
Abstract
A series of sixteen curcumin derivatives (CURC-D) were designed and evaluated for their antibacterial/antifungal activity. POM analyses showed that lipophilicity and presence of (X–Y) pharmacophore site (X, Y = O, N) are the major factors that governed the orientation in determining antibacterial and/antifungal activity. Furthermore, it was also found that some of the POM analyzed CURC-derivatives have a closed pharmacophore sites which might be responsible of low bioactivity. To confirm the electronic, steric, and hydrophobic requirements for future modifications, we have also carried out receptor-based electrostatic analysis. Therefore, we conclude that POM analyses may prove to be a suitable method to correlate structural features of CURC-D with their promising combined antibacterial/antifungal activity and may contribute to the development of novel antimicrobial agents against drug-resistant human pathogens.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Curcumin and its derivatives (CURC-D) have been extensively studied in past due to their promising activity as protectors in cardiovascular (Wongcharoen and Phrommintikul, 2009), Alzheimer’s and Parkinson’s diseases (Maher et al., 2010; Shen et al., 2005; Gomez et al., 2007 and Ringman et al., 2005), and cancer inhibition (Lopez-Lazaro, 2008; Aggarwal et al., 2003; Patel et al., 2008; Anand et al., 2008; Lin and Lin, 2008). These derivatives also used as antibacterial, fungi (Sahu et al., 2012), antiviral (Rai et al., 2008), antidiabetic (Kuhad and Chopra, 2007), anti-inflammatory (Chattopadhyay et al., 2004 and Chainani-Wu, 2003), and antioxidant (Sharma, 1976; Balogun et al., 2003). Examples of success involving the Curcumin family which attracted our attention are CNBOO1 as protector/activator of memory, and chicoric acid and 3,5-dicaffeoylquinic acid (Fig. 1) are two specific selective HIV-Integrase inhibitors (Santo et al., 2003).

Some important curcumin derivatives with attractive industrial applications
Although CURC-D have been used in different biological evaluations by various groups around the world while in all these high experimental screening, POM bioinformatics approaches like petra/osiris/molinspiration (POM) analyses to predict molecular properties for these compounds with multi potential bioactivity were missing throughout in previous studies. Once again and curiously, little attention has been paid to the identification of type of pharmacophore sites of CURC-D, especially the bioavailability and toxicity risks of the series 1–16 (Scheme 1) have never been established.

Synthesis of curcumin derivatives 1–16
For our group, it becomes necessary to elucidate the origin of this poly antibacterial/antifungal/antiviral/antitumor/antioxidant properties of CURC-D drugs and to establish the real link among such activities. It is evident for our Computational group that this poly-activity can be generated from CURC-D tautomerism/isomerism/conformism or co-existence of two independent pharmacophore sites (Hadda et al., 2013; Sheikh and Hadda, 2013; Chohan et al. 2010) in the same molecule. Most of drugs are the subject of pharmacological properties change in solution. The bioinformatist/pharmacologist should take in consideration the resulting principal active tautomer/isomer/conformer which is the real responsible of bioactivity not always the parent molecule. To clarify and support this hypothesis, we have analyzed, for example, the series of CUTC-D 1-16 (Scheme 1).
Chemistry
Synthesis of compounds
In our previous communication, we have report the novel modification on two free carbonyl moieties (4H-Pyrimido[2,1-b]Benzothiazole and pyrazole derivatives, 1–8 and 9–12), and active methylene (benzylidene derivatives, 13–16) of curcumin, respectively (Sahu et al. 2012). Curcumin allows for the exploration of various curcumin derivatives 1–16 (Scheme 1).
Pharmacology
Antimicrobial activity of 1–16
In the present study, ciprofloxacin (CIPRO) and fluconazole (FLUC) were used as standard for antibacterial and antifugal activity, respectively. Results of antimicrobial activity were compared with these standard drugs and parent molecule (curcumin). Activities of these compounds have been executed on MH agar plates and it was done on bacteria Staphylococcus aureus (ATCC 11632), Pseudomonas aeruginosa (ATCC 15499), Salmonella typhi (ATCC 23564), Escherichia coli (ATCC 35218), Bacillus cereus (MTCC 7350), and Providencia rettgeri (Table 1). The antifungal activity of CURC-D 1-16 has been evaluated against three isolated fungi viz. A. niger, A. fumigates, and A. flavus (Table 2).
Compound 11 with (R = isoniazide moiety) showed maximum activity against S. aureus (1.25 μM/mL) and P. aeruginosa (1.25 μM/mL) and compound 12 (R = 2,4-dinitro phenyl) showed excellent activity against both S. aureus and B. cereus bacteria. From the Table 1, it is clear that thiosemicarbazide (entry 10) showed promising antibacterial impact on all tested bacterial strains except E. Coli. Of the 16 CURC-D 1-16 screened for antibacterial activity, the compounds 14 and 15 were found to exhibit good antibacterial activity (Table 1). This can be easily explained by the in situ formation of antibacterial [metal-(O,O-ligand)-(diaquo)] complexes (Hadda et al., 2009).
Interestingly, we note from the screening results (Tables 1, 2) that both electron donating and electron withdrawing R groups on the phenyl ring of the compounds 1–8 influenced the antibacterial activity. However, overall screening revealed that hydroxyl derivatives (derivatives 6 and 11) showed the better antibacterial activity against most of tested bacterial strains. The antifungal screening results of 1–8 also suggest that the tested compounds have shown mild to moderate activity against A. niger only but no significant activity against other two fungi strains (Table 2), compared to the standard employed. These teaches us that the acidic milieu of bacteria is responsible for something (hydrolysis of drugs).
The data of Tables 1 and 2 revealed that a comparison of the antibacterial and antifungal activity of all compounds 1–16 follow the order given in Table 3.
Curcumin derivatives have emerged as nonsteroidal anti-inflammatory drugs but also for their chemopreventative properties; however, the mechanisms of action are only a beginning to be investigated rigorously. These computational POM studies were executed to investigate the molecular properties and mechanisms by which curcumin derivatives inhibit both bacteria and viruses, using the (X–Y) pharmacophore site as representative model.
What hydrogen bonds may be found within structures 1–16?
Petra/osiris/molinspiration analysis (POM) is one of the well-known approach that has been used regularly to produce the two dimensional models to identify and to indicate the type of pharmacophore site that affects biological activity with a change in the chemical substitution (Hadda et al. 2013; Parvez et al., 2010a, b; Jarrahpour et al. 2012). The advantages of POM are the ability to predict the biological activities of the molecules and to represent the relationships between steric/electrostatic property as well as biological activity in the form of pharmacophore site, which gives key features on not only the ligand-receptor interaction, but also on the topology of the receptor. Hence to find out the structural features for the bacterial inhibitory activity, we have carried out POM analyses of series 1–16.
Results and discussion
Tautomerism study of 1–16
Tautomerism is an important and under-appreciated phenomenon in the drug design process. Therefore, the purpose of this study is important and has a potential to improve how the descriptor-based POM analyses are performed. Our study is unique, as we have to choose a simplistic approach to the problem. Tautomers equilibria in homologous structures depend on structure and the fractions of individual tautomer in the equilibrium mixture. It will vary from compound to compound in the set. These fractions are a key component in the correct bioactivity attribution. So we suggest that the study is re-worked with the multi-species formalism (Figs. 2, 3, 6).

Curcumin and tautomerism

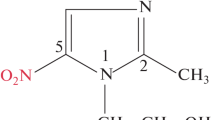
Possible tautomerism of series 9–12
The DFT and TD-DFT calculations (Shen and Ji, 2007) support that curcumin exists predominantly in enol form in solution. Secondly, the calculated absorption spectra of curcumin anions provide direct evidence that the lowest pK of curcumin corresponds to the dissociation of enolic proton, which not only reconciles the controversy on this topic, but also has important implications on the proton-transfer/dissociation-associated radical-scavenging mechanisms of curcumin (Fig. 2).
The 1,3-diketo tautomerism with majority keton/enol form is observed for example in the CURC (Figs. 2), constituting precursor of compounds 1–16. This fact is also observed with derivatives 9–12 (Figs. 3). In contrast to series 9–12, the two series 1–8 and 13–16 represent more rigid structures (Fig. 4).

Rigid structures of series 1–8 and 13–16
Molecular properties calculations
The objective of this study is to investigate the problematic of the potential pharmacophore sites of 1–16 species using antibacterial and antifungal screenings dependence on pH and comparison with the calculated molecular properties.
Pi-charge calculations
The CURCUM-D series 1–16 have been subjected to delocalised-charge calculations using Petra method of the non-hydrogen common atoms (Fig. 5), obtained from the partial pi-charge of the heteroatoms, has been used to model the bioactivity against bacteria and fungi.

Identification of pharmacophores sites of series 1–16
It is found that the negative charges of the nitrogen of N–N group and amide and thioamide groups CXN-H (X = O, S) contribute positively in favor of an antibacterial activity, more, and this is in good agreement with the mode of antibacterial action of the compounds bearing (Xδ−–Yδ+) pharmacophore site. It was hypothesized that difference in charges between two heteroatoms of the same dipolar pharmacophore site (Xδ−–Yδ+) may facilitate the inhibition of bacteria, more than viruses. The same theoretical approach has been recently used to evaluate the antifungal properties of various drugs. In contrast to first antibacterial pharmacophore site (Xδ−–Yδ+), it was verified that the same negative charge of terminal heteroatoms led to potential antiviral/antifungal pharmacophore site (Xδ−–Yδ−) (Parvez et al., 2010a, b; Jarrahpour et al., 2012; Hadda et al. 2012a, b).
It is further observed that the activity increases with increase in negative charge of one heteroatom of the common pharmacophore fragment of the potential tautomers. On the basis of the analog system described above, in compounds 1–16, sets of isomeric and tautomeric 9–12 tautomers 01–02 could be generated in situ in the presence of bacteria or fungi. This synergistic and streamlined working procedure led to highly active isomeric/tautomeric Gram (±) and fungi receptor ligands. However, a little difference in their respective binding affinities was consistently found for all tautomeric pairs 01–02. The analysis of conformational differences due to heteroatom interactions in tautomers 01–02 revealed a favorable (CONH2–N) interaction in tautomer-01, whereas conformet/tautomer-01 showed a repulsive (C=O/C=N) repulsion.
In contrast to the apparent good in general bioactivity of series 9–12, this can be satisfactorily explained by tautomerization/resonance stabilization. The origin of bioactivity of series 13–16 is a mystery query. How series 13–16 could be active without any apparent (Xδ+–Yδ−) or (Xδ−–Yδ−) pharmacophore site? How does this series work? How to control its bioactivity? Which type of bioactivity is dominant? Any reply to these important queries will be of great help as guide in further objective screening.
In aqueous media, 2-substituted Imidazo[1,2a]pyrimidines exist as an equilibrium mixture of the 2- and 3-substituted isomers. However, because these species cannot be detected simultaneously, the variation of the intensities of these species with pH cannot be estimated accurately concurrently. Thus, accurate estimation of the pKa of these amides is not possible.
The Dimroth rearrangement is an opening/closing ring taking place with certain heterocyclic compounds where endocyclic and exocyclic nitrogen atoms switch place. This in situ pharmacophore site regeneration was discovered since 1902 by Otto Dimroth (Dimroth, 1902, 1909; Dimroth and Michaelis, 1927). Unfortunately, this important chemical process was taken in considerations by few and limited some chemists/biologists (Rozhkov et al., 2004; Lauria et al., 2008; Cao and Wang 2009). Among this missing estimation from part of chemists, the Dimroth rearrangement is ignored or not enough explored in drug design by pharmacologist groups over the world. The Dimroth rearrangement deserves more attention and high consideration than it was done until now because it is the crucial and fundamental piece of pharmaceutical puzzle to interpret, predict, and modulate the bioactivity of various heterocycles as pro-drugs not as like drugs.
Osiris calculations
With our recent publications of the drug design combination of various pharmacophore sites (Parvez et al., 2010a, b; Jarrahpour et al., 2012), it becomes now more easy for us to predict the type of bioactivity of candidate drugs.
From the data evaluated in Table 4 indicate that, all structures are supposed to be non-mutagenic when run through the mutagenicity assessment system and as far as irritating and reproductive effects are concerned, all the compounds are at low risk comparable with standard drugs used (except 12 and 14). The clogP value of a compound, which is the logarithm of its partition coefficient between n-octanol and water, is a well-established measure of the compound’s hydrophilicity. Low hydrophilicity, and therefore high cLogP values may cause poor absorption or permeation. It has been shown for compounds to have a reasonable probability of being well absorb their cLogP value must not be greater than 5.0.
On this basis, all the series of compounds 9–16 is having clogP values under the acceptable criteria should be active. The geometrical parameter and the aqueous solubility of a compound significantly affect its absorption, distribution characteristics, and bioactivity.
Typically, a low solubility goes along with a bad absorption, and therefore the general aim is to avoid poorly soluble compounds. Further, the Table 4 shows drug-likeness of compounds 1–16 which is not in general in the comparable zone with that of standard drugs used (0.10–0.23, 0.04–0.55, and 0.09–0.50, respectively for series 1–8, 9–12, and 13–16; DS of references = 0.39–0.46).
We have calculated overall drug-score (DS) for the compounds 1–16 and compared with that of standard drugs CURC and FLUC used as shown in Tables 4 and 5. The DS combines drug-likeness, clogP, logS, molecular weight, and toxicity risks in one handy value that may be used to judge the compound’s overall potential to qualify for a drug. The reported compounds 1–16 showed 6/16 compounds have good DS but the rest (10/16) of series 1–16 have low to moderate DS as compared with standard drugs used. That indicates that some parameters in drug design should be taken in consideration.
There is one repetitive fundamental error accomplished by many reputed groups involved in computational chemistry and docking, it is the docking and virtual screening of pro-drugs. Is it credible and true to continue to accept that? No, it is time to clarify and to establish that both Docking and virtual screening should be done on metabolites of pro-drugs, not on pro-drug itself. Of course, the transition of pro-drug to its bioactive metabolite(s) should be taken in consideration.
Molinspiration calculations
The method is very robust and is able to process practically all organic and most organometallic molecules. Molecular Polar Surface Area TPSA is calculated based on the methodology previously published (Ertl et al., 2000). O- and N- centered polar fragments are considered. TPSA has been shown to be a very good descriptor characterizing drug absorption, including intestinal absorption, bioavailability, and blood–brain barrier penetration. Prediction results of compounds 1–16 molecular properties (TPSA, GPCR ligand, and ICM) are valued (Table 5).
A number of important points emerged concerning the electronic and steric factors which have direct impact on bioactivity properties (Fig. 6). The positive POM results we have recorded, while encouraging for purposes of new drug design, confirm that very likely most of these compounds could be used as potential antimicrobial agents after major modifications. Based on their structural properties, these compounds may be useful as chelating agents with potential anti-tumoral activity. These results prompt several pertinent observations: (i) This type of CURD can furnish an interesting model for studying the interaction of metal-antibiotics complexes (Fig. 7) with cancer target because the possible charge modification of pi-charge pharmacophore group; (ii) The future flexible O, O/N,N/O,N-pharmacophore site (s) geometric conformation enables us to prepare molecules for multi-therapeutic materials with high antiviral/antifungal activity. It was reported that curcumin and quercetin combined with cis-platin are able to induce apoptosis in human laryngeal carcinoma Hep-2 cells through the mitochondrial pathway (Kuhar et al., 2007). The interest of curcumin-metal complexes as potential antiviral agents have been indicated since 1993 (Sui et al., 1993).

Plausible Dimroth mechanism or opening/closing pyridine ring for 1–8

Molecules designed as new perspectives
Conclusions
In our findings, we have observed that most of the tested CURC-D (series 1–16) showed moderate to high combined antibacterial/antifungal activity against multi-drug-resistant pathogenic microorganisms. Among the synthesized derivatives, 9–12 and 13–16 showed excellent activity against both bacteria and fungus. The results of this virtual screening investigation support the suggested models for antibacterial and antifungal activity; we developed the past 16 years in collaboration with NCI and TAACF of USA. It has been suggested that any modification of central functional group of curcumin present in this natural compound displayed crucial role of biological activity that may be responsible for the observed dual antibacterial and antifungal activity. The aryl group of compounds increases the hydrophobic character and liposolubility of the molecules. This in turn, enhances activity of the compounds and biological absorbance, so as, all the synthesized THBT should have good antibacterial and antifungal properties but the tautomerism in carboxamide/thioamide group to hydroxyimine/thiolateimine generates one other concurrent important (OH–N)/(SH–N) intramolecular interaction with imino group of adjacent position. This has a synergic good and direct impact on availability of pharmacophore site which shows promising bioactivity (Fig. 3). These findings supported the selection of POM analyses to be used as suitable method in determination of pharmacophore sites in relation to bioactivity of designed CURCUM-D series.
On other hand, it was revealed that Cu (II)-curcumin complexes were more active than the parent curcumin in Alzheimer’s disease (AD) by scavenging radicals with donating proton or electron (Shen et al., 2005; Barik et al., 2005). Therefore, the series 9–12 could be multi-potent agents to combat AD, with the activities of scavenging ROS, blocking Ab aggregation, and chelating various transition metals.
References
Aggarwal BB, Kumar A, Bharti AC (2003) Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res 23:363–398
Anand P, Sundaram C, Jhurani S, Kunnumakkara AB, Aggarwal BB (2008) Curcumin and cancer: an ‘‘old-age” disease with an ‘‘age-old” solution. Cancer Lett 267:133–164
Balogun E, Hoque M, Gong P, Killeen E, Green CJ, Foresti R, Alam J, Motterlini R (2003) Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem J 371:887–895
Barik A, Mishra B, Shen L, Mohan H, Kadam RM, Zhang HY, Priyadarsini KI, Dutta S (2005) Evaluation of a new copper(II)–curcumin complex as superoxide dismutase mimic and its free radical reactions. Free Radic Bio Med 39:811–822
Cao H, Wang Y (2009) Fragmentation of isomeric intrastrand cross-link lesions of dna in an ion-trap mass spectrometer. J Am Soc Mass Spectrom 20:611–617
Chainani-Wu N (2003) Safety and anti-inflammatory activity of curcumin: a component of tumeric (Curcuma longa). J Altern Complement Med 9:161–168
Chattopadhyay I, Biswas K, Bandyopadhyay U, Banerjee RK (2004) Turmeric and curcumin: biological actions and medicinal applications. Curr Sci 87:44–53
Chohan ZH, Youssoufi MH, Jarrahpour A, Hadda TB (2010) Identification of inhibition: indolenyl sulphonamide derivatives. Eur J Med Chem 45:1189–1199
Dimroth O (1902) Mittheilungen Ueber eine Synthese von Derivaten des 1.2.3-Triazols. Ber Dtsch Chem Ges 35:1029–1038
Dimroth O (1909) Ueber intramolekulare Umlagerungen. Umlagerungen in der Reihe des 1, 2, 3-Triazols. Justus Liebig’s Annalen der Chemie 364:183–226
Dimroth O, Michaelis W (1927) Intramolekulare Umlagerung der 5-Amino-1,2,3-triazole. Justus Liebig’s Annalen der Chemie 459:39–46
Ertl P, Rohde B, Selzer P (2000) Fast calculation of molecular polar surface area (PSA) as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 43:3714–3717
Gomez G, Mansouraty G, Gardea J, Narayan M (2007) Acceleration of oxidative protein folding by curcumin through novel non-redox chemistry. Biochem Biophys Res Co 364:561–566
Hadda TB, Akkurt M, Baba MF, Daoudi M, Bennani B, Kerbal A, Chohan ZH (2009) Anti-tubercular activity of ruthenium (II) complexes with polypyridines. J Enzym Inhib Med Chem 24:457–463
Hadda TB, Mouhoub R, Jawarkar R, Masand V, Warad I (2012a) POM analyses of antitrypanosomal activity of 2-iminobenzimidazoles: favourable and unfavourable parameters for drugs optimization. Med Chem Res. doi:10.1007/s00044-012-0238-0
Hadda TB, Fergoug T, Warad I (2012b) POM theoretical calculations and experimental verification of antibacterial potential of 5-hydroxy-4-(substituted-amino)-2(5H)-furanones. Res Chem Intermed. doi:10.1007/s11164-012-0729-0
Hadda TB, Ali MA, Masand V, Gharby S, Fergoug T, Warad I (2013) Tautomeric origin of dual effects of N1-nicotinoyl-3-(4′-hydroxy-3′-methyl phenyl)-5-[(sub)phenyl]-2-pyrazolines on bacterial and viral strains: POM analyses as new efficient bioinformatics’ platform to predict and optimize bioactivity of drugs. Med Chem Res 22:1438–1449
Jarrahpour A, Fathi J, Mimouni M, Hadda TB, Sheikh J, Chohan ZH, Parvez A (2012) Petra, osiris and molinspiration (POM) together as a successful support in drug design: antibacterial activity and biopharmaceutical characterization of some azo schiff bases. Med Chem Res 21:1984–1990
Kuhad A, Chopra K (2007) Curcumin attenuates diabetic encephalopathy in rats: behavioral and biochemical evidences. Eur J Pharmacol 576:34–42
Kuhar M, Imran S, Singh N (2007) Curcumin and quercetin combined with cisplatin to induce apoptosis in human laryngeal carcinoma Hep-2 cells through the mitochondrial pathway. J Cancer Mol 3:121–128
Lauria A, Abbate I, Patella C, Gambino N, Silvestri A, Barone G, Almerico AM (2008) Pyrazolo[3,4-d][1,2,3]triazolo[1,5-a]pyrimidine: a new ring system through Dimroth rearrangement. Tetrahedron Lett 49:5125–5128
Lin CL, Lin JK (2008) Curcumin: a potential cancer chemopreventive agent through suppressing NF-κB signaling. J Cancer Mol 4:11–16
Lopez-Lazaro M (2008) Anticancer and carcinogenic properties of curcumin: considerations for its clinical development as a cancer chemopreventive and chemotherapeutic agent. Mol Nutr Food Res 52:103–127
Maher P, Akaishi T, Schubert D, Abe K (2010) A pyrazole derivative of curcumin enhances memory. Neurobiol Aging 31:706–709
Parvez A, Meshram J, Tiwari V, Sheikh J, Dongre R, Youssoufi MH, Hadda TB (2010a) Pharmacophores modeling in terms of prediction of theoretical physicochemical properties and verification by experimental correlations of novel coumarin derivatives produced via Betti’s protocol. Eur J Med Chem 45:4370–4378
Parvez A, Jyotsna M, Youssoufi MH, Hadda TB (2010b) Theoretical calculations and experimental verification of the antibacterial potential of some monocyclic beta-lactames containing two synergetic buried antibacterial pharmacophore sites. Phosphorus Sulfur 185:1500–1510
Patel BB, Sengupta R, Qazi S, Vachhani H, Rishi AK, Majumdar APN, Yu Y (2008) Curcumin enhances the effects of 5-fluorouracil and oxaliplatin in mediating growth inhibition of colon cancer cells by modulating EGFR and IGF-1R. Int J Cancer 122:267–273
Rai D, Yadav D, Bamzarini J, De Clercq E, Singh RK (2008) Design and development of curcumin bioconjugates as antiviral agents. Nucleic Acids Symp 52:599–600
Ringman JM, Frautschy SA, Cole GM, Masterman DL, Cummings JL (2005) A potential role of the curry spice curcumin in Alzheimer’s disease. Curr Alzheimer Res 2:131–136
Rozhkov VY, Batog LV, Shevtsova EK, Struchkova MI (2004) Synthesis and Dimroth rearrangement of 3-amino-4-(5-amino-1H-1,2,3-triazol1-yl)-1,2,5-oxadiazoles. Mendeleev Commun 14:76–77
Sahu PK, Gupta SK, Thavaselvam D, Agarwal DD (2012) Synthesis and evaluation of antimicrobial activity of 4H-pyrimido[2,1-b]benzo. Eur J Med Chem 54:366–378
Santo RD, Costi R, Artico M, Tramontano E, Colla PL, Pani A (2003) HIV-1 integrase inhibitors that block HIV-1 replication in infected cells. Planning synthetic derivatives from natural products. Pure Appl Chem 75:195–206
Sharma OP (1976) Antioxidant activity of curcumin and related compounds. Biochem Pharmacol 25:1811–1812
Sheikh J, Hadda TB (2013) Antibacterial, antifungal and antioxidant activity of some new water-soluble b-diketones. Med Chem Res 22:964–975
Shen L, Ji HF (2007) Theoretical study on physicochemical properties of curcumin. Spectrochim Acta A 67:619–623
Shen L, Zhang HY, Ji HF (2005) A theoretical study on Cu(II)-chelating properties of curcumin and its implications for curcumin as a multipotent agent to combat Alzheimer’s disease. J Mol Struct THEOCHEM 757:199–202
Sui Z, Salto R, Li J, Craik C, Ortiz de Montellano PR (1993) Inhibition of the HIV-1 and HIV-2 proteases by Curcumin and curcumin Boron Complexes. Bioorg Med Chem 1: 415–422
Wongcharoen W, Phrommintikul A (2009) The protective role of curcumin in cardiovascular diseases. Int J Cardiol 133:145–151
Acknowledgments
The authors would like to extend their sincere appreciation to the Deanship of Scientific at King Saud University for its funding of this research through the Research Group Project no RGP-VPP-222.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Youssoufi, M.H., Sahu, P.K., Sahu, P.K. et al. POM analyses of antimicrobial activity of 4H-pyrimido[2,1-b]benzothiazole, pyrazole, and benzylidene derivatives of curcumin. Med Chem Res 24, 2381–2392 (2015). https://doi.org/10.1007/s00044-014-1297-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-014-1297-1