to afford pure compound 4a (Scheme 1).

Synthetic routes of target compound
The same procedure was performed with 3-chloropropionyl chloride, 4-bromobutyryl chloride, 5-bromopentanoyl chloride, and 6-bromohexanoyl chloride to afford compounds 4b–4e.
2-Chloro-N-(4-(4-methoxyphenyl)thiazol-2-yl)acetamide 4a, 3-chloro-N-(4-(4-methoxyphenyl)thiazol-2-yl)propanamide 4b characterization data were consistent with that in the previous literature (Ma et al. 2014).
4-Bromo-N-(4-(4-methoxyphenyl)thiazol-2-yl)butanamide (4c)
It was obtained as a white solid; yield: 86%; mp 178–180 °C; ν (cm−1): 3147, 1683, 1581, 1261, 912, 885; proton nuclear magnetic resonance (1H NMR) (600 MHz, DMSO-d6): δ= 12.13 (s, 1H, -CONH-), 7.53 (d, J= 7.6 Hz, 2H, -ArH), 7.38 (s, 1H, = CHS-), 7.03 (d, J= 7.6 Hz, 2H, -ArH), 3.87 (s, 3H,-OCH3), 3.74 (t, J= 6.8 Hz, 2H, -CH2Br), 2.71 (t, J= 6.8 Hz, 4H, -CH2-). Carbon-13 nuclear magnetic resonance (13C NMR) (151 MHz, DMSO-d6): δ = 171.36, 158.49, 157.41, 148.61, 129.25, 127.37, 114.13, 105.72, 55.11, 31.66, 27.88, 21.70; ESI-MS calcd for C14H16BrN2O2S 357.0095 [M + H]+, found 357.0090 [M + H]+.
5-Bromo-N-(4-(4-methoxyphenyl)thiazol-2-yl)pentanamide (4d)
It was obtained as a white solid; yield: 83%; mp 182–184 °C; IR (KBr), ν (cm−1): 3143, 1683, 1588, 1270, 915, 893; 1H NMR (600 MHz, DMSO-d6) δ = 12.09 (s, 1H, -CONH-), 7.55 (d, J= 7.6 Hz, 2H, -ArH), 7.36 (s, 1H, =CHS-), 7.03 (d, J= 7.6 Hz, 2H, -ArH), 3.88 (s, 3H, -OCH3), 3.74 (t, J= 6.8 Hz, 2H, -CH2Br), 2.71 (t, J= 6.8 Hz, 2H, -CH2-), 1.82 (t, J= 6.8 Hz, 2H, -CH2-), 1.53. (t, J= 6.8 Hz, 2H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 171.07, 158.49, 157.68, 148.61, 129.25, 126.95, 114.05, 105.81, 55.11, 34.58, 31.86, 23.32, 21.38; ESI-MS calcd for C15H18BrN2O2S 369.0272 [M + H]+, found 369.0264 [M + H]+.
6-Bromo-N-(4-(4-methoxyphenyl)thiazol-2-yl)hexanamide (4e)
It was obtained as a white solid; yield: 85%; mp 186–188 °C; IR (KBr), ν (cm−1):3145, 1683, 1590, 1275, 920, 890; 1H NMR (600 MHz, DMSO-d6) δ = 12.17 (s, 1H, -CONH-), 7.82 (d, J = 8.5 Hz, 2H, -ArH), 7.43 (d, J = 4.6 Hz, 1H, =CHS-), 7.01 (dd, J = 33.5, 8.6 Hz, 2H, -ArH), 3.80 (d, J = 11.9 Hz, 3H, -OCH3), 3.54 (t, J = 6.7 Hz, 2H, -CH2Br), 3.17 (s, 2H, -CH2-), 2.46 (t, J = 7.5 Hz, 2H, -CH2-), 1.86–1.79 (m, 2H, -CH2-), 1.64 (dt, J = 15.2, 7.4 Hz, 2H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 171.28, 158.92, 157.72, 148.60, 127.17, 126.94, 114.05, 105.78, 55.11, 34.92, 34.68, 31.90, 27.07, 23.82; ESI-MS calcd for C16H20BrN2O2S 383.0429 [M + H]+, found 383.0425 [M + H]+.
General procedure for the synthesis of N-(4-(4-methoxyphenyl)thiazol-2-yl)-2-(pyridin-4-ylthio)amide and N-(4-(4-methoxyphenyl)thiazol-2-yl)-2-((1-benzylpiperidin-4-yl)amino)-amide (5a–5j)
To a solution of (2-amino-4-phenylthiazole) amide derivatives 4a–4e (5 mmol) in acetonitrile (5 mL), 4-mercaptopyridine (6 mmol), DIEA (10 mmol), and potassium iodide (0.5 mmol) were added, and the resulting solution was stirred at 110 °C for 12 h. The completion of the reaction was assessed by TLC. The organic layer was separated, washed with saturated NaHCO3 solution (100 mL), water, and brine, the aqueous phase was extracted with CH2Cl2 (60 mL), and the organic phases were collected, dried over anhydrous MgSO4, and evaporated in vacuo. The product was separated and purified by column chromatography on silica gel using an ethyl acetate/petroleum ether mixture (1/2) as the eluent to afford pure compounds 5a–5e.
The same procedure was performed with 4-amino-1-benzylpiperidine using a methanol/dichloromethane mixture (1/20) as the eluent to afford pure compounds 5f–5j (Scheme 1).
N-(4-(4-methoxyphenyl)thiazol-2-yl)-2-(pyridin-4-ylthio)acetamide (5a)
It was obtained as a white solid; yield: 50%; mp 208–210 °C; IR (KBr), ν (cm−1): 3112, 1674, 1580, 1260, 885; 1H NMR (600 MHz, DMSO-d6) δ = 12.61 (s, 1H, -CONH-), 8.41 (d, J = 5.8 Hz, 2H, -ArH), 7.83 (d, J = 8.6 Hz, 2H, -ArH), 7.49 (s, 1H, =CHS-), 7.36 (d, J = 6.0 Hz, 2H, -ArH), 7.00 (d, J = 8.7 Hz, 2H, -ArH), 4.17 (s, 2H, -CH2-), 3.79 (s, 3H, -OCH3). 13C NMR (151 MHz, DMSO-d6) δ = 166.59, 159.02, 157.39, 149.20, 147.44, 126.99, 125.30, 120.49, 114.10, 106.36, 55.13, 33.39; ESI-MS calcd for C17H16N3O2S2 358.0684 [M + H]+, found 358.0675 [M + H]+.
N-(4-(4-methoxyphenyl)thiazol-2-yl)-3-(pyridin-4-ylthio)propanamide (5b)
It was obtained as a white solid; yield: 47%; mp 214–216 °C; IR (KBr), ν (cm−1): 3110, 1681, 1583, 1265, 890; 1H NMR (600 MHz, DMSO-d6) δ = 12.31 (s, 1H, -CONH-), 8.40 (d, J = 5.9 Hz, 2H, -ArH), 7.82 (d, J = 8.5 Hz, 2H, -ArH), 7.45 (s, 1H, =CHS-), 7.33 (d, J = 6.0 Hz, 2H, -ArH), 6.99 (d, J = 8.6 Hz, 2H, -ArH), 3.79 (s, 3H, -OCH3), 3.35 (t, J = 7.0 Hz, 2H, -CH2-), 2.88 (t, J = 7.0 Hz, 2H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 169.22, 158.91, 157.09, 149.09, 147.91, 127.09, 126.95, 120.61, 114.07, 106.01, 55.12, 34.11, 24.95; ESI-MS calcd for C18H18N3O2S2 372.0840 [M + H]+, found 372.0839 [M + H]+.
N-(4-(4-methoxyphenyl)thiazol-2-yl)-4-(pyridin-4-ylthio)butanamide (5c)
It was obtained as a white solid; yield: 51%; mp 220–222 °C; IR (KBr), ν (cm−1): 3168, 1681, 1582, 1262, 887; 1H NMR (600 MHz, DMSO-d6) δ = 12.27 (s, 1H, -CONH-), 8.38 (d, J = 4.2 Hz, 2H, -ArH), 7.83 (d, J = 8.5 Hz, 2H, -ArH), 7.43 (s, 1 H, =CHS-), 7.30 (d, J = 5.8 Hz, 2H, -ArH), 6.99 (d, J = 8.5 Hz, 2H, -ArH), 3.79 (s, 3H, -OCH3), 3.12 (t, J = 7.3 Hz, 2H, -CH2-), 2.65 (t, J = 7.2 Hz, 2H, -CH2-), 1.98 (p, J = 7.3 Hz, 2H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 170.65, 158.93, 157.74, 149.17, 148.13, 127.16, 126.95, 120.57, 114.05, 105.84, 55.11, 33.65, 28.90, 23.67; ESI-MS calcd for C19H20N3O2S2 386.0997 [M + H]+, found 386.0997 [M + H]+.
N-(4-(4-methoxyphenyl)thiazol-2-yl)-5-(pyridin-4-ylthio)pentanamide (5d)
It was obtained as a white solid; yield: 49%; mp 225–227 °C; IR (KBr), ν (cm−1): 3163, 1681, 1581, 1266, 887; 1H NMR (600 MHz, DMSO-d6) δ = 12.21 (s, 1H, -CONH-), 8.35 (d, J = 5.9 Hz, 2H, -ArH), 7.82 (d, J = 8.7 Hz, 2H, -ArH), 7.43 (s, 1H, =CHS-), 7.26 (d, J = 6.0 Hz, 2H, -ArH), 6.99 (d, J = 8.7 Hz, 2H, -ArH), 3.79 (s, 3H, -OCH3), 3.09 (t, J = 7.3 Hz, 2H, -CH2-), 1.81-1.74 (m, 2H, -CH2-), 1.71–1.63 (m, 4H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 171.14, 158.50, 157.41, 149.13, 148.36, 127.16, 126.95, 120.53, 114.05, 105.82, 55.11, 34.23, 29.06, 27.52, 23.83; ESI-MS calcd for C20H22N3O2S2 400.1153 [M + H]+, found 400.1155 [M + H]+.
N-(4-(4-methoxyphenyl)thiazol-2-yl)-6-(pyridin-4-ylthio)hexanamide (5e)
It was obtained as a white solid; yield: 47%; mp 230–232 °C; IR (KBr), ν (cm−1): 3112, 1681, 1585, 1260, 889; 1H NMR (600 MHz, DMSO-d6) δ = 12.18 (s, 1H, -CONH-), 8.35 (d, J = 6.1 Hz, 2H, -ArH), 7.82 (d, J = 8.7 Hz, 2H, -ArH), 7.42 (s, 1H, =CHS-), 7.25 (dd, J = 4.9, 1.3 Hz, 2H, -ArH), 6.99 (d, J = 8.8 Hz, 2H, -ArH), 3.79 (s, 3H, -OCH3), 3.06 (t, J = 7.3 Hz, 2H, -CH2-), 2.46 (t, J = 7.3 Hz, 2H, -CH2-), 1.69–1.62 (m, 2H, -CH2-), 1.48–1.41 (m, 4H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 171.31, 158.92, 157.41, 149.12, 148.50, 127.18, 126.94, 120.51, 114.05, 105.77, 55.11, 34.72, 29.30, 27.78, 27.66, 24.18; ESI-MS calcd for C21H24N3O2S2 414.1310 [M + H]+, found 414.1306 [M + H]+.
2-((1-Benzylpiperidin-4-yl)amino)-N-(4-(4-methoxyphenyl)thiazol-2-yl)acetamide (5f)
It was obtained as a white solid; yield: 47%; mp 232–234 °C; IR (KBr), ν (cm−1): 3307, 1699, 1581, 1261, 890; 1H NMR (600 MHz, DMSO-d6) δ = 7.83 (d, J = 8.6 Hz, 2H, -ArH), 7.45 (s, 1H, =CHS-), 7.33–7.21 (m, 5H, -ArH), 6.99 (d, J = 8.7 Hz, 2H, -ArH), 3.78 (s, 3H, -OCH3), 3.47 (s, 2H, -CH2-), 2.74 (d, J = 11.3 Hz, 2H, -CH2-), 2.43 (dd, J = 11.8, 8.0 Hz, 1H, -NH-), 1.98–1.90 (m, 5H, -CH2-), 1.77 (d, J = 11.4 Hz, 2H, -CH2-), 1.34–1.24 (m, 2H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 170.91, 158.96, 157.08, 148.70, 138.61, 128.06, 126.75, 125.40, 125.20, 114.04, 105.97, 62.29, 55.11, 51.58, 48.86, 31.53, 21.05; ESI-MS calcd for C24H29N4O2S 437.2011 [M + H]+, found 437.2008 [M + H]+.
3-((1-Benzylpiperidin-4-yl)amino)-N-(4-(4-methoxyphenyl)thiazol-2-yl)propan- amide (5g)
It was obtained as a white solid; yield: 40%; mp 235–237 °C; IR (KBr), ν (cm−1): 3392, 1681, 1585, 1263, 886; 1H NMR (600 MHz, DMSO-d6) δ = 7.83 (d, J = 8.7 Hz, 2H, -ArH), 7.45 (s, 1H, =CHS-), 7.34–7.22 (m, 5H, -ArH), 6.99 (d, J = 8.7 Hz, 2H, -ArH), 3.79 (s, 3H, -OCH3), 3.03 (t, J = 6.5 Hz, 2H, -CH2-), 2.84–2.66 (m, 5H, -CH2-), 2.00–1.83 (m, 6H, -CH2-), 1.46 (dd, J = 20.7, 10.6 Hz, 2H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 169.12, 158.93, 157.49, 148.62, 138.01, 128.72, 126.84, 125.33, 124.91, 114.05, 105.88, 61.85, 55.12, 54.89, 51.34, 33.36, 29.98, 22.01; ESI-MS calcd for C25H31N4O2S 451.2168 [M + H]+, found 451.2165 [M + H]+.
6-((1-Benzylpiperidin-4-yl)amino)-N-(4-(4-methoxyphenyl)thiazol-2-yl)hexanamide (5j)
It was obtained as a white solid; yield: 40%; mp 235–237 °C; IR (KBr), ν (cm−1): 3147, 1679, 1582, 1259, 888; 1H NMR (600 MHz, DMSO-d6) δ = 7.82 (d, J = 8.5 Hz, 2H, -ArH), 7.41 (s, 1H, =CHS-), 7.34–7.20 (m, 5H, -ArH), 6.98 (t, J = 8.1 Hz, 2H, -ArH), 3.78 (s, 3H, -OCH3), 3.45 (d, J = 8.5 Hz, 2H, -CH2-), 2.79 (dd, J = 18.4, 10.5 Hz, 4H, -CH2-), 2.43 (dt, J = 41.0, 13.9 Hz, 2H, -CH2-), 1.97–1.81 (m, 4H, -CH2-), 1.67–1.19 (m, 8H, -CH2-), 1.03 (dt, J = 21.0, 7.0 Hz, 1H, -CH-). 13C NMR (151 MHz, DMSO-d6) δ = 171.35, 158.92, 157.76, 148.22, 138.02, 128.69, 128.12, 126.94, 126.86, 114.05, 105.68, 61.74, 55.12, 54.88, 51.20, 34.68, 34.52, 28.97, 28.89, 25.81, 24.23; ESI-MS calcd for C28H37N4O2S 493.2637 [M + H]+, found 493.2637 [M + H]+.
General procedure for the synthesis of N-(4-(4-methoxyphenyl)thiazol-2-yl)-2-(pyridin-4-ylthio)amide derivative (6a–6e)
A solution of 5a–5e (5 mmol) and benzyl chloride (5 mmol) in acetonitrile reacted at 110 °C for 24 h. Subsequently, the precipitates were filtered and washed with dichloromethane to give the solid salts 6a–6e (Scheme 1).
1-Benzyl-4-((2-((4-(4-methoxyphenyl)thiazol-2-yl)amino)-2-oxoethyl)thio)pyri- din-1-ium chloride (6a)
It was obtained as a white solid; yield: 40%; mp 282–284 °C; IR (KBr), ν (cm−1):3103, 1679, 1579, 1263, 889; 1H NMR (600 MHz, DMSO-d6) δ = 12.87 (s, 1H, -CONH-), 8.99 (d, J = 6.5 Hz, 2H, -ArH), 8.09 (d, J = 6.5 Hz, 2H, -ArH), 7.84 (d, J = 8.6 Hz, 1H, =CHS-), 7.56–7.49 (m, 5H, -ArH), 7.47–7.39 (m, 2H, -ArH), 7.00 (d, J = 8.7 Hz, 2H, -ArH), 4.53 (s, 4H, -CH2-), 3.80 (s, 3H, -OCH3). 13C NMR (151 MHz, DMSO-d6) δ = 165.53, 161.86, 159.04, 157.09, 148.63, 142.41, 134.70, 129.15, 128.20, 127.01, 126.86, 125.31, 123.25, 114.10, 106.45, 61.77, 55.15, 33.97; ESI-MS calcd for C24H22ClN3O2S2 448.1148 [M-Cl]+, found 448.1155 [M-Cl]+.
1-Benzyl-4-((3-((4-(4-methoxyphenyl)thiazol-2-yl)amino)-3-oxopropyl)thio)pyridin-1-ium chloride (6b)
It was obtained as a white solid; yield: 35%; mp 286–288 °C; IR (KBr), ν (cm−1): 3392, 1687, 1583, 1260, 885; 1H NMR (600 MHz, DMSO-d6) δ = 12.41 (s, 1H, -CONH-), 8.98 (d, J = 6.9 Hz, 2H, -ArH), 8.05 (d, J = 7.0 Hz, 2H, -ArH), 7.82 (d, J = 8.7 Hz, 1H, =CHS-), 7.54 (d, J = 6.9 Hz, 5H, -ArH), 7.49–7.38 (m, 2H, -ArH), 6.99 (d, J = 8.8 Hz, 2H, -ArH), 3.79 (s, 3H, -OCH3), 3.59 (t, J = 6.9 Hz, 4H, -CH2-), 2.97 (t, J = 6.9 Hz, 2H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 168.92, 162.20, 158.97, 157.37, 148.71, 142.53, 134.69, 129.14, 128.31, 127.11, 126.96, 125.40, 123.18, 114,07, 106.08, 61.59, 55.14, 33.43, 25.97; ESI-MS calcd for C25H24ClN3O2S2 462.1304 [M-Cl]+, found 462.1307 [M-Cl]+.
1-Benzyl-4-((4-((4-(4-methoxyphenyl)thiazol-2-yl)amino)-4-oxobutyl)thio)pyridin-1-ium chloride (6c)
It was obtained as a white solid; yield: 30%; mp 290–293 °C; IR (KBr), ν (cm−1): 3112, 1679, 1585, 1265, 891; 1H NMR (600 MHz, DMSO-d6) δ = 12.35 (s, 1H, -CONH-), 8.98 (d, J = 6.6 Hz, 2H, -ArH), 8.04 (d, J = 6.4 Hz, 2H, -ArH), 7.83 (d, J = 8.7 Hz, 1H, =CHS-), 7.55 (d, J = 7.6 Hz, 5H, -ArH), 7.43 (ddd, J = 13.3, 10.9, 5.5 Hz, 2H, -ArH), 6.99 (d, J = 8.7 Hz, 2H, -ArH), 3.79 (s, 3H, -OCH3), 3.33 (d, J = 7.5 Hz, 4H, -CH2-), 2.69 (t, J = 7.1 Hz, 2H, -CH2-), 2.03 (p, J = 7.2 Hz, 2H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 170.53, 162.55, 158.94, 157.61, 148.63, 142.01, 134.69, 129.12, 128.42, 127.13, 126.86, 125.40, 123.17, 114.06, 105.88, 61.52, 55.13, 33.46, 29.87, 23.01; ESI-MS calcd for C26H26ClN3O2S2 476.146 [M-Cl]+, found 476.1468 [M-Cl]+.
1-Benzyl-4-((5-((4-(4-methoxyphenyl)thiazol-2-yl)amino)-5-oxopentyl)thio)pyridin-1-ium chloride (6d)
It was obtained as a white solid; yield: 28%; mp 296–298 °C; IR (KBr), ν (cm−1): 3109, 1679, 1583, 1265, 886; 1H NMR (600 MHz, DMSO-d6) δ = 12.26 (s, 1H, -CONH-), 8.95 (dd, J = 17.7, 6.8 Hz, 2H, -ArH), 8.01 (t, J = 11.0 Hz, 2H, -ArH), 7.82 (d, J = 8.3 Hz, 1H, =CHS-), 7.51 (dd, J = 21.0, 7.5 Hz, 5H, -ArH), 7.48–7.36 (m, 2H, -ArH), 6.99 (d, J = 8.4 Hz, 2H, -ArH), 3.78 (s, 3H, -OCH3), 3.32 (t, J = 7.0 Hz, 4H,-CH2-), 2.54 (d, J = 6.8 Hz, 2H, -CH2-), 1.82-1.69 (m, 4H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 171.11, 162.78, 158.93, 157.64, 148.22, 142.43, 134.69, 129.13, 128.53, 127.13, 126.88, 125.42, 123.13, 114.05, 105.83, 61.55, 55.13, 34.14, 30.15, 26.90, 23.66; ESI-MS calcd for C27H28ClN3O2S2 490.1617 [M-Cl]+, found 490.1627 [M-Cl]+.
1-Benzyl-4-((6-((4-(4-methoxyphenyl)thiazol-2-yl)amino)-6-oxohexyl)thio)pyridin-1-ium chloride (6e)
It was obtained as a white solid; yield: 25%; mp 300–302 °C; IR (KBr), ν (cm−1): 3107, 1677, 1589, 1266, 892; 1H NMR (600 MHz, DMSO-d6) δ = 12.22 (s, 1H, -CONH-), 8.91 (d, J = 7.0 Hz, 2H, -ArH), 7.97 (d, J = 7.0 Hz, 2H, -ArH), 7.82 (d, J = 8.6 Hz, 1H, =CHS-), 7.52 (d, J = 6.9 Hz, 5H, -ArH), 7.46–7.39 (m, 2H, -ArH), 6.99 (d, J = 8.7 Hz, 2H, -ArH), 3.79 (s, 3H, -OCH3), 3.28 (t, J = 7.3 Hz, 2H, -CH2-), 2.48 (t, J = 7.3 Hz, 2H, -CH2-), 1.74–1.64 (m, 4H, -CH2-), 1.47 (dd, J = 15.0, 7.9 Hz, 4H, -CH2-). 13C NMR (151 MHz, DMSO-d6) δ = 171.30, 162.84, 158.93, 157.69, 148.22, 142.41, 134.28, 129.13, 128.56, 127.15, 126.89, 125.42, 123.11, 114.06, 105.79, 61.55, 55.13, 34.66, 30.29, 27.54, 27.15, 24.11; ESI-MS calcd for C28H30ClN3O2S2 504.1774[M-Cl]+, found 504.1773 [M-Cl]+.
Results and discussion
AChE and BuChE inhibition assay
In vitro AChE inhibition test
The inhibitory activity of 4-methoxy-phenylthiazole-2-amine derivatives 5a–6e against electroporated AChE was determined using Ellman’s test (Abbas-Mohammadi et al. 2018) with huperzine-A and rivastigmine as reference compounds. The compounds (5a–6e) were dissolved in dimethyl sulfoxide. ACTI (acetylthiocholine iodide) was used as a substrate, and 5,5′-dithiobis [2-nitrobenzoic acid] (DTNB) was used for the measurement of AChE activity. Electroporated AChE was used as a source of AChE enzyme. Tris buffer’s pH was 7.2, and the concentrations of substrate ACTI, chromogenic reagent DTNB, and AChE were 0.6 mmol/L, 3 mmol/L, and 0.35 U/mL, respectively. The concentrations of compounds 5a–6e were configured as 1 × 10−4, 1 × 10−5, 1 × 10−6, 1 × 10−7, and 1 × 10−8 μmol/L, respectively. Tris buffer B, AChE, and each concentration of compounds were taken as 20 μL, 10 μL, and 10 μL, respectively, and were then added to a 96-well plate reader. The mixtures were incubated for 12 min at 37 °C. The hydrolysis of acetylthiocholine was monitored by the formation of a yellow-colored enzymatic product, 5-thio-2-nitrobenzoate anion, which was determined by the variation in absorbance measured at 405 nm, with a 96-well plate reader. The blank group without the substrate and compounds 5a–6e was added to 10 μL Tris buffer B two times, respectively. The standard group without compounds 5a–6e was added to 10 μL Tris buffer B. Each compound was tested two times at different concentrations; then, with the inhibition rate I [I = 100 × (Astandard − Aexperiment)/(Astandard − Ablank)%] was plotted against inhibitor concentration C to perform an S-curve linear fit to obtain the IC50 corresponding to the concentration Cx.
In order to get an accurate IC50, select the nearest 1 × 10−x μmol/L from 1 × 10−5 to 1 × 10−9 μmol/L, set 1 × 10−x μmol/L as less than Cx. The compound for which the activity was best was configured at concentrations 1 × 10−x, 3 × 10−x, 5 × 10−x, 7 × 10−x, and 9 × 10−x μmol/L; the enzyme inhibition experiment chosen was the same method. In order to find the accurate IC50, the inhibition rate I was plotted against the inhibitor concentration C to a perform linear fit (Table 1) and the results were expressed as mean ± standard error mean (SEM).
In vitro BuChE inhibition test
Five milliliters of venous blood was collected from a rat and kept at room temperature for 1 h and then at 4 °C for 2 h. The supernatant obtained after centrifugation (2000 r/min for 10 min) was used as the butyrylcholinesterase (BuChE) enzyme. The method used used was the same as that used for determining the activity of AChE enzyme, except that the substrate acetylthiocholine iodide was replaced by butyrylthiocholine iodide in the AChE activity measurement (Table 2).
From Table 1, it can be seen that 10 of 13 target compounds had certain acetylcholinesterase inhibitory activity in vitro. Among them, compound 5g showed better AChE inhibitory activity with IC50 = 5.84 μmol/L, and the activity was higher than that of the reference compound, rivastigmine, but lower than that of huperzine-A. From Tables 1 and 2 it can be seen that the compound having a better inhibitory effect on acetylcholinesterase had almost no inhibitory effect on BuChE.
A careful insight and analysis of the AChE inhibition activity results presented in Tables 1 and 2 led to two eventual SAR. By comparing the structure and bioactivity of the compounds, it was found that: (1) the difference of RX (X = 1, 2) and R3 had an important effect on the activity of compounds. When R2 was 4-amino-1-benzylpiperidinyl, the activity is better than other groups and that (2) the difference of -(CH2)n- (n = 1, 2, 3, 4, 5) had an important effect on the activity of compounds. When carbon chain length was -(CH2)2-, the activity is better than others. It was shown that the group of (-NH-CO-CH2-CH2-NH-) was necessary for the active compound.
Kinetic analysis of lead AChEI 5g
To determine the inhibition type of these compounds against electroporated AChE, a kinetic study was carried out with inhibitor 5g as the representative AChEI. In this process, the concentrations of inhibitors 0.5 × IC50, IC50, and 2 × IC50 were 2.92, 5.84, and 11.68 μmol/L, respectively. This type of inhibition was established from the analysis of Lineweaver–Burk reciprocal plots (Fig. 3).

Compound 5g V–[S] and double-reciprocal V−1–[S]−1
It can be found from V (reaction rate)–[S] (substrate concentration) (Fig. 3a) that at the same inhibitor concentration, V increased as the [S] increased; at the same [S], V decreased as the concentration of the inhibitor increased, which indicated that there was a reversible mode of enzyme inhibition. According to the characteristics of enzymatic kinetics, the double-reciprocal curve of the competitive inhibition type is on the Y-axis; the double-reciprocal curve of the non-competitive inhibition type is on the negative semi-axis of the X-axis; the double-reciprocal curve of the mixed inhibition type is assigned to the second quadrant. The double-reciprocal V−1–[S]−1 curve of compound 5g intersected in the second quadrant, which indicated that there was a mixed inhibition type (Cavdar et al. 2019) (Fig. 3b).
In silico molecular property analysis and ADMET prediction
Unfavorable ADME and toxicity properties have been identified as the major cause of failure for new drug molecules moving towards the higher phases of drug development (Zhong et al. 2018). Thus, with the objective of increasing the success rate of compounds reaching development, in silico molecular property analysis and ADMET prediction studies were performed using Molinspiration software 2015 (www.molinspiration.com) online property calculation toolkit and PreADMET online server (www.preadmet.bmdrc.org), respectively.
All the structures reported herein show suitable MW values (MW < 500) necessary for a successful penetration to central nervous system, with the exception of compounds 6c, 6d, and 6e. All compounds exhibit moderate lipophilicity (expressed as MiLogP (octanol–water partition coefficient)), except 5j, 6c, 6d, and 6e. The number of hydrogen bond acceptors and hydrogen bond donors for all the compounds were in accordance with the Lipinski’s rule of five (Lipinski et al. 2012). Moreover, the topological polar surface area for all the compounds was observed to fall within the range. In addition, the predicted values for all compounds fall into the appropriate range, indicating good bioavailability of the ligand molecules, except 5j, 6c, 6d, and 6e (Table 3). Thus, it was predicted that most of the compounds are likely to be orally active.
In addition, the results of in silico ADMET properties were listed in Tables 4 and 5 (Lipinski et al. 2012).
Human intestinal absorption (HIA) property is the determinant for those drugs that purport oral administration. All the compounds expressed >70% HIA values, indicating good permeation across the membrane, which exhibited moderate permeation.
The Caco-2 cell permeability in vitro is an important parameter to assess intestinal absorption of the drug. The results indicated that all compounds exhibited moderate permeation (Table 4).
The MDCK cell permeability test in vitro utilizes canine kidney cells for the analysis of permeability. All the compounds 5a–6e showed permeation <25 nm/s, indicating low permeability.
The skin permeability is an important factor for the delivery of drug via transdermal administration. All the compounds 5a–6e exhibited negative permeability values, which indicated that transdermal mode of administration was not the suitable means to administer these drugs.
In addition, apart from the compounds 5a, 5e, 5f, 5g, 5j, and 6a, all the compounds were predicted to possess >90% plasma protein binding (PPB), indicating decreased excretion and increased half-life.
The Ames test assesses mutagenicity of the compounds and all the compounds were predicted as mutagen. Moreover, carcinogenicity test was performed to identify the tumerogenic possibility of compounds in animals (mouse and rat). Analyzing carcinogenicity in animals (mouse and rat), all the compounds were predicted as negative. Human ether-a-go-go-related gene (hERG) encodes potassium channels, which are responsible for normal repolarization of cardiac action potentials (Macdonald et al. 2018). Blockage or other impairment of these channels in the heart can lead to fatal cardiac problems, so it is a major concern with regard to drug-induced blockage of potassium channels. Compounds 6b, 6c, and 6d presented low risk; 5a, 5b, 5c, 5d, 5e, 5j, 6a, and 6e presented medium risk; compounds 5f and 5g presented high risk (Table 5).
In summary, although other compounds showed good characteristics, it can be concluded that the lead AChEI compound 5g presented satisfactory drug-like characteristics and ADME properties.
Docking studies
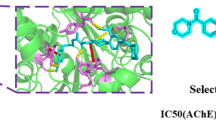
In order to achieve the binding mode of the synthesized compounds and provide more insights into the interactions, the molecular docking was conducted to investigate the potential binding mode of compound 5g with the catalytic domain of AChE (PDB: 4EY7) using Autodock 4.2. The complex of protein with best docked pose of inhibitor showed that the docked ligand was almost superimposed on the native co-crystallized ligand donepezil (Cheung et al. 2012) (Fig. 4). In this position, the 4-methoxythiazole ring moiety oriented toward the bottom of the active site similar to the benzylpiperidine moiety of ligand donepezil. As represented in Fig. 4, a closer look at the binding mode showed that the 4-methoxyphenylthiazole ring adjusted in parallel with indole ring of Trp-286 and made π–π interaction. The amide group also fitted in the midgorge of AChE active site and bound to the active site via hydrogen bonding interaction with the amino group of Phe-295 residue. Moreover, the benzylpiperidine amino ion and benzyl moiety of the compound 5g also established amino ion and π of benzene ring interaction with Tyr-337 and another π–π stacking interaction with Trp-86 in the peripheral anionic site, respectively.

Three-dimensional representation of best pose of compound 5g obtained from docking simulation in the active site of AChE
Conclusion
The designed 4-methoxy-phenylthiazole-2-amine derivatives as AChEIs were successfully synthesized, and their inhibitory activity test results showed that most of 4-methoxy-phenylthiazole-2-amine derivatives had a certain acetylcholinesterase inhibitory activity in vitro; moreover, their IC50 values were in micromolar range. Specially, 3-((1-benzylpiperidin-4-yl)amino)-N-(4-(4-ethoxyphenyl)-thiazol-2-yl)propanamide (5g) showed a strong inhibitory activity against AChE (IC50 = 5.84 ± 0.01 μmol/L), which was found to be ~0.83-fold active compared to huperzine-A and ~2.02-fold stronger compared to rivastigmine. Kinetic studies revealed that compound 5g exhibited a mixed type and reversible mode of enzyme inhibition. Molecular docking stimulation of the representative compound 5g performed using Autodock 4.2 achieved the binding mode of the synthesized compounds with AChE and provided more insights into the interactions. Furthermore, the representative compound 5g basically conformed to four-point pharmacophore model, which is essential for the AChE inhibition. Finally, it can be concluded that the lead AChEI compound 5g presented satisfactory drug-like characteristics and ADME properties by in silico molecular property analysis and ADMET prediction. In summary, the compound 5g having excellent acetylcholinesterase inhibitory activity in vitro was found in this study, while the compound had good selectivity, and structural modifications can be further made on the basis of compound 5g to seek compounds having higher inhibitory activity against acetylcholinesterase. These results encourage medicinal chemists to explore more structures based on 4-methoxy-phenylthiazole-2-amine derivatives for high-efficiency cholinesterase inhibitor.
References
Ibrar A, Khan A, Ali M, Sarwar R, Mehsud S, Farooq U, MA Halimi S, Khan I, Al-Harrasi A (2018) Combined in vitro and in silico studies for the anticholinesterase activity and pharmacokinetics of coumarinyl thiazoles and oxadiazoles Front Chem 6:61–72
Atri A, Frölich L, Ballard C, Tariot PN, Molinuevo JL, Boneva N, Windfeld K, Raket LL, Cummings JL (2018) Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease. JAMA 319:130–142
Bag S, Tulsan R, Sood A, Datta S, Torok M (2013) Pharmacophore modeling, virtual and in vitro screening for acetylcholinesterase inhibitors and their effects on amyloid-β self- assembly. Curr Comput Aided Drug Des 9:2–14
Cheung J, Rudolph MJ, Burshteyn F, Cassidy MS, Gary EN, Love J, Frankin MC, Height JJ (2012) Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J Med Chem 55:10282–10286
Grozeva D, Saad S, Menzies GE, Sims R (2019) Benefits and challenges of rare genetic variation in Alzheimer’s disease Curr Genet Med Rep 7:53–62
Zhong F, Xing J, Li X, Liu X, Fu Z, Xiong Z, Lu D, Wu X, Zhao J, Tan X, Li F, Luo X, Li Z, Chen K, Zheng M, Jiang H (2018) Artificial intelligence in drug design Sci China Life Sci 61:59–72
Hiremathad A, Chand K, Keri RS (2018) Development of coumarin and benzofuran conjugated hybrids as the versatile multi-targeted compounds for the treatment of Alzheimer’s disease. Chem Biol Drug Des 92:1497–1503
Cavdar H, Senturk M, Guney M, Durdagi S, Kayik G, Supuran CT, Ekinci D (2019) Inhibition of acetylcholinesterase and butyrylcholinesterase with uracil derivatives: kinetic and computational studies J Enzyme Inhib Med Chem 34:429–437
Kaushik V, Smith ST, Mikobi E, Raji MA (2018) Acetylcholinesterase inhibitors: beneficial effects on comorbidities in patients with Alzheimer’s disease. Am J Alzheimers Dis Other Demen 33:73–85
Yurttaş L, AsımKaplancıklı Z, Özkay Y (2013) Design, synthesis and evaluation of new thiazole-piperazines as acetylcholinesterase inhibitors J Enzyme Inhib Med Chem 28:1040–1047
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2012) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 64:4–17
Lopes RM, Filho MVS, Salles JBD, VLFC Bastos, Bastos JC (2014) Cholinesterase activity of muscle tissue from freshwater fishes: characterization and sensitivity analysis to the organophosphate methyl-paraoxon. Environ Toxicol Chem 33:1331–1336
Macdonald LC, Kim RY, Kurata HT, Fedida D (2018) Probing the molecular basis of hERG drug block with unnatural amino acids. Sci Rep 8:289–298
Abbas-Mohammadi M, Moridi Farimani M, Salehi P, Nejad Ebrahimi S, Sonboli A, Kelso C, Skropeta D (2018) Acetylcholinesterase-inhibitory activity of Iranian plants: combined HPLC/bioassay-guided fractionation, molecular networking and docking strategies for the dereplication of active compounds. J Pharm Biomed 158:471–479
Ma ZY, Yang Q, Zhang YG, L JJ, Yang GL (2014) Design, synthesis and evaluation of N-acyl-4-phenylthiazole-2-amines as acetylcholinesterase inhibitors. Acta Pharm Sin 49:813–818
Pepeu G, Grazia Giovannini M (2017) The fate of the brain cholinergic neurons in neurodegenerative diseases. Brain Res 1670:173–184
Zhang Q, Zhao H, Liu W, Zhang Z, Qin H, Luo F, Leng S (2016) Developmental perfluorooctane sulfonate exposure results in tau hyperphosphorylation and β-amyloid aggregation in adults rats: incidence for link to Alzheimer’s disease Toxicology 347–349:40–46
Schelke MW, Attia P, Palenchar DJ, Kaplan B, Mureb M, Ganzer CA, Scheyer O, Rahman A, Kachko R, Krikorian R, Mosconi L, Isaacson RS (2018) Mechanisms of risk reduction in the clinical practice of Alzheimer’s disease prevention. Front Aging Neurosci 10:96–109
Tian YF, Chen JT, Li JJ, Zhang YC, Cao TT, Ma ZY (2015) Design, synthesis and evaluation of new l-proline as acetylcholinesterase inhibitors. Acta Pharm Sin 50:719–724
Tripathi RKP, M Sasi V, Gupta SK, Krishnamurthy S, Ayyannan SR (2018) Design, synthesis, and pharmacological evaluation of 2-amino-5-nitrothiazole derived semicarbazones as dual inhibitors of monoamine oxidase and cholinesterase: effect of the size of aryl binding site. J Enzyme Inhib Med Chem 33:37–57
Acknowledgements
This work was supported by grants from Natural Science Foundation of Hebei Province (No. H2017201075).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, XZ., Xu, Y., Jian, MM. et al. Synthesis, in vitro assays, molecular docking, theoretical ADMET prediction, and evaluation of 4-methoxy-phenylthiazole-2-amine derivatives as acetylcholinesterase inhibitors. Med Chem Res 28, 1683–1693 (2019). https://doi.org/10.1007/s00044-019-02405-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-019-02405-6