
Abstract
Design and synthesis of some pyrazole derivatives 4–11 of expected anti-inflammatory and analgesic activities. In addition, docking of the tested compounds into cyclooxygenase II using (MOE) was performed to rationalize the obtained biological results and their mechanism of action. The structures of the new compounds were elucidated by spectral and elemental analyses. All the newly synthesized compounds were evaluated for their anti-inflammatory activity using the carrageenan-induced rat paw edema method. Analgesic activity of the target compounds was measured using the p-benzoquinone writhing-induced method, and their ability to induce gastric toxicity was also evaluated. Results showed that the newly synthesized compounds exhibited weak to good activities compared to ibuprofen and celecoxib as reference drugs. Some compounds, such 4a and 11b exhibited significant anti-inflammatory activity with gastric ulcerogenic potential less than that of ibuprofen. Results of the analgesic activity showed that compounds possessing good anti-inflammatory activity showed also good analgesic. Substitution of pyrazole ring with at least one aryl moiety was found to be essential for anti-inflammatory and analgesic activities. Free NH (of pyrazole ring) and/or acidic group (COOH) will improve the anti-inflammatory activity.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammation is a normal and essential response to any noxious stimulus, which threatens the host and may vary from a localized response to a more generalized one (Fylaktakidou et al., 2004). The inflammatory process is designed to provide a rapid mechanism by which the host can respond to the invasion of foreign materials and return to homeostatic equilibrium. Acute inflammation is mediated by the release of autacoids such as histamine, serotonin, bradykinin, prostaglandins, and leukotrienes (Lacerda et al., 2009). On the other hand, the chronic inflammatory process involves the release of diverse mediators, as interleukins, interferon, and tumor necrosis factor α (TNF-α), a cytokine that plays a major role in this kind of inflammatory process and whose production is associated with some inflammatory diseases such as rheumatoid arthritis, Crohn’s disease, and others (Lacerda et al., 2009).
Non-steroidal anti-inflammatory drugs (NSAIDs) are one of the most useful clinical therapies for the treatment of pain, fever, and inflammation (Laufer and Luik, 2010). The major mechanism by which NSAIDs exert their anti-inflammatory activity is by the inhibition of cyclooxygenase-derived prostaglandin synthesis, which is also responsible for the gastrointestinal (Sostres et al., 2010; Naesdal and Brown, 2006; Cryer, 2005; Lazzaroni and Bianchi Porro, 2004; James and Hawkey, 2003), renal (Schneider et al., 2006; Mounier et al., 2006; Zadrazil, 2006), and hepatic (Yanai et al., 2009) side effects that are observed mainly in chronic use of NSAIDs. Therefore, the challenge still exists for the pharmaceutical industry to develop effective anti-inflammatory agents with enhanced safety profile. Literature survey revealed that many 2-pyrazoline derivatives have been reported to exhibit various pharmacological activities such as antimicrobial (Karthikeyan et al., 2007; Ramiz et al., 2010), anti-inflammatory (Barsoum et al., 2006; Shoman et al., 2009; Rathish et al., 2009; Abbas et al., 2010), and antihypertensive (Turan-Zitouni et al., 2000). Among the highly marketed COX-2 inhibitors that comprise the pyrazole nucleus, celecoxib is the one which is treated as a safe anti-inflammatory and analgesic agent. Some examples of pyrazole derivatives as NSAIDs are mefobutazone, morazone, famprofazone, and ramifenazone (Reynold, 1993; Menozzi et al., 2003; Sakya et al., 2007; Patel et al., 2004).
Motivated by the aforementioned findings, it was designed to synthesize novel series of pyrazole derivatives that would act as anti-inflammatory agents with reduced gastric toxicity. The synthesized compounds were tested in vivo for their anti-inflammatory and analgesic activities as well as their ulcerogenic liability. In order to rationalize the pharmacological results obtained and the mechanism of action, MOE (Molecular Operating Environment, 2005) docking studies of the synthesized compounds were performed using murine cyclooxygenase-2 co-crystallized with celecoxib (Protein Data Bank code PDB ID: 6COX) as a template.
Result and discussion
Chemistry
The synthesis routes for the preparation of the target compounds were illustrated in Schemes 1, 2, 3, and 4. Synthesis of the appropriate 1-(2,4-dimethoxyphenyl)alkyl/aryl ketones 2a and 2b was carried out via the reaction of 1,3-dimethoxybenzene with propionic acid or phenyl acetic acid in polyphosphoric acid (Slotta and Heller, 1930). The structure of 2b was confirmed by appearance of a singlet at δ 4.29 ppm corresponding to methylene protons. Compounds 3a and 3b were synthesized from 2a and 2b, respectively, using Mannich reaction/elimination sequence. 1H-NMR spectrum of 3a had two doublets due to geminal coupling at δ 2.93 (J: 7.4 Hz) and 3.04 ppm (J: 7.4 Hz) corresponding to the methylene protons and that of 3b appeared at δ 4.21 (J: 8.2 Hz) and 4.86 ppm (J: 8.2 Hz). Moreover, cyclocondensation of 3a and 3b with hydrazine hydrate yielded the corresponding pyrazoline derivatives 4a and 4b. IR spectra of the prepared pyrazolines 4a and 4b showed υ (C=N) stretching at 1609–1604 cm−1 due to the ring closure. In addition, a sharp band at 3422–3393 cm−1 due to the υ (NH) stretching was also observed. 1H-NMR spectra recorded for the prepared compounds in CDCl3 clearly supported the proposed structures. Protons of pyrazoline ring in compounds 4a and 4b showed a prominent ABX system, with protons Ha, Hb, and Hx seen as doublets of doublets at δ 2.49–2.80, 3.39–3.81, and 5.00–5.10 ppm (JHa–Hb: 4.2–4.4, J Ha–Hx: 7.4–8.2, J Hb–Hx: 7.6 Hz), respectively. On the other hand, Hx of 4a appeared as multiplet as a result of CH3 substituent (Scheme 1).

Reagents and solvents: a RCH2COOH/PAA, b HCHO/piperdine/acetic acid, and c NH2NH2·H2O/ethanol

Reagents and solvents: a toluene

Reagents and solvents: a CH3CN, 0°C

Reagents and solvents: a (COOC2H5)2/NaH/toluene, b R’C6H4NHNH2·HCl/triethylamine/ethanol, and c KOH/methanol
Coupling of 3,4-disubstituted pyrazolines 4a and 4b with sulfonylated carbamic acid methyl esters 5a and 5b (Lange et al., 2004) afforded 6a–d. The structures of the prepared compounds were confirmed by the appearance of an additional strong absorption band of (C=O) stretching at υ 1683–1656 cm−1 in addition to absorption bands at υ 1380–1327 and 1163–1155 cm−1 that were attributed to (SO2) stretching. Also, 1H-NMR spectra showed an increase in the integration of the aromatic protons compared to the parent pyrazolines 4a and 4b and an additional singlet at δ 2.16–2.21 ppm corresponding to the three protons of the CH3 in compounds 6a and 6c (Scheme 2).
Nucleophilic addition of pyrazoline derivatives 4a and 4b to 4-chlorobenzoyl isothiocyanate 7 (Cho and Shon, 1991) gave the adduct 8a and 8b. IR spectra of 8a and 8b revealed the presence of (C=O) at υ 1669 and 1656 cm−1. 1H-NMR of these compounds showed downfield shifting of the NH proton to δ 6.30 and 6.11 ppm (Scheme 3).
Reaction of propan-1-one derivative 2a or phenyl ethanone derivative 2b with diethyl oxalate in the presence of sodium hydride gave 4-aryl-3-substituted-2,4-dioxobutanoic acid ethyl esters 9a and 9b. IR spectra of these key intermediates 9a and 9b showed three strong absorption bands at υ 1665–1612 and 1730–1712 cm−1 (C=O of ketones and ester). 1H-NMR of the 1,3-diketone structures 9a and 9b showed a signal for the CH proton that was observed as a quartet at nearly δ 6.00 ppm in 9a or as a singlet at δ 6.40 ppm in 9b, in addition to the appearance of ethyl protons at the expected chemical shifts (Scheme 4).
Condensation of 9a and 9b with hydrazine salts in ethanol and triethylamine afforded pyrazol-3-carboxylic acid ethyl esters 10a–d. The structures of the prepared compounds 10a–d were confirmed by the appearance of only one strong absorption band at υ 1738–1717 cm−1 corresponding to the carbonyl ester moiety. 1H-NMR also confirmed the formation of the pyrazole ring from the disappearance of the characteristic signal for the CH proton that was observed in 9a and 9b (Scheme 4). Alkaline hydrolysis of the ester containing compounds 10b and 10d gave the corresponding free carboxylic acid derivatives 11a,b. IR spectra showed a broad absorption band at υ 2950–2830 cm−1 for OH of the carboxylic group. 1H-NMR confirmed the structures of the pyrazole derivatives 11a,b from the appearance of an exchangeable proton at δ 11.00 ppm corresponding to COOH and disappearance of the characteristic pattern of the ethyl protons (Scheme 4). Mass spectra of the newly synthesized compounds were concomitant with their molecular weight.
Pharmacological evaluation
The experimental tests on animals have been performed in accordance with the Institutional Ethical Committee approval, Faculty of Pharmacy, Cairo University.
LD50
LD50 of each compound was determined using Finney’s method (Finney, 1964). Intraperitoneal injection of the tested compounds in doses less than 75 mg/kg body weight failed to kill the mice within 24 h. LD50 celecoxib was 250 mg/kg body weight, and 2b, 3a, 4b, 9a, 9b, 10a, and 10d were 277.5 mg/kg body weight. While of 4a, 6c, 8b, and 10c were 292.5 mg/kg body weight. LD50 of 6b, 6d, and 10b was 247.5 mg/kg body weight, and 3b, 6a, 8a, 11a, and 11b were 232.5 mg/kg body weight. As a result, the chosen dose was 25 mg/kg body weight which is similar to celecoxib and at the same time it is safe (Buck and Osweiter, 1976).
Anti-inflammatory activity (Table 1)
All the newly synthesized compounds 2–11 were evaluated for their anti-inflammatory activity using carrageenan-induced paw edema described by Winter et al. (Winter et al., 1962). The tested compounds and reference drugs ibuprofen and celecoxib were intraperitoneal injected at a dose level of 25 mg/kg, 30 min before carrageenan injection at the right hind paw of albino male rats, the thickness of both paws was measured at different time intervals of 1, 2, 3, and 4 h after carrageenan injection. Anti-inflammatory activity of the tested compounds and reference drugs was calculated as the percentage decrease in edema thickness induced by carrageenan with the following formula (Alam et al., 2009).
where V R represents the mean right paw thickness, V L represents the mean left paw thickness, (V R − V L) control represents the mean increase in paw thickness in the control group of rats, and (V R − V L) treated represents the mean increase in paw thickness in rats treated with the tested compounds. The results listed in Table 1 revealed that some of the synthesized compounds showed moderate to weak anti-inflammatory activity. Compounds 4a and 11b induced considerable anti-inflammatory activity relative to ibuprofen, and celecoxib that reached to 67% after 3 h. Another important observation was the decline in activity of compounds 6d and 8b upon replacement of SO2 by C=O. On the other hand, the triaryl carboxylic acid derivative 11b was found to be twice as active as diaryl analog 11a.
Analgesic activity (Table 2)
All the synthesized compounds as well as celecoxib were tested for their analgesic activity using the p-benzoquinone-induced writhing test in mice (Collier et al., 1968). The results showed that compounds possessing good anti-inflammatory activity induced also good analgesic, except 10b exhibited only analgesic activity (Table 2).
Ulcerogenic effect (Table 3)
The ulcerogenic effect of the most active compounds 3a, 4a, 11b, and the reference drugs (ibuprofen, and celecoxib) was evaluated according to Meshali’s method (Meshali et al., 1983). The ulcer index was calculated according to Robert’s method (Robert et al., 1968). The results showed that compounds 3a, 4a, and 11b exhibited little gastric ulceration; about 50–60% that of ibuprofen. On the other hand, these compounds showed higher gastric ulceration than celecoxib (Table 3).
Molecular modeling study
Molecular docking of celecoxib and the synthesized compounds were performed to rationalize the obtained biological results. Besides, molecular docking studies were helped in understanding the various interactions between the ligand and enzyme active site. Docking studies of the inhibitors were performed by MOE (Molecular Operating Environment, 2005) using murine COX-2 co-crystallized with celecoxib (PDB ID: 6COX) as a template. We performed 100 docking iterations for each ligand, and the top-scoring configuration of each of the ligand–enzyme complexes was selected on energetic grounds.
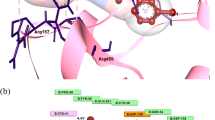
Docking of celecoxib in the active site of murine COX-2 showed hydrogen bonds interactions between the SO2 group and His90 (distances = 2.69 Å). Also, hydrogen bonds were observed between Gln192 and Leu352 with NH2 group (distances = 3.01 and 3.43 Å, respectively). Ten hydrophobic bonds with Val116, Val349 (distance 3.70 Å), Leu352, Leu359, Phe381, Leu384, Try387, Phe518, and Val523 (distance 3.10 Å).
Docking of non pyrazole derivatives 2a,b, 3a,b, and 9a,b revealed the appearance of hydrogen bonds between His90 and the 4-methoxy group of 2b, 3b, 9b, or the carbonyl of the ester group of 9a (distances = 3.48, 2.02, 1.73, and 3.39 Å). Also, hydrogen bonds were detected between Arg120 and 4-methoxy group of 2a and 3a (distances = 2.68 and 3.37 Å). In addition to, the presence of hydrophobic interactions especially with Val523, Leu352 (except 3a), Val349 (except 2b) and Phe518 for 2a, 2b, and 3a.
Concerning 3-(2,4-dimethoxyphenyl)-4-methyl/phenyl-4,5-dihydro-1H-pyrazole 4a,b, it was found that the hydrogen bonds were between Arg120, Leu352, and try355 with the 4-methoxy group, NH group, and 4-methoxy group for 4a or 2-methoxy group for 4b, respectively (distances = 2.64 and 2.68, 2.51 and 2.44, and 3.44 and 3.09 Å for 4a and 4b, respectively). The presence of the hydrophobic interactions with Val349, Val523 were observed, and an extra hydrophobic interaction with Leu352 was found in the case of 4b.
For compounds 6a–d, it was observed the presence of hydrogen bond between Arg120 and 4-methoxy group of compound 6d (distances = 2.38 Å). On the other hand, hydrogen bonds were formed between His90 and SO2 group of 6a, 6b, and 6c or with the carbonyl group of 6d (distances = 3.17, 2.60, 3.68, and 2.67 Å). Also, more hydrogen bonds were found between Arg513 and 4-methoxy group of them (distances = 2.51, 2.52, 3.07, and 3.07 Å for 6a–d). Additional hydrogen bonds were observed between Leu352 and NH group of 6a, 6b, or between Ser530 and NH group of 6c. Besides, the hydrophobic interactions with Val523, Val349, Phe518, Leu352 (except 6a), Ile517 (for 6a, 6b) and Try387 (for 6c).
Thiocarboxamide derivatives 8a,b showed hydrogen bonds between Ser530 and NH group (distances = 2.98 and 2.63 Å). Another bond was detected between His90 and 4-methoxy group of 8b (distance = 3.44 Å). In addition to, the hydrophobic interactions with Val523, Val349, Leu359, and Leu352.
Concerning compounds 10a–d and 11a,b, hydrogen bonds were appeared between Arg120 and 4-methoxy group of 10b, 10d, and 11a (distances = 2.92, 2.86 and 2.75 Å) and between tyr355 and 4-methoxy group of 10a, 10c or 2-methoxy group of 11b (distances = 3.01, 2.78, and 2.68 Å). Also, the same pattern of the hydrophobic bonds with Val523 and Val349 (Figs. 1, 2).

3D Docked structure of compound 4a in the active site of murine COX-2 showed two hydrogen bonds: 4-OCH3 and Arg120 (2.64 Å) and pyrazole N and Leu352 (3.44 Å). Also, two hydrophobic bonds with Val349 and Val523 (3.72 and 3.17 Å, respectively)

3D Docked structure of compound 11b in the active site of murine COX-2 showed four hydrogen bonds: Arg120, Val523, Gly526, and Ala527 (2.31, 3.22, 3.64, and 3.50 Å). Also, six hydrophobic bonds: Val349, Leu352, Ile517, Phe518, Val523, and Leu531 (3.87, 3.87, 3.70, 2.81, 2.71, and 4.32 Å, respectively)
Conformational alignment of celecoxib from the crystal structure of celecoxib-murine COX-2 complex and that of compound 4a from the docking simulation is shown in Fig. 3. The superposition showed overlapping through heterocyclic and one aryl regions. While, that of compound 11b is shown in Fig. 4 showed complete overlapping through heterocyclic and two aryl regions.

Conformational alignment of celecoxib from the crystal structure of celecoxib-murine COX-2 complex and that of compound 4a from the docking simulation

Conformational alignment of celecoxib from the crystal structure of celecoxib-murine COX-2 complex and that of compound 11b from the docking simulation
Conclusions
Various substituted pyrazole derivatives were synthesized and screened for anti-inflammatory, analgesic activities as well as for their ulcerogenic effect. Results showed that compounds 3a, 4a, 6a, 6d, 9a, 11a, and 11b exhibited anti-inflammatory and analgesic activities with the exception of 10b that established only analgesic activity.
SAR: Substitution of the pyrazole ring with at least one aryl moiety is essential for activity. Moreover, the presence of acidic center expressed by NH (of pyrazole ring) “compound 4a” or (COOH) “compound 11b” improves the anti-inflammatory activity. On the other hand, the ulcerogenic effect of 4a and 11b is slightly higher than that of celecoxib due to the primary insult effect.
Experimental
Chemistry
All melting points were determined by the open capillary method using Gallen Kemp melting point apparatus (MFB-595-010M) and were uncorrected. Microanalyses were carried out at the Micro analytical Unit, Faculty of Science, and Cairo University. IR (KBr) was determined using Shimadzu Infrared Spectrometer (IR-435) and FT-IR 1650 (Perkin Elmer). 1H-NMR Spectra were carried using Fourier transform EM-390, 200 MHz NMR Spectrometer and Varian Mercury VX-300 MHz NMR Spectrometer. Mass spectra were carried using Fining SSQ 7000 Gas Chromatograph Mass spectrometer and Shimadzu Gas Chromatograph Mass spectrometer-QP 1000 EX. Nomenclature of presented compounds was according to IUPAC system. Compounds 2a, 5a, and 5b were prepared according to the literature procedures (Slotta and Heller, 1930; Lange et al., 2004; Cho and Shon, 1991), respectively.
1-(2,4-Dimethoxyphenyl)-2-phenyl ethanone 2b
A solution of 1,3-dimethoxybenzene 1 (2.76 g, 20 mmol) and phenylacetic acid (2.72 g, 20 mmol) in polyphosphoric acid (50 ml) was heated in boiling water bath for 30 min. After cooling, the red residue was decomposed by pouring on ice-water and extracted with chloroform. The organic layer was washed twice with water, dried over Na2SO4, and concentrated. The residue was crystallized from MeOH to give 2b (60% yield), mp 72–73°C. 1H-NMR (CDCl3) δ: 3.78 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 4.29 (s, 2H, CH2), 6.43 (s, 1H, ArH-3), 6.53 (m, 2H, Ar), 7.30 (m, 3H, Ar), 7.80 (d, 1H, ArH-5, J: 8.8 Hz), 8.12 (d, 1H, ArH-6, J: 8.8 Hz). IR (KBr) cm−1: 3050 (CH Ar), 2964, 2939, 2834 (CH aliphatic), 1668 (C=O). MS m/z: 256 [M+, 4%], 165 [C6H3(OCH3)2CO, 100%]. Anal. calcd for C16H16O3 (256.20): C 74.98; H 6.29. Found: C 74.90; H 6.20.
1-(2,4-Dimethoxyphenyl)-2-methyl prop-2-en-1-one 3a
To a solution of 1-(2,4-dimethoxyphenyl) propan-1-one 2a (1.95 g, 10 mmol) in MeOH (50 ml), piperdine (0.12 ml, 1.21 mmol), acetic acid (0.12 ml, 2.08 mmol), and formalin (4 ml: 37% aqueous solution, 5.32 mmol) were successively added, and the resulting mixture was refluxed for 4 h. The mixture was concentrated in vacuum. Water was added to the residue, and the mixture was extracted with chloroform. The organic layer was separated, washed with water (3×), dried over Na2SO4, filtered, and concentrated to give 3a (70% yield) as an oil. 1H-NMR (CDCl3) δ: 1.21(s, 3H, CH3), 2.93 (d, 1H, CH, methylene, J: 7.4 Hz), 3.04 (d, 1H, CH, methylene, J: 7.4 Hz), 3.80 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 6.48 (s, 1H, ArH-3), 6.56 (d, 1H, ArH-5, J: 8.8 Hz), 7.86 (d, 1H, ArH-6, J: 8.4 Hz). IR (KBr) cm−1: 3050 (CH Ar), 2934, 2840 (CH aliphatic), 1661 (C=O). MS m/z: 208 [M+ + 2, 1%], 206 [M+, 1%], 165 [C6H3(OCH3)2CO, 100%]. Anal. calcd for C12H14O3 (206.24): C 69.89; H 6.84. Found: C 69.77; H 6.74.
1-(2,4-Dimethoxyphenyl)-2-phenyl prop-2-en-1-one 3b
Compound 3b was prepared from 2b as oil (60% yield) by the same procedure as described for 3a. 1H-NMR (CDCl3) δ: 3.79 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 4.21 (d, 1H, CH, methylene, J: 8.2 Hz), 4.86 (d, 1H, CH, methylene, J: 8.2 Hz), 6.12 (s, 1H, ArH-3), 6.30–7.42 (m, 5H, Ar), 7.59 (d, 1H, ArH-5, J: 8.6 Hz), 8.31 (d, 1H, ArH-6, J: 8.6 Hz). IR (KBr) cm−1: 3030 (CH Ar), 2930, 2840 (CH aliphatic), 1660 (C=O). MS m/z: 268 (M+, 14%), 165 [C6H3(OCH3)2CO, 100%]. Anal. calcd for C17H16O3 (268.31): C 76.10; H 6.01. Found: C 76.37; H 5.81.
3-(2,4-Dimethoxypheny)-4-methyl-4,5-dihydro-1H-pyrazole 4a
A solution of 3a (2.06 g, 10 mmol), hydrazine hydrate (6.0 ml, 120 mmol) in absolute ethanol (50 ml) was refluxed for 3 h. The mixture was concentrated in vacuum; water was added to the residue and extracted with chloroform. The organic layer was twice washed with water, dried over Na2SO4, and concentrated. The residue was oil (70% yield). 1H-NMR (CDCl3-D2O) δ: 1.20 (d, 3H, CH3, J: 4.6 Hz), 2.49 (dd, 1H, CH, methylene, J: 4.2, 7.4 Hz), 3.81 (dd, 1H, CH, methylene, J: 42, 7.4 Hz), 3.86 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 5.00 (m, 1H, CH py), 5.29 (broad, 1H, NH exch.), 6.51 (s, 1H, ArH-3), 6.96 (d, 1H, ArH-5, J: 8.2 Hz), 7.45 (d, 1H, ArH-6, J: 8.2 Hz). IR (KBr) cm−1: 3393 (NH), 3050 (CH Ar), 2936, 2836 (CH aliphatic), 1609 (C=N). MS m/z: 220 [M+, 14%], 177 [C6H3 (OCH3)2CH=NNH, 100%]. Anal. calcd for C12H16N2O2 (220.27): C 65.43; H 7.32; N 12.72. Found: C 65.10; H 7.60; N 12.78.
3-(2,4-Dimethoxypheny)-4-phenyl-4,5-dihydro-1H-pyrazole 4b
Compound 4b was prepared from 3b as oil (75% yield) using the same procedure described for 4a. 1H-NMR (CDCl3–D2O) δ: 2.80 (dd, 1H, CH, methylene, J: 4.4, 8.2 Hz), 3.39 (dd, 1H, CH, methylene, J: 4.4, 8.2 Hz), 3.80 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 5.10 (d, 1H, CH J:7.6 Hz), 5.53 (s, 1H, NH exch.), 6.14 (s, 1H, ArH-3), 6.30–7.42 (m, 5H, Ar), 7.61 (d, 1H, ArH-5, J: 8.4 Hz), 8.31 (d, 1H, ArH-6, J: 8.4 Hz). IR (KBr) cm−1: 3422 (NH), 3058 (CH Ar), 2934, 2837 (CH aliphatic), 1604 (C=N). MS m/z: 282 [M+, 5%], 165 [C6H3(OCH3)2CHNH, 100%]. Anal. Calcd for C17H18N2O2 (282.34): C 72.32; H 6.43; N 9.92. Found: C 72.27; H 6.22; N 9.97.
3-(2,4-Dimethoxyphenyl)-N-[(4-methylphenyl)sulfonyl]-4-methyl-4,5-dihydro-1H-pyrazol-1-carboxamide 6a
To a solution of 4a (2.20 g, 10 mmol) in toluene (50 ml), 5a (2.75 g, 12 mmol) was added, and the resulting mixture was refluxed for 4 h. After cooling of the solution to room temperature, the mixture was concentrated in vacuum, water was added and extracted with chloroform. The organic layer was washed twice with water, dried over Na2SO4, and concentrated. The product was obtained as oil (80% yield). 1H-NMR (CDCl3–D2O) δ: 1.21 (d, 3H, CH3, J: 4.6 Hz), 2.21 (s, 3H, CH3), 3.70 (d, 1H, CH, methylene, J: 4.4 Hz), 3.80 (d, 1H, CH, methylene, J: 4.4 Hz), 3.85 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 4.11 (m, 1H, CH py), 5.27 (s, 1H, NH exch.), 6.51 (s, 1H, ArH-3), 7.20 (d, 1H, ArH-5, J: 8.4 Hz), 7.33 (d, 1H, ArH-6, J: 8.7 Hz), 7.80 (d, 2H, Ar, J: 8.4 Hz), 7.90 (d, 2H, Ar, J: 8.1 Hz). IR (KBr) cm−1: 3328 (NH), 3030 (CH Ar), 2969, 2928 (CH aliphatic), 1683 (C=O), 1610 (C=N), 1327, 1155 (SO2). MS m/z: 417 [M+, 1%], 165 [C6H3(OCH3)2CHNH, 8%], 91 [C7H7, 100%]. Anal. calcd for C20H23N3O5S (417.48): C 57.54; H 5.55; N 10.06. Found: C 57.40; H 5.77; N 9.96.
N-[(4-Chlorophenyl)sulfonyl]-3-(2,4-dimethoxyphenyl)-4-methyl-4,5-dihydro-1H-pyrazol-1-carboxamide 6b
Compound 6b was prepared from 4a and 5b as white crystals from methanol, m.p. 225°C (78% yield) using the same procedure described for 6a. 1H-NMR (CDCl3–D2O) δ: 1.45 (d, 3H, CH3, J: 4.6 Hz), 3.07 (d, 1H, CH, methylene, J: 4.6 Hz), 3.16 (d, 1H, CH, methylene, J: 4.4 Hz), 3.70 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 4.60 (broad, 1H, CH), 4.78 (broad, 1H, NH exch.), 7.28 (s, 1H, ArH-3), 7.53 (d, 3H, Ar, J: 8.0 Hz), 7.91 (d, 3H, ArH, J: 8.2 Hz). IR (KBr) cm−1: 3300 (NH), 3030 (CH Ar), 2900, 2800 (CH aliphatic), 1680 (C=O), 1600 (C=N), 1380, 1160 (SO2). MS m/z: 423 [M+-CH3, 3%], 165 [C6H3 (OCH3)2CHNH, 48%], 57 [C2H5N2, 100%]. Anal. calcd for C19H20ClN3O5S (437.90): C 52.11; H 4.60; N 9.65. Found: C 52.18; H 5.20; N 9.57.
3-(2,4-Dimethoxyphenyl)-N-[(4-methylphenyl)sulfonyl]-4-phenyl-4,5-dihydro-1H-pyrazol-1-carboxamide 6c
Compound 6c was prepared from 4b and 5a as oil (84% yield) using the same procedure described for 6a. 1H-NMR (CDCl3–D2O) δ: 2.16 (s, 3H, CH3), 3.60 (d, 1H, methylene, J: 4.0 Hz), 3.77 (d, 1H, methylene, J: 4.0 Hz), 3.82 (s, 3H, OCH3), 4.01 (s, 3H, OCH3), 4.20 (d, 1H, CH J:7.6 Hz), 5.30 (s, 1H, NH exch.), 6.20 (s, 1H, ArH-3), 6.60 (d, 1H, ArH-5, J: 8.4 Hz), 6.80 (d, 1H, ArH-6, J: 8.2 Hz), 7.00–7.65 (m, 5H, Ar), 7.77 (d, 2H, Ar, J: 8.0 Hz), 7.95 (d, 2H, Ar, J: 8.0 Hz). IR (KBr) cm−1: 3261 (NH), 3063 (CH Ar), 2939, 2841 (CH aliphatic), 1660 (C=O), 1605 (C=N), 1343, 1162 (SO2). MS m/z: 479 [M+, 1%], 165 [C6H3(OCH3)2CHNH, 100%]. Anal. calcd for C25H25N3O5S (479.55): C 62.62; H 5.25; N 8.76. Found: C 62.62; H 5.25; N 8.83.
N-[(4-Chlorophenyl)sulfonyl]-3-(2,4-dimethoxyphenyl)-4-phenyl-4,5-dihydro-1H-pyrazol-1-carboxamide 6d
Compound 6d was prepared from 4b and 5b as oil (76% yield) adopting the same procedure described for 6a. 1H-NMR (CDCl3–D2O) δ: 3.40 (d, 1H, methylene, J: 4.0 Hz), 3.60 (d, 1H, methylene, J: 4.0 Hz), 3.81 (s, 3H, OCH3), 4.04 (s, 3H, OCH3), 4.20 (d, 1H, CH J: 7.6 Hz), 5.20 (s, 1H, NH exch.), 6.15 (s, 1H, ArH-3), 6.605 (d, 1H, ArH-5, J: 8.2 Hz), 6.85 (d, 1H, ArH-6, J: 8.2 Hz), 7.00–7.70 (m, 5H, Ar), 7.88 (d, 2H, Ar, J: 8.4 Hz), 7.99 (d, 2H, Ar, J: 8.2 Hz). IR (KBr) cm−1: 3363 (NH), 3090 (CH Ar), 2937, 2842 (CH aliphatic), 1656 (C=O), 1604 (C=N), 1346, 1163 (SO2). MS m/z: 501 [M++2, 1%], 499 [M+, 1%], 165 [C6H3(OCH3)2CHNH, 100%]. Anal. calcd for C24H22ClN3O5S (499.97): C 57.65; H 4.44; N 8.41. Found: C 57.91; H 4.62; N 8.48.
4-Chlorobenzoylisothiocynate 7
Compound 7 was prepared according to the reported procedure (Cho and Shon, 1991) from 4-chlorobenzoyl chloride and ammonium thiocyanate and immediately reacted with 4.
N-(4-Chlorobenzoyl)-3-(2,4-dimethoxyphenyl)-4-methyl-4,5-dihydro-1H-pyrazol-1-thiocarboxamide 8a
Compound 4a (2.20 g, 10 mmol) was added to a cold (0°C) solution of 7 (2.37 g, 12 mmol) in anhydrous acetonitrile (20 ml), and the resulting mixture was stirred at room temperature for 3 h. The precipitate was removed by filtration and thoroughly washed with acetonitrile. The filtrate was concentrated in vacuum, and the residue was collected and further purified by extraction with chloroform. The organic layer was twice washed with water, dried over Na2SO4, and concentrated. The product was obtained as oil (61% yield). 1H-NMR (CDCl3–D2O) δ: 1.27 (d, 3H, CH3), 3.20 (d, 1H, methylene, J: 4.2 Hz), 3.40 (d, 1H, methylene, J: 4.2 Hz), 3.70 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 4.49 (m, 1H, CH), 6.30 (s, 1H, NH exch.), 6.40(s, 1H, ArH-3), 7.26 (d, 1H, ArH-5, J: 8.6 Hz), 7.33 (d, 1H, ArH-6, J: 8.6 Hz), 7.76 (d, 2H, Ar, J: 8.8 Hz), 7.85 (d, 2H, Ar, J: 8.8 Hz). IR (KBr) cm−1: 3360 (NH), 3050 (CH Ar), 2934, 2878 (CH aliphatic), 1669 (C=O), 1620 (C=N), 1098 (C=S). MS m/z: 417 [M+, 2%], 385 [M+-S, 3%], 177 [C6H3(OCH3)2CNNH, 100%]. Anal. calcd for C20H20ClN3O3S (417.91): C 57.48; H 4.82; N 10.05. Found: C 57.68; H 4.84; N 10.43.
N-(4-Chlorobenzoyl)-3-(2,4-dimethoxyphenyl)-4-phenyl-4,5-dihydro-1H-pyrazol-1-thiocarboxamide 8b
Compound 8b was prepared from 4b and 7 as oil (73% yield) by the same procedure as described for 8a. 1H-NMR (CDCl3–D2O) δ: 3.20 (d, 1H, methylene, J: 4.2 Hz), 3.36 (d, 1H, methylene, J: 4.2 Hz), 3.89 (s, 3H, OCH3), 4.04 (s, 3H, OCH3), 4.59 (d, 1H, CH J: 7.5 Hz), 6.11 (s, 1H, NH exch.), 6.53 (s, 1H, ArH-3), 7.26–7.96 (m, 9H, Ar), 8.05 (d, 1H, Ar, J: 9.0 Hz), 8.35 (d, 1H, Ar, J: 8.6 Hz). IR (KBr) cm−1: 3368 (NH), 3050 (CH Ar), 2853, 2772 (CH aliphatic), 1656 (C=O), 1619 (C=N), 1087 (C=S). MS m/z: 481 [M++2, 35%], 479 [M+, 1%], 281 [M+–C6H4(p-Cl)CONHCS, 50%], 165 [C6H3(OCH3)2C+NH, 100%]. Anal. calcd for C25H22ClN3O3S (479.98): C 62.56; H 4.62; N 8.75. Found: C 62.50; H 4.60; N 8.76.
3-Methyl-2,4-dioxo-4-(2,4-dimethoxyphenyl) butanoic acid ethyl ester 9a
To compound 2a (1.94 g, 10 mmol) in toluene (20 ml) was added sodium hydride (0.48 g, 20 mmol), and the mixture was stirred for 10 min. Then, diethyl oxalate (3.29 g, 3.0 ml, 15 mmol) was added dropwise, and the mixture was stirred at reflux (1 h). The mixture was cooled to room temperature, washed with ether, acidified with acetic acid, and extracted with chloroform. The organic layer was washed with water, and concentrated. The product was obtained as oil (60% yield). 1H-NMR (CDCl3) δ: 1.35 (t, 3H, CH2CH 3 , J: 4.2 Hz), 2.35 (d, 3H, COCHCH 3 , J: 2.2 Hz), 3.79 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 4.30 (q, 2H, CH 2 CH3), 6.00 (q, 1H, CH), 7.41 (s, 1H, ArH-3), 7.66 (d, 1H, ArH-5, J: 8.0 Hz), 8.08 (d, 1H, ArH-6, J: 8.0 Hz). IR (KBr) cm−1: 3030 (CH Ar), 2969, 2843 (CH aliphatic), 1712, 1612 (3 C=O). MS m/z: 417 [M+, 1%], 165 [C6H3(OCH3)2CHNH, 8%], 91 [C7H7, 100%]. Anal. calcd. for C15H18O6 (294.31): C 61.22; H 6.12. Found: C 61.49; H 5.88.
2,4-Dioxo-3-phenyl-4-(2,4-dimethoxyphenyl) butanoic acid ethyl ester 9b
Compound 9b was prepared from 2b as oil (72% yield) following the same procedure described for 9a. 1H-NMR (CDCl3) δ: 1.38 (t, 3H, CH2CH 3 , J: 7.2 Hz), 3.77 (s, 3H, OCH3), 3.98 (s, 3H, OCH3), 4.28 (q, 2H, CH 2 CH3), 6.44 (s, 2H, COCH & ArH-3), 6.54 (d, 1H, ArH-5, J: 8.6 Hz), 7.15–7.60 (m, 5H, Ar), 7.83 (d, 1H, ArH-6, J: 8.4 Hz). IR (KBr) cm−1: 3060 (CH Ar), 2940, 2838 (CH aliphatic), 1730, 1665 (3 C=O). MS m/z: 417 [M+, 1%], 165 [C6H3(OCH3)2CHNH, 8%], 91 [C7H7, 100%]. Anal. calcd for C20H20O6 (356.38): C 67.41; H 5.66. Found: C 67.69; H 5.40.
5-(2,4-Dimethoxyphenyl)-4-methyl-1-phenyl-1H-pyrazole-3-carboxylic acid ethyl ester 10a
To a solution of 9a (2.94 g, 10 mmol) in absolute ethanol (50 ml), phenyl hydrazine hydrochloride (1.45 g, 10 mmol) and triethylamine (1.01 g, 10 mmol) were added. The reaction mixture was then heated under reflux for 9 h. It was then cooled, washed with water, and extracted with chloroform (3× 20 ml). The organic layer was then separated, dried over sodium sulfate, and evaporated to give 10a. The product was oil (84% yield). 1H-NMR (CDCl3) δ: 1.39 (t, 3H, CH2CH 3 , J: 4.0 Hz), 2.96 (s, 3H, CH3), 3.83 (s, 3H, OCH3), 4.02 (s, 3H, OCH3), 4.20 (q, 2H, CH 2 CH3), 6.42 (s, 1H, ArH-3), 6.60 (d, 1H, ArH-5, J: 8.4 Hz), 7.10–7.80 (m, 5H, Ar), 8.11 (d, 1H, ArH-6, J: 8.4 Hz). IR (KBr) cm−1: 3061 (CH Ar), 2935, 2843 (CH aliphatic), 1717 (C=O). MS m/z: 366 [M+, 2%], 165 [C6H3(OCH3)2CO, 100%]. Anal. calcd for C21H22N2O4 (366.42): C 68.84; H 6.05; N 7.68. Found: C 68.65; H 6.24; N 7.59.
1-(4-Chlorophenyl)-5-(2,4-dimethoxyphenyl)-4-methyl-1H-pyrazole-3-carboxylic acid ethyl ester 10b
Compound 10b was prepared from 9a (2.94 g, 10 mmol), 4-chlorophenyl hydrazine hydrochloride (1.79 g, 10 mmol) and triethylamine (1.01 g, 10 mmol) following the same procedure described for 10a. The product was obtained as oil (80% yield). 1H-NMR (CDCl3) δ: 1.29 (t, 3H, CH2CH 3 , J: 4.0 Hz), 2.92 (s, 3H, CH3), 3.75 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 4.34 (q, 2H, CH 2 CH3), 6.38 (s, 1H, ArH-3), 6.43 (d, 1H, ArH-5, J: 8.2 Hz), 7.40 (d, 1H, ArH-6, J: 8.2 Hz), 7.80 (d, 2H, Ar, J: 8.4 Hz), 8.06 (d, 2H, Ar, J: 8.4 Hz). IR (KBr) cm−1: 3050 (CH Ar), 2933, 2843 (CH aliphatic), 1720 (C=O). MS m/z: 402 [M++2, 1%], 400 [M+, 1%], 165 [C6H3(OCH3)2CO, 100%]. Anal. calcd for C21H21ClN2O4 (400.98): C 62.90; H 5.28; N 7.02. Found: C 63.14; H 5.46; N 7.32.
5-(2,4-Dimethoxyphenyl)-1,4-diphenyl-1H-pyrazole-3-carboxylic acid ethyl ester 10c
It was prepared from 9b (3.56 g, 10 mmol), phenyl hydrazine hydrochloride (1.45 g, 10 mmol) and triethylamine (1.01 g, 10 mmol) using the same procedure described for 10a. The product was obtained as oil (74% yield). 1H-NMR (CDCl3) δ: 1.37 (t, 3H, CH2CH 3 , J: 4.2 Hz), 3.78 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 4.30 (q, 2H, CH 2 CH3), 6.23 (s, 1H, ArH-3), 6.40 (d, 1H, ArH-5, J: 8.2 Hz), 6.80–8.00 (m, 11H, Ar). IR (KBr) cm−1: 3057 (CH Ar), 2927, 2843 (CH aliphatic), 1738 (C=O). MS m/z: 428 [M+, 1%], 165 [C6H3(OCH3)2CO, 100%]. Anal. calcd for C26H24N2O4 (428.49): C 72.88; H 5.65; N 6.54. Found: C 72.87; H 5.30; N 6.25.
1-(4-Chlorophenyl)-5-(2,4-dimethoxyphenyl)-4-phenyl-1H-pyrazole-3-carboxylic acid ethyl ester 10d
It was prepared from 9b (3.56 g, 10 mmol), 4-chlorophenyl hydrazine hydrochloride (1.79 g, 10 mmol) and triethylamine (1.01 g, 10 mmol) following the same procedure described for 10a. The product was obtained as oil (88% yield). 1H-NMR (CDCl3) δ: 1.26 (t, 3H, CH2CH 3 , J: 4.0 Hz), 3.79 (s, 3H, OCH3), 4.00 (s, 3H, OCH3), 4.28 (q, 2H, CH 2 CH3), 6.30 (s, 1H, ArH-3), 6.45 (d, 1H, ArH-5, J: 8.2 Hz), 6.65 (d, 1H, ArH-6, J: 8.2 Hz), 7.00–7.60 (m, 5H, Ar), 7.80 (d, 2H, Ar, J: 8.6 Hz), 8.10 (d, 2H, Ar, 8.6 Hz). IR (KBr) cm−1: 3059 (CH Ar), 2937, 2839 (CH aliphatic), 1728 (C=O). MS m/z: 464 [M++2, 1%], 462 [M+, 1%], 165 [C6H3(OCH3)2CO, 100%]. Anal. calcd for C26H23ClN2O4 (462.93): C 67.46; H 5.01; N 6.05. Found: C 67.44; H 5.21; N 5.83.
1-(4-Chlorophenyl)-5-(2,4-dimethoxyphenyl)-4-methyl-1H-pyrazole-3-carboxylic acid 11a
Compound 10b (2.00 g, 5 mmol) was dissolved in methanol (20 ml) and to this a solution of KOH (0.56 g, 10 mmol) in methanol (5 ml) was added. The reaction mixture was heated to reflux for 3 h, cooled, and poured into ice-water. It was acidified with acetic acid and extracted with chloroform. The organic layer was washed with water, dried over Na2SO4, and concentrated. The residue was obtained as oil (51% yield). 1H-NMR (CDCl3–D2O) δ: 3.08 (s, 3H, CH3), 3.70 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 6.23 (s, 1H, ArH-3), 6.50 (d, 1H, ArH-5, J: 8.0 Hz), 7.10 (d, 1H, ArH-6, J: 8.0 Hz), 7.35 (d, 2H, Ar, J: 8.6 Hz), 7.78 (d, 2H, Ar, J: 8.6 Hz), 11.00 (s, 1H, COOH, exch.). IR (KBr) cm−1: 3030 (CH Ar), 2950–2850 (CH aliphatic and OH), 1720 (C=O). MS m/z: 372 [M+, 8%], 165 [C6H3(OCH3)2CO, 100%]. Anal. calcd for C19H17ClN2O4 (372.81): C 61.21; H 4.60; N 7.51. Found: C 61.35; H 4.80; N 7.57.
1-(4-Chlorophenyl)-5-(2,4-dimethoxyphenyl)-4-phenyl-1H-pyrazole-3-carboxylic acid 11b
It was prepared from 10d as oil (57% yield) by using the same procedure described for 11a. 1H-NMR (CDCl3–D2O) δ: 3.83 (s, 3H, OCH3), 4.27 (s, 3H, OCH3), 6.35 (s, 1H, ArH-3), 6.50 (d, 1H, ArH-5, J: 8 Hz), 6.62 (d, 1H, ArH-6, J: 8.2 Hz), 7.60–7.78 (m, 5H, Ar), 7.88 (d, 2H, Ar, J: 8.4 Hz), 8.20 (d, 2H, ArH, J: 8.7 Hz), 11.00 (s, 1H, COOH, exch.). IR (KBr) cm−1: 3060 (CH Ar), 2940–2830 (CH aliphatic and OH), 1730 (C=O). MS m/z: 436 [M++2, 56%], 434 [M+, 20%], 151 [C6H3(OCH3)2CH2, 100%]. Anal. calcd for C24H19ClN2O4 (434.88): C 66.29; H 4.40; N 6.44. Found: C 66.41; H 4.79; N 6.07.
Pharmacological evaluation
The experimental tests on animals have been performed in accordance with the Institutional Ethical Committee approval, Faculty of Pharmacy, Cairo University.
LD50, anti-inflammatory, and analgesic activities of all the newly synthesized compounds were performed. The tested compounds were dissolved in dimethyl sulfoxide and water.
Determination of LD50
LD50 determined using Finney’s method (Finney, 1964). For this purpose, albino mice (25–30 g) were divided into groups each of five animals. Preliminary experiments were done for each compound to determine the minimal dose that kills all mice and the maximal dose that fails to kill any animal. Several increasing intraperitoneal doses were given in between these doses. Animals were kept under observation for 24 h during which symptoms of toxicity and rate of mortality in each group were recorded.
Anti-inflammatory activity (in vivo screening, carrageenan-induced edema in rats)
The anti-inflammatory activity of the newly synthesized compounds was evaluated according to the method described by Winter et al. (Winter et al., 1962). One-hundred and five rats of both sexes weighing 150–180 g were divided into 21 groups. The thickness of the left hind paw of each rat was measured in millimeter using a vernier caliber. The first group was kept as a control, while the second was intraperitoneal injected with celecoxib as a reference in a dose of 25 mg/kg body weight. Other groups were intraperitoneal injected with the tested compounds in a dose 25 mg/kg body weight which nearly equal to 1/10 their LD50. After 30 min, inflammation was induced by subcutaneous injection of 50 μl of 1% carrageenan in a normal saline into the left hind paw. Degree of inflammation (mm) is measured as the difference between thickness after carrageenan treatment and thickness before carrageenan treatment. The paw thickness was measured hourly for a period of 4 h. The anti-inflammatory efficacy of the tested compounds was assessed by comparing the magnitude of the paw swelling in the treated animals with that of the control then calculates percent inhibition with the following formula (Alam et al., 2009).
Analgesic activity
The analgesic activity of the tested compounds was evaluated using the writhing method (Collier et al., 1968). One-hundred and five mice of both sexes weighing 25–30 g were divided into 21 groups. The first group was kept as a control, while the second was intraperitoneal injected with celecoxib as standard drug in a dose of 25 mg/kg body weight. Other groups were intraperitoneal injected with the tested compounds in a dose 25 mg/kg body weight. After 30 min, each mouse was intraperitoneal injected with 0.25 ml of p-benzoquinone aqueous solution (0.1 mg/ml). Thereafter, mice in all groups were observed for writhing hourly for 4 h. Animals devoid of writhing in each group were counted, and the analgesic potency of the tested compounds was determined as percent protection against writhing.
Ulcerogenic effect
The ulcerogenic effect of the most active compounds 3a, 4a, and 11b as well as both celecoxib and ibuprofen was evaluated according to Meshali’s method (Meshali et al., 1983). Thirty adult male albino rats weighing 120–150 g were used in this study. Animals were divided into six groups and received the drug orally. The first group received 2% tween 80 and kept as control, while the second and third groups received celecoxib and ibuprofen in a dose of 25 mg/kg body weight. The other groups were received 3a, 4a, and 11b in the same dose. Animals were fed 2 h after administration of the drug. Rats received the given dose orally for three successive days. Two hours following the last dose, rats were sacrificed; the stomach of each rat was removed, opened along the greater curvature, and rinsed with 0.9% sodium chloride (isotonic solution). The stomach was stretched, by pins, on a corkboard. Examination with a magnifying lens (10×) was done for the presence of ulcers and erosions. The ulcer index was calculated according to Robert’s method (Robert et al., 1968). The degree of ulcerogenic effect was expressed in term of the percentage incidence of ulcers in each group of animals divided by ten, the average number of ulcers per stomach, and the average severity of ulcers by visual observation. The ulcer index is the value that result from the sum of the above three values.
References
Abbas SE, Awadallah FM, Ibrahim NA, Gouda AM (2010) Novel substituted and fused pyrrolizine derivatives: synthesis, anti-inflammatory and ulcerogenecity studies. Eur J Med Chem 45:482–491
Alam MI, Baboota S, Kohli K, Ali J, Ahuja A (2009) Development and evaluation of transdermal patches of celecoxib. PDA J Pharm Sci Technol 63:429–437
Barsoum FF, Hosni HM, Girgis AS (2006) Novel bis(1-acyl-2-pyrazolines) of potential anti-inflammatory and molluscicidal properties. Bioorg Med Chem 14:3929–3937
Buck WB, Osweiter GD, Van Gelder AG (1976) Clinical and diagnostic veterinary toxicology, 2nd edn. Kendall/hunt Publishing Co., Iowa, p 5211
Cho NS, Shon HI (1991) Synthesis of 5-(aroylamino)-2-methyl-2H–1,2,4-thiazoliazol-3-ones by oxidative cyclisation of 1-aroyl-5-methyl-2-thiobiurets. J Heterocycl Chem 28:1645–1649
Collier HD, Dinnin LC, Johnson CA, Schneider C (1968) The abdominal response and its suppression by analgesic drugs in the mouse. Br J Pharmacol Ther 32:295–310
Cryer B (2005) NSAID-associated deaths: the rise and fall of NSAID-associated GI mortality. Am J Gastroenterol 100:1694–1695
Finney DJ (1964) Statistical methods in biological assay. Charles Griffin and Company Ltd, London, p 597
Fylaktakidou KC, Hadjipavlou-Litina DJ, Litinas KE, Nicolaides DN (2004) Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr Pharm Des 10:3813–3833
James MW, Hawkey CJ (2003) Assessment of non-steroidal anti-inflammatory drug (NSAID) damage in the human gastrointestinal tract. Br J Clin Pharmacol 56(2):146–155
Karthikeyan MS, Holla BS, Kumari NS (2007) Synthesis and antimicrobial studies on novel chloro-fluorine containing hydroxy pyrazolines. Eur J Med Chem 42:30–36
Lacerda RB, de Lima CK, da Silva LL, Romeiro NC, Miranda AL, Barreiro EJ, Fraga CA (2009) Discovery of novel analgesic and anti-inflammatory 3-arylamine-imidazo[1,2-a]pyridine symbiotic prototypes. Bioorg Med Chem 17:74–84
Lange JHM, Coolen HKAC, van Stuivenberg HH, Dijksman JAR, Herremans AHJ, Ronken E, Keizer HG, Tipker K, McCreary AC, Veerman W, Wals HC, Stork B, Verveer PC, den Hartog AP, de Jong NMJ, Adolfs TJP, Hoogendoorn J, Kruse CG (2004) Synthesis, biological properties, and molecular modeling investigations of novel 3,4-diarrypyrazolines as potent and selective CB1 cannabinoid receptor antagonists J. Med. Chem 17:627–643
Laufer S, Luik S (2010) Different methods for testing potential cyclooxygenase-1 and cyclooxygenase-2 inhibitors. Methods Mol Biol 644:91–116
Lazzaroni M, Bianchi Porro G (2004) Gastrointestinal side-effects of traditional non-steroidal anti-inflammatory drugs and new formulations. Aliment Pharmacol Ther 2:48–58
Menozzi G, Merello L, Fossa P, Mosti L, Piana A, Mattioli F (2003) 4-substituted 1,5-diarylpyrazole, analogues of celecoxib: synthesis and preliminary evaluation of biological properties. Farmaco 58:795–808
Meshali M, El-Sabbah E, Foda A (1983) Effect of encapsulation of flufenamic acid with acrylic resins on its bioavailability and gastric ulcerogenic activity in rats. Acta Pharm Technol 29:217–230
Molecular Operating Environment (MOE) (2005) Version 2005.06, Chemical Computing Group, Inc., Montreal, Quebec, Canada. http://www.chemcomp.com
Mounier G, Guy C, Berthoux F, Beyens MN, Ratrema M, Ollagnier M (2006) Severe renal adverse events with arylcarboxylic non-steroidal anti-inflammatory drugs: results of an eight-year French national survey. Therapie 61:255–266
Naesdal J, Brown K (2006) NSAID-associated adverse effects and acid control aids to prevent them: a review of current treatment options. Drug Saf 29:119–132
Patel MV, Bell R, Majest S, Henry R, Kolasa T (2004) Synthesis of 4,5-diaryl-1H-pyrazole-3-ol derivatives as potential COX-2 inhibitors. J Org Chem 69:7058–7065
Ramiz MM, El-Sayed WA, El-Tantawy AI, Abdel-Rahman AA (2010) Antimicrobial activity of new 4,6-disubstituted pyrimidine, pyrazoline, and pyran derivatives. Arch Pharm Res 33:647–654
Rathish IG, Javed K, Ahmad S, Bano S, Alam MS, Pillai KK, Singh S, Bagchi V (2009) Synthesis and anti-inflammatory activity of some new 1,3,5-trisubstituted pyrazolines bearing benzene sulfonamide. Bioorg Med Chem Lett 19:255–258
Reynold JEF (1993) Martindale, the extra pharmacopia, 30th edn. Pharmaceutical Press, London, p 1310
Robert A, Nezamis JE, Phillips JP (1968) Effect of prostaglandin E1 on gastric secretion and ulcer formation in the rat. Gastroenterology 55:481–487
Sakya SM, Hou X, Minich ML, Rast B, Shavnya A, DeMello KM, Cheng H, Li J, Jaynes BH, Mann DW, Petras CF, Seibel SB, Haven ML (2007) 5-Heteroatom substituted pyrazoles as canine COX-2 inhibitors. Part III: molecular modeling studies on binding contribution of 1-(5-methylsulfonyl)pyrid-2-yl and 4-nitrile. Bioorg Med Chem Lett 17:1067–1072
Schneider V, Lévesque LE, Zhang B, Hutchinson T, Brophy JM (2006) Association of selective and conventional nonsteroidal anti-inflammatory drugs with acute renal failure: a population-based, nested case-control analysis. Am J Epidemiol 164:881–889
Shoman ME, Abdel-Aziz M, Aly OM, Farag HH, Morsy MA (2009) Synthesis and investigation of anti-inflammatory activity and gastric ulcerogenicity of novel nitric oxide-donating pyrazoline derivatives. Eur J Med Chem 44:3068–3076
Slotta KH, Heller H (1930) Über β-phenyl-äthylamine, I. Mitteil.: Mezcalin und mezcalin-ähnliche substanzen. Ber 63:3029–3044
Sostres C, Gargallo CJ, Arroyo MT, Lanas A (2010) Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract Res Clin Gastroenterol 24:121–132
Turan-Zitouni G, Chevallet P, Kiliç FS, Erol K (2000) Synthesis of some thiazolyl-pyrazoline derivatives and preliminary investigation of their hypotensive activity. Eur J Med Chem 35:635–641
Winter CA, Risley EA, Nuss GW (1962) Proceedings of the society for experimental biology and medicine. Proc Soc Exp Biol 544–547
Yanai H, Furutani N, Yoshida H, Tada N (2009) Myositis, vasculitis, hepatic dysfunction in adult-onset still’s disease. Case Rep Med 2009:504897
Zadrazil J (2006) Nonsteroidal anti-inflammatory drugs and the kidney. Vnitr Lek 52:686–690
Acknowledgment
The authors wish to offer their deep gratitude to Prof. Dr. Gamal. A. Soliman, Department of Pharmacology, Faculty of Veterinary Medicine, Cairo University for carrying out the pharmacological evaluation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abd-El Gawad, N.M., Hassan, G.S. & Georgey, H.H. Design and synthesis of some pyrazole derivatives of expected anti-inflammatory and analgesic activities. Med Chem Res 21, 983–994 (2012). https://doi.org/10.1007/s00044-011-9606-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-011-9606-4