
Abstract
The growing evidence for the role of introgression in evolution is challenging for species delimitation, taxonomy, and conservation. Here, we examine the isolation of Saxifraga delphinensis, a south-west alpine narrow endemic, from its sympatric congeners Saxifraga exarata and Saxifraga moschata. Our analyses, based on morphological, molecular and genome-wide data, were extended to taxa from Jura, Massif Central, north-east Iberian mountains and Pyrenees to broaden the phylogenetic scope of the study. Phylogenetic results support the close relationship between (i) S. delphinensis, Saxifraga cebennensis and one population of S. exarata from Queyras and (ii) between S. lamottei, S. giziana and S. moschata. Saxifraga exarata, which was extensively sampled, is composed of several clades and is paraphyletic to other taxa. While morphological and genetic data support the evolutionary distinctiveness of S. delphinensis, introgression with S. exarata is shown by various data and analyses. In addition, ABBA-BABA analyses and plastome phylogeny reveal a possible contribution of the north-east Iberian endemic Saxifraga vayredana into the history of S. delphinensis. Overall, the results show a complex evolutionary history with frequent hybridization and spanning several mountain ranges in Europe.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Long before the development of molecular markers and omics, the terms “incipient species” (Cuénot 1917) or “semispecies” (Grant 1981) were proposed to refer to cases of incomplete isolation between taxa. Today, molecular and genomic evidence confirms that continued gene flow and recombination between species is widespread in nature (Mallet et al. 2016; Edelman and Mallet 2021). Hybridization and introgression are important for speciation and diversification (Abbott et al. 2013) as well as for adaptation (Suarez-Gonzalez et al. 2018). In plants, hybridization challenges our understanding of clustered biological diversity (i.e., species) and reproductive isolation (Rieseberg et al. 2006; Christie et al. 2022). Hybridization can also have been facilitated in climate refugia during Quaternary climate-driven shifts in species range (Nieto Feliner 2014; Voisin et al. 2023). Lastly, hybridization is clearly of conservation significance when dealing with rare or restricted endemic taxa. In practice, incomplete differentiation and introgression can lead to taxonomic problems and confusion that weaken our knowledge of the biogeography and ecology of species, which also undermines biodiversity conservation strategies.
In the iconic genus Saxifraga, Ebersbach et al. (2020) reported that more than 40 papers or book chapters were devoted to hybridization and estimated that 84 hybrid taxa have been described. They also showed that the rate of hybridization varied among mountain regions and within the genus and that several reproductive barriers counteract gene flow. The clade corresponding to the section Saxifraga in Tkach et al. (2015), has a high rate of hybridization causing incongruities between morphological and molecular data (Vargas 2000) or between nuclear ribosomal and plastid data (Mas De Xaxars et al. 2015; Carnicero et al. 2023). In this clade, Saxifraga exarata Villars, widespread in Alps, and Saxifraga moschata Wulf., widespread in Pyrenes and Alps, are known to be interfertile with at least 10 other taxa (Ebersbach et al. 2020). In Pyrenees and north-east Iberian mountains, S. moschata hybridizes with several endemic taxa, such as Saxifraga pubescens Pourr, Saxifraga vayredana Luizet or Saxifraga geraniodes Host (Mas De Xaxars et al. 2015; Carnicero et al. 2023) but the taxa maintain their genetic integrity (Carnicero et al. 2023). However, in the Alps this issue was never addressed to the relationships among the “Saxifrage du Dauphiné” (Saxifraga delphinensis Ravaud) and its widespread congeners. Saxifraga delphinensis is a narrow endemic taxon restricted to the calcareous massifs situated at the south-west margin of the European Alps (Fig. 1a). In the nineteenth century, Ravaud described it in Vercors from the Grand Veymont summit and the Grimone Pass in his “Guide du botaniste dans le Dauphiné” (Ravaud 1885), naming it Saxifraga delphinensis. Based on a general aspect of the plant but without morphological study, it was renamed S. exarata subsp. delphinensis Ravaud (Kerguélen) in Kerguélen (1993) and Flora Alpina (Aeschimann et al. 2004), before being finally renamed S. delphinensis Ravaud by Garraud (2004) and this name is used in Flora Gallica (Tison and Foucault 2014). Ravaud (1885) underlined the similarity of S. delphinensis with two taxa endemic to the Pyrenees Saxifraga pubescens Pourr (which shares similar leaves) and Saxifraga iratiana Schullz (which shares similar flowers). Until recently, S. delphinensis has not been the subject of any molecular study but a recent plastome-based phylogeny (Smyčka et al. 2022) reported its closest relationship not with S. exarata nor S. pubescens but with S. vayredana, a north-east Iberian endemic (Catalanidic mountains). Interestingly, in Smyčka et al. (2022) S. pubescens and Saxifraga moschata Wulf are the closest taxa of S. exarata whereas in Tkach et al. (2015), the sister species of S. exarata is Saxifraga cebennensis Rouy and Camus, an endemic from the Cévennes range, located at the south-east margin of the Massif Central (Fig. 1a). Similarly, various geographical and ecological isolates have been separated over time from S. moschata, such as Saxifraga lamottei Luizet (Luizet 1913), a mid-altitude isolate on basaltic siliceous rocks restricted to the Massif Central (Fig. 1a), and Saxifraga giziana Mouly and Ferrez (Bouchard 1947; Mouly and Ferrez 2020), a low altitude isolate on limestone from the southern Jura Massif (Fig. 1a). This latter region also harbors low altitude isolates of Saxifraga rosacea Moench. and S. moschata.

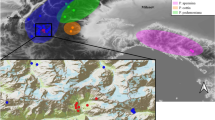
Map of the 57 sampling sites A with a focus on south-west Alps B where S. delphinensis, S. exarata, and S. moschata were more intensively sampled
In this study, we obtained morphological, molecular and genome-wide sequencing data to examine the distinctiveness and reproductive isolation between S. delphinensis, S. exarata and S. moschata in south-west Alps where they often occur in sympatry. Acknowledging previous studies (Vargas 2000; Mas De Xaxars et al. 2015; Smyčka et al. 2022) that established phylogenetic links between taxa of different mountain ranges in the section Saxifraga, we broaden the scope of the study by including several endemic taxa to contribute to the knowledge of their phylogenetic relationships. We therefore include S. giziana from Jura; S. lamottei and S. cebennensis from Massif Central, Saxifraga pubescens and S. geraniodes from Eastern Pyrenees; Saxifraga vayredana and Saxifraga genesiana Vargas from the Catalan Pre-Coastal Range; Saxifraga moncayensis Webb from the Moncayo Range and Saxifraga hariotii Luizet & Soulié from Western Pyrenees.
Material and methods
Sampling and general strategy
The sampling strategy was focused on S. delphinensis and S. exarata and S. moschata in the range of S. delphinensis in southwestern PreAlps. Apart from this area, Saxifraga exarata was more intensively sampled in eastern Queyras (France), because several forms were described by Villars (1779) in the higher parts of Queyras (in high mountains, above the village of Ristolas). In the locality of Pic de Bure (Devoluy), we sampled more intensively S. delphinensis and S. exarata because both taxa are abundant on this plateau, sharing a similar ecology and occurring in close sympatry. The three narrow endemics, S. lamottei, endemic from Auvergne, S. cebennensis, endemic from Cevennes, and S. giziana endemic from Jura, were less sampled but some material was obtained from several populations whenever possible. In Jura, samples from S. moschata and S. rosacea occurring in the same area as S. giziana were also included. Two populations of S. vayredana were sampled from north-east Iberian mountains (Catalanidic mountains, Catalan Pre-Coastal Range) and two populations of S. pubescens in Eastern Pyrenees. Only one locality was sampled for the other taxa, namely S. geraniodes, S. genesiana, S. moncayensis and S. hariotii. The geographical coordinates of sampling locations are listed in Sup. Mat. Table 1. A map of the 57 localities sampled is shown in Fig. 1.
We generated three data sets: morphometric measurements, rDNA internal transcribed spacer (ITS) sequences and genome-wide genotypes by double digest restriction site associated DNA sequencing (RADseq). The accessions used to generate molecular data are listed in Sup. Mat. Table 1. Morphometric measurements were made on 9 taxa and a subset of localities and all the data are summarized in Sup. Mat Table 2. In total, the morphological data, the ITS data and the RADseq data are based on 242, 105 and 168 individuals respectively. Sixty-four specimens, representing various sampled populations, were deposited in the GAP herbarium (voucher list in Sup. Mat. Table 1). ITS haplotypes sequences were deposited in Genbank (accession list in Sup. Mat. Table 5) and the raw RADseq reads were deposited at NCBI SRA database under Bioproject accession PRJNA935954.
DNA extraction
Total DNA was extracted from leaves stored with silica-gel using NucleoSpin Plant II kit (Macherey Nagel) following the kit instructions with slight modifications. From 40 to 50 mg of dry leaves were grinded (Qiagen Tissu Lyser), centrifuged and resuspended into 700 µl PL1 + 10 µl of RNase A for 45 min of lysis at 65 °C. After filtration, the lysate was collected taking care not to mix the solution and the last 100 µl, at the bottom of the tube, were abandoned. This modification made it possible to remove contaminants that caused abnormal 260/230 ratio values, which were measured to assess DNA purity. After this modification, we followed the manufacturer’s protocol and elution of DNA was done with 90 µl of PE solution. DNA concentrations were measured with a Nanodrop, and concentration results were trusted only if 260/230 ratio was greater than 1. Only DNA extracts with at least 20 ng/µl of DNA in 80 µl were used for RADseq.
ITS sequencing and analysis
The ITS region (ITS1 + 5.8S + ITS2) was amplified using the ITSp5 and ITSp4 primers of Chen et al. 2016. PCR was performed in a 25 μL reaction volume containing 5 μL 5X buffer (kit GoTaq G2 Flexi DNA polymerase, Promega), 1.5 mM MgCl2, 1.25 mM each PCR Nucleotide (Promega, U1420), 0.3 μM each primer, 0.2 units Taq polymerase (GoTaq G2 Flexi DNA Polymerase, M7805, Promega), and 10 ng DNA. Amplifications were carried out in an Eppendorf thermocycler. The following thermal profile was used: initial denaturation at 94 °C for 5 min, followed by 30 cycles of 45 s at 94 °C, 45 s at 55 °C and 1 min at 72 °C, and finishing with 8 min extension at 72 °C. Sanger sequencing was done by Eurofins Genomics; sequences were examined and aligned in Mega-X (Kumar et al. 2018). When the chromatograms revealed polymorphic peaks we tried to identify the ribotypes or we replaced the ambiguous site by the appropriate IUPAC code. Haplotypes were identified with Fabox (Villesen 2007) and a network was built using the haploNet function of R pegas package (Paradis 2010).
Plastome phylogeny
To include plastid data in our study, we used the plastome phylogeny of Smyčka et al. (2022). Briefly, complete circular plastomes from several species of Saxifraga sect. Saxifraga were retrieved using a genome skimming approach, then dated phylogenies were done with BEAST 2 (Bouckaert et al. 2014) with fossil calibrations in accordance with Ebersbach et al. (2017). We used only the clade including the taxa studied here.
RADseq, assembly optimization, data filtering
Genomic library preparation and sequencing were conducted by Microsynth ecogenics GmbH (Blagach, Switzerland). DNA samples (with a minimum of 1 µg input and on average 3.6 µg of DNA) were digested with the restriction enzymes EcoRI/MSeI following heat inactivation according to the manufacturer’s protocol (New England Biolabs, NEB). Fragments between 500 and 600 bp were selected by automated gel cutting (Pippin Prep system, sage science®), Illumina Y-shaped adaptors were ligated, and ligation products were bead purified. Each library was then individually barcoded by PCR using a dual-indexing strategy. Individually barcoded libraries were pooled and subsequently purified before sequencing. Three runs of paired-end sequencing 2 × 75 bp on an Illumina NextSeq platform were performed on 60, 60 and 48 samples respectively leading to an average of 4.2 million raw reads per sample. Demultiplexing and trimming of Illumina adaptor residuals was done by Ecogenics.
Ipyrad was used for reads trimming and assembly (Eaton and Overcast 2020). A pilot study was done on 18 samples to optimize the level of sequence similarity for the clustering threshold (steps 3 and 6 of ipyrad); the assembly was repeated for 7 values of this parameter from 84 to 96%. The beginning and the end of each read were trimmed (5 bp), the minimum length of reads was set on 35 bp and the quality filters were left at the default values. The minimal depth for base calls was set to 6. To limit the presence of repetitive loci we set the maximum depth, above which cluster were removed, to 1000. The parameters helping to limit paralogs were set to default values: maximum number of SNPs in a final locus (20%), the maximum number of indel by locus (4) and the maximum proportion of shared polymorphic sites in a locus (50%). The other parameters were also set to default values. After assembly, to estimate the efficiency of the assembly we first used the following ipyrad summary statistics: the number of RAD loci filtered by the maximum % of shared heterozygous sites in a locus, the sequencing error rate, the number of RAD loci, and the sum of parsimony informative sites. Then we used a custom R script to estimate the inertia of the two first axes of a principal coordinates analysis to roughly estimate the information of the dataset. According to this pilot analysis (Sup. Mat. Table 4) we found that cluster thresholds of 92% or 94% were optimal. Because the number of parsimony informative sites was slightly higher, we proceed to the assembly of the whole data set with a 92% clustering threshold. To account for the impact of the minimum number of individuals by loci (Min_ind) we produce two assemblies by setting this parameter to 24 (Min_24) and 48 (Min_48). Below these values the file sizes were difficult to handle and above these values a large amount of data was lost. We used ITS sequence as a filter to remove reads that could correspond to this sequence. The other parameters of ipyrad were set to be the same as those used during the pilot study.
IQ-TREE maximum-likelihood phylogeny of 166 RADseq genotypes
The concatenated matrix of RADseq loci was used to infer a phylogenetic tree of all samples by maximum likelihood, using the software IQ-TREE (Nguyen et al. 2015) for a maximum likelihood phylogenetic analysis. The best model of molecular evolution was determined by ModelFinder implemented in IQ-TREE (Kalyaanamoorthy et al. 2017). Branch support was estimated using the ultrafast bootstrap method (1000 iterations). Default parameters were applied. This analysis was performed on Min_24 and Min_48 data-sets and the two phylogenetic trees were compared with the comparePhylo function of ape R packages (Paradis and Schliep 2019). Tree were drawn, rooted and edited with FigTree (Rambaut 2009).
Genetic admixture analyses
Evidence for introgression were searched by estimating a co-ancestry matrix among all individuals performing RADpainter (Malinsky et al. 2018a) on the Min_24 datasets. RADpainter, is based on haplotype sequence differences and therefore uses all of the data. Because RAD loci were not mapped on a reference genome, we used the LD script, provided with RADpainter, to reduce, as recommended, the effect of linkage disequilibrium. After this step, we used RADpainter with default settings to compute a co-ancestry matrix among all individuals. The matrix was re-organized with respect to the phylogenetic tree obtained with IQ-TREE and then used to build a heat map displaying shared ancestry among individuals with a custom R script. Under a scenario of strict divergence, the RADpainter heat map organized in this way will show a contrasting color pattern with squares in warm colors (i.e. clusters of high co-ancestry genotypes) aligned on the diagonal and corresponding to clades. Incomplete lineage sorting will produce cool to warm color gradients around the squares, but these will still be aligned on the diagonal, respecting the phylogenetic structure, whereas introgression will result in warm squares appearing off the diagonal.
Genetic admixture was also estimated with snmf (R LEA package, Frichot and François 2015) which has the merit of relaxing all assumptions of the Hardy–Weinberg structure. We used the Min_24 data sets that were thinned and filtered with an R custom script to keep one SNP by locus, the one having the minimum allelic frequency, and only SNP with the less frequent allele observed in at least 3 individuals. Snmf was run for K = 2 to 6, 400 repetitions, 100 iterations, regularization parameter set to 25 and 20% of the genotypes masked to compute the cross-entropy criterion. Barplots showing ancestry coefficients were obtained with the compoplot function in adegenet R package (Jombart and Ahmed 2011), with genotypes sorted according to their order in the phylogenetic tree obtained with IQ-TREE. A second genetic admixture analysis was restricted to S. delphinensis, S. exarata, and S. moschata with the same parameters as indicated above, but we filtered individual and SNPs to have an overall missing rate below 20% and Snmf was run for 3 groups.
SVDQuartet coalescent-based phylogeny
Concatenation methods such as IQ-TREE may potentially be affected by incomplete lineage sorting and overconfidence in node support. This is a likely issue here because of the recent diversification of the taxa studied (Smyčka et al. 2022). To explicitly address this issue, we perform a coalescent-based phylogenetic inference. To reduce the influence of hybridization and missing data, we selected genotypes to maximize loci coverage and minimize admixture level (see Snmf analysis below). The ipyrad assembly was repeated with this new set of genotypes and was filtered with vcftools (Danecek et al. 2011) to retain only biallelic SNPS, a maximum of two SNP by loci, only SNPs with the minimum frequency allele present in at least 3 individuals and with a missing data rate below 50%. Then we use the vcf2phylip script (Ortiz 2019) to obtain a nexus file that was analyzed with the SVDQuartet method (Chifman and Kubatko 2014), as implemented in PAUP (Swofford 2018) to obtain a coalescent-based phylogenetic tree. The analysis was conducted at the genotype and at the species level, using all possible quartets, the multi-species coalescent and 200 replicates for the bootstrap.
Test of introgression
Introgression was tested with the Dsuite software (Malinsky et al. 2021) implementing ABBA-BABA tests and related statistics. The interpretations of ABBA-BABA tests are difficult because (i) the number of possible trios (P1, P2, P3) increase with the number of populations or species analyzed, (ii) it is not always possible to use a phylogenetic tree to eliminate non-relevant tests (iii) and the D statistics can be correlated among the branches of a phylogeny thus causing possible confusion between recent and ancient gene flow. To overcome these difficulties, we performed two analyses.
The objective of the first analysis was to confirm by a test the significant prevalence of introgression in the whole data set (Min_24). We conducted ABBA-BABA tests at the level of the RADpainter clusters of genotypes. This analysis was not guided by a phylogenetic tree and we summarize the results by analyzing the distribution of significant D statistics. We filtered the Min_24 data set with vcftools to keep one SNP per locus and a minimum of 12 individuals per SNP. Then we used the genotypes of four taxa that were grouped as a basal lineage in the maximum likelihood tree as the outgroup. Dtrios was conducted according to the BBAA pattern to organize the P1 and P2 taxa. Significant Dstat scores indicate a phylogenetic conflict due to an excess of ABBA pattern with P2 as receiver and P3 as donor. To avoid the risk of false positives we considered only Zcores above 3. We reported these results as violin plots of Dstat distributions (for Zscores > 3) to show overall patterns of the contribution, as donor or receiver, of each of the three main taxa sampled in the study: S. delphinensis, S. exarata and S. moschata.
The second analysis examined the significance of introgression. For this we re-used the SNPs dataset obtained for the SVDQuartet phylogeny in which admixed individuals were deleted. The genotypes were assigned to their species and the analysis was guided by the species-level phylogeny. We developed the f-branch method which assigns hypotheses of gene flow to internal or external branches of the phylogeny (Malinsky et al. 2018b, 2021) and therefore facilitates the interpretation by reducing the number of possible donor or receptor lineages. The f-branch metric, derived from f4-ratios, reflects excessive sharing of alleles between the P3 species and the descendants of a branch. The graphical output assigns gene flow between the branches of the phylogeny.
Morphometry
To investigate morphological distinctiveness of study taxa, we estimated or measured vegetative characters. The cushion compactness was estimated from 1 (open and loose) to 5 (compact and domed cushion). Based on leaf pictures, hairiness was estimated from 1 (total or nearly total absence of trichomes) to 4 (dense hairiness); leaf furrows were estimated as following: 1 (absence), 2 (incomplete furrow) and 3 (complete furrow from the basis to the tip of the leaf). All characters were coded by the same person. Three leaves collected per individual were scanned in the lab in order to count the blade segments and to capture the leaf shape with positioning 12 pseudo-landmarks on each leaf (R geomorph package, Adams and Otárola‐Castillo 2013) (Sup. Mat. Figure 1a). Generalized Procrustes analysis was performed, and the leaf values were averaged to obtain an estimated leaf shape for each individual. A total of 242 individuals were scored for 10 taxa (Sup. Mat. Table 3). Multivariate analyses were conducted in two steps. First, the principal components of leaf shapes variation were retrieved by a discriminant analysis with the taxa as the discriminant factor (R ade4 package, Dray and Dufour 2007). Secondly, we performed a Hill and Smith analysis (R ade4 package) to analyze the variation of all traits: hairiness, leaf furrow, cushion compactness, leaf shapes principal components and blade segments number.
Results
rDNA ITS network versus plastid phylogeny
The alignment of 105 ITS sequences (686 bp) revealed 59 variables sites and 27 haplotypes that were present together in a network (Fig. 2a). The distribution of ITS haplotypes among the samples, the taxa and their localities are shown in Sup. Mat Tables 5 and 6. Saxifraga exarata is present in almost all the network and has the main central haplotypes 1, 2, 3 and 5. By contrast S. moschata is represented by only three haplotypes, the most frequent being shared with other taxa and two singletons. The four endemics present different patterns. S. delphinensis and S. cebennensis have evolved original haplotypes but S. delphinensis shares haplotypes 3 and 5 with S. exarata, S. moschata, S. giziana (haplotype N°3) and with S. exarata (haplotype N°5) respectively. These haplotypes are observed in several locations (Sup. Mat Table 6). By contrast S. giziana and S. lamottei have only one haplotype each. Saxifraga giziana has the haplotype 3 shared with 3 other taxa (S. exarata, S. moschata, S. delphinensis) and S. lamottei has the haplotype 6 deriving from haplotype 3.

Nuclear ribosomal and plastid data report different histories for Saxifraga. A Parsimony network of ITS haplotypes, the size of the circles is proportional to the number of samples and the colors indicate the taxa. The strokes on lines indicate longer branches that were shortened. B Plastome-based time tree adapted from Smyčka et al. (2022). Blue squares indicate boostrap values above 95%. In (B) taxa names are those used by Smyčka et al. (2022), S. exarata subsp. moschata corresponds to S. moschata here
The plastome-based time tree (Fig. 2B) shows that the taxa studied here are forming two clades diverging from 3.2 Ma (1.3–5.7). It shows also the close relationship between S. moschata and S. exarata, but that S. cebennensis and S. delphinensis belong to different clades; S. cebennensis being related to S. rosacea and S. pentadactylis while S. delphinensis is at the base of the second clade which includes S. exarata and S. moschata. The sister species of S. delphinensis could be S. vayredana but their node is not robust (posterior 50%).
IQ-TREE maximum-likelihood phylogeny and admixture analyses
After filtering, the mean number of reads was of 3.6 M reads per individual. Two samples having less than one million raw reads were removed (too few loci after the assembly) and we performed loci assemblies on 166 individuals. After the assembly, the Min_24 data set retained 32,398 loci with 73% missing data and 58,138 parsimony informative sites. The mean number of loci per individual was of 9291 loci, the mean individual heterozygosity of 2.2%, and the mean sequencing error rate of 0.7% (Sup. Mat. Table 7). The Min_48 had 10931 loci, with 58% missing data and 22,519 parsimony informative sites. As expected, the mean number of loci per individual was lower (5039 loci) but the mean individual heterozygosity was identical to that of the Min_24 assembly (Sup. Mat. Table 7).
The IQ-TREE analysis performed on the Min_24 and Min_48 alignments revealed very similar trees (Sup. Res. Fig. S1). The main differences produced by changing the minimum number of individuals per locus concerned a few genotypes but didn’t change the overall phylogenetic structure. All of the following results are thus based on the Min_24 data set (summary stats of this assembly are given in Sup. Mat. Table 7). The IQ-TREE phylogeny of 166 samples (Fig. 3; Sup. Res. Fig. S2) reveals three main robust clades (ultrafast bootstrap values > 95%). Saxifraga delphinensis, S. cebennensis and S. exarata genotypes form the first clade, S. moschata, S. giziana, S. lamottei and S. rosacea the second clade and the genotypes from Aragon and Catalan Pyrenees, including S. pubescens, S. vareydana and S. moschata, the third clade.

Genome-wide phylogenetic relationships among 166 Saxifraga individuals revealed by an IQ-TREE maximum likelihood tree based on a concatenated alignment of 264,183 bp (58,138 parsimony informative sites, 73% of missing data) and a TVM + F + I + G4 model. Blue squares are indicating ultrafast bootstrap values > 95%. Numbers correspond to groups of high co-ancestry (RADpainter; Fig. 4). The tree with each genotype ID is in Sup. Res. Fig. S2
Interestingly, although S. delphinensis and S. cebennensis are not well supported, and S. delphinensis is not monophyletic, these two taxa form a robust clade with S. exarata genotypes from the Traversette Pass (Queyras) that were morphologically different from other S. exarata (see morphometry).
When compared, the IQ-TREE phylogeny and the RADpainter co-ancestry matrix match for 17 groups (Figs. 3 and 4). Their geographic distribution is summarized in the Fig. 3. Within these groups, genotypes have high co-ancestry (warm colors along the diagonal, Fig. 4) but the overall pattern of RADpainter reveal the presence of incomplete differentiation and probable gene flow among the groups, indicating a loose phylogenetic structure. For example, there is clear gradient of decreasing co-ancestry from the group “1” to group “9” that is congruent with the paraphyletic relationships between S. exarata, S. delphinensis and S. cebennensis. Interestingly, the S. exarata genotypes from Pic de Bure site (“5”) have clear co-ancestry with S. delphinensis (“1”) and S. moschata (“10”). This pattern is already noted in the Snmf admixture analysis (see below). Saxifraga moschata, S. lamottei and S. giziana are closely related as shown by their high genetic co-ancestry and they are less isolated from S. exarata than from S. delphinensis.

RADpainter co-ancestry matrix of 166 Saxifraga genotypes. The heat map was ordered according to the IQ-TREE phylogeny (left and Fig. 3) obtained on the same dataset. Genetic similarity between pairs of genotypes increases from blue to orange values. The black squares with numbers were reported for comparison with the phylogeny
As with the RADpainter heat map, the genetic admixture analysis by Snmf performed on the 166 genotypes (Fig. S2, 566 SNPs, 33% missing data) shows a general correspondence between the genetic and phylogenetic structures but without complete differentiation between clades or between taxa. The Snmf analysis (Fig. 5, 560 SNPs, 12% missing data) confirmed the clustering of the S. delphinensis, S. exarata and S. moschata into 3 genetic groups and the genetic admixture involving mostly S. exarata and S. delphinensis and to a lesser degree S. moschata. On the plateau of Pic de Bure, 10 individuals (group “5”, “P” Fig. 5) were identified in the field as S. exarata but they are genetically admixed with genetic S. delphinensis and with S. moschata to a lesser extent. The S. delphinensis and S. moschata from this locality were almost non-admixed. Genetic admixture is also supported in localities where the taxa do not occur in sympatry. In such cases it involves a contribution of the genetic group corresponding mainly to S. delphinensis sometimes distant from hundreds of kilometers away. This is very clear for S. exarata genotypes sampled from the Traversette Pass (“T” Fig. 5), and in other populations of S. exarata or S. moschata from the inner Alps, although to a lesser extent.

Admixture analysis (Snmf) performed on 99 RAD genotypes of 560 independent SNPs and 12% of missing data. Genotypes are ordered and colored according to their membership in 3 genetic clusters corresponding to S. delphinensis, S. exarata and S. moschata respectively. “P” and “T” indicate S. exarata genotypes from Pic de Bure and Traversette Pass respectively
SVDQuartet coalescent-based phylogeny
This analysis was designed to reduce the influence of incomplete lineage sorting and genetic admixture in the phylogenetic inference. It was performed on 7625 biallelic SNPs with the minimum frequency allele present in at least 3 individuals. The SVDQuartet phylogeny, supported by 76% of the quartets (Fig. 6), revealed a different phylogenetic structure than the IQ-TREE phylogeny. Indeed, a robust clade unites S. delphinensis and S. cebennensis in one clade and S. moschata, S. lamottei and S. giziana in a second. This clade derives from S. exarata which is again paraphyletic.

Coalescent-based phylogeny by SVDQuartet method of 32 Saxifraga genotypes. Exhaustive quartet sampling was conducted in a matrix of 7625 biallelic SNPs. The boostrap values near the nodes were obtained with 200 replicates. Branch lengths are not meaningful
Tests of introgression
ABBA-BABA tests, performed on the 166 genotypes dataset assigned to the 17 groups (Fig. 4) and without phylogenetic guide tree (Sup. Mat. Table 8; Fig. 7B, C), revealed 313 out of 680 trios with Zcores greater than 3 (all pval ***, Sup. Mat. Table 8) with a Dstat ranging from 3 to 60% and a median of 22%, supporting a high excess of ABBA pattern in the data set. Such result indicates that introgression, rather than incomplete lineage sorting, is at the source of the incomplete differentiation and phylogenetic conflicts revealed previously. The median Dstat is higher when S. exarata is the receiver (P2, 30%, Fig. 7B) and lower when it is the donor (P3, 19%, Fig. 7C). The second analysis (Fig. 7D, E) was performed on the 32 genotypes used for the SVDQuartet phylogeny. The genotypes were assigned to their taxa and the analysis was guided by the SVDQuartet phylogeny. The ABBA-BABA tests revealed 11 out of 56 trios with Zscores greater than 3 (all pval **, Fig. 7D, Sup. Mat. Table 9) with Dstat ranging from 11 to 25%, thus indicating significant excess of ABBA pattern. The detected introgressions involve (i) S. delphinensis as a receiver (P2) with S. exarata or S. vayredana as a donor (P3), (ii) S. exarata as a receiver with S. vayredana as a donor, (iii) S. moschata and S. giziana as receivers with S. exarata as a donor, and (iv) S. pubescens as a receiver and S. vayredana as a donor. The f-branch metric (Fig. 7E) confirmed gene flow between S. exarata and S. delphinensis, S. vayredana and S. delphinensis and between S. exarata and the ancestor of S. moschata and S. giziana.

Results of Dsuite introgression tests. A–C ABBA-BABA tests performed on 166 genotypes assigned to 17 groups (Fig. 4), only Dstat with Zscores > 3 were kept. A Positive Dstat scores indicate an excess of ABBA pattern and an introgression from P3 to P2. B and C Violin plots of the Dstat distribution according to P2 or P3 for the three main species sampled here, S. delphinensis (DEL), S. exarata (EXA) and S. moschata (MOS). D–E Dsuite analyses guided by a coalescent-based species phylogenetic tree and performed on individuals with low genetic admixture. D Results of ABBA-BABA tests with z-scores > 3. E Results of f-branch analysis. The phylogenetic tree is shown on x and y axes, colored squares indicate significant excess of SNPs sharing caused by gene flow. White or grey squares represent non-significant values or trios incompatible with the phylogeny
Morphological differences with overlap
The ten taxa show largely overlapping vegetative phenotypes (Fig. 8). The main axis is opposing, left to right, phenotypes with absent or incomplete furrow to phenotypes with complete furrow, strong hairiness, and 3 to 5 or more leaf blade segments. The second axis is explained by compact, dense cushions with stocky leaves versus flat, loose cushions with slender leaves.

Multivariate Hill & Smith analysis of vegetative polymorphism of Saxifraga based on principal components of leaf shapes (Leaf_PC), number of leaf blade segments (Leaf_tips), absence of leaf furrow (Furrow 1), incomplete leaf furrow (Furrow 1.5), complete leaf furrow (Furrow 2), multiple leaf furrow (Furrow3), leaf hairiness from nearly absent (Hairiness 1) to very dense (Hairiness 2) and cushion form from loose and open (Cushion 1) to compact and domed cushion (Cushion 4)
The leaf furrow is always present and complete in S. delphinensis, S. cebennensis, S. exarata, S. pubescens and S. vayredana. It is rarely present or it is incomplete in S. moschata, S. giziana and S. lamottei. The leaf hairiness is very dense in S. delphinensis, S. cebennensis, S. pubescens, S. vayredana and the Traversette Pass (“TP”) population of S. exarata. In contrast it is either sparse or absent in S. moschata and S. lamottei. Note that S. giziana differs from S. moschata through its stronger hairiness (not seen in Fig. 8, see Sup. Mat Table 3). For S. exarata, the leaf hairiness can be either dense or sparse. The cushion is very compact and domed in most S. delphinensis; compact but not domed in S. cebennensis, often loose and flat in S. exarata. Both loose and compact cushions were observed in S. moschata. Note that it is not possible to distinguish S. vayredana from S. pubescens using the morphological traits measured in this study, but both have a high number of leaf blade segments and slender leaves. These traits are also characteristic of S. exarata phenotypes from the Traversette Pass, which are well differentiated from other S. exarata.
Discussion
With 400–500 species, Saxifraga displays its greatest diversity in mountain areas and has one of the highest endemism rates in the Alps (Aeschimann et al. 2011). According to Ebersbach et al. (2017) and Folk et al. (2019), Saxifraga, and related genera, originated at the end of the Cretaceous period in rare cold habitats, probably restricted to arctic or alpine North America and then colonized Europe during the late Eocene (ca 33 Ma). Appearing in the early Miocene (ca 19 Ma), the Saxifraga sect. Saxifraga evolved with a constant rate of diversification, mainly driven by geographic isolation and bedrock shifts (Smyčka et al. 2022). The lineage studied here originated at the end of the Pliocene (3.8 million years ago; Fig. 2, Smyčka et al. 2022) and now forms several species, sometimes rare and endemic, in the European mountains.
We examined the distinctiveness of S. delphinensis, based on morphological, molecular, and genome-wide sequencing data. This narrow endemic is considered in the literature as morphologically well differentiated from closely related species occurring in the same region (Tison and Foucault 2014). Apart from the high-altitude areas in the Vercors and Devoluy, its distribution mainly corresponds to unglaciated areas during the last glacial maximum (Seguinot et al. 2018). This historical fact reinforces the hypothesis of a differentiation acquired over a long period. Our results support an origin probably earlier than the Würm and Riss glaciations. Indeed, the differentiation of S. delphinensis is significant with a characteristic leaf and cushion morphology, a plastid lineage deeply rooted in the Pleistocene, the existence of original and specific ITS haplotypes, and the grouping of almost all its RADseq genotypes into one clade. However, despite this evolutionary distinctiveness, our various analyses further reveal a gradient of genetic admixture between S. exarata and S. delphinensis that supports fertility of F1 hybrids allowing for backcrosses. The introgression between S. delphinensis and S. exarata is particularly frequent in high-altitude sites in the Vercors (Grand Veymont, Royou) or in the Devoluy (Pic de Bure). Morphologically, this introgression is difficult to discern but some hybrids resemble S. delphinensis (strong hairiness, stocky leaf shape and compact cushion) but without having a dense, domed cushion. The final rebuttal to the complete isolation of S. delphinensis came from the Diois area (Eysahut and Saou populations) where introgression causes S. delphinensis to be polyphyletic in the RADseq phylogeny or the ITS network. The genotypes from Eysahut (Diois) population are situated at an altitude of only 700 m of elevation, on the extreme western margin of the Alps. At this site, S. delphinensis shows introgression from S. exarata with which it also shares an ITS haplotype. As far as we know, the closest S. exarata population is situated approximately 20 kms away. In Saou, S. delphinensis, living in sympatry with S. moschata, shows clear signs of introgression from S. moschata, with a shared ITS haplotype. This result is rather surprising since the introgression between S. moschata and S. delphinensis was not expected because of their very rather distinct morphologies in the field.
Our analysis also detected hybrids between S. exarata and S. moschata but at a lesser extent, despite their frequent sympatry. Recently Carnicero et al. (2023) also reported genetic admixture and nuclear-plastid mismatches for Pyrenean taxa, among which S. moschata and S. pubescens, but they concluded that the consequence of introgression was limited due to the partial sterility of hybrids and the absence of allopolyploidy that could fix hybrid genomes. By contrast to S. moschata from the Pyrenees (Mas de Xaxars et al. 2015), our study revealed a reduced ITS haplotypes diversity for S. moschata in south-west Alps and for its relatives S. giziana and S. lamottei. This indicates a recent diversification maintained by the geographical or ecological isolation. In the Jura Massif, the very low altitude (c. 350 to 500 m), favouring an earlier reproduction period than in the higher massifs, probably contributed to the genetic isolation in the periphery of S. moschata range. The present relative isolation of these populations combined with their low pollen-dispersal capacity or pollen limitation (García-Camacho and Totland 2009) indicates that introgression took place very early in their divergence and explain why S. moschata and S. giziana maintain their morphological differences (Mouly and Ferrez 2020).
Limits and perspectives
A limit of our study was the lack of chromosome counts and genome size estimations, which are known to be very variable in these taxa and more particularly in S. exarata (Vargas 2000) and S. moschata (Mas de Xaxars et al. 2015; Carnicero et al. 2023). Polyploidy is an important factor of evolution and speciation in plants and the separation of diploids from polyploids helps to optimize bioinformatics and phylogenomics by reducing the effect of paralogy (Karbstein et al. 2020; Carnicero et al. 2023). Paralogs, if they are present in the data, can lead to false phylogenetic relationships. However, we found the main phylogenetic clades rather robust in the face of changing of assembly parameters or when we repeat the assembly and analysis by limiting admixture. Even if we cannot reject the possible effect of paralogy, we are confident that our phylogenetic results are valuable in the context of studying S. exarata, S. moschata and their relatives in the western Alps. In the phylogenies shown here, some relationships are robust and allow to confirm some differences among taxa. For example, the complete leaf furrow is a reliable characteristic to differentiate S. moschata from S. exarata and we observed that hybrids have partial leaf furrow. Leaf furrow, which is easier to observe on fresh material, can help to correct several confusions between these taxa. For example, Saxifraga exarata still appears in the Pyrenees in the GBIF database (https://www.gbif.org consulted 2/02/2023) or the POWO database (https://powo.science.kew.org consulted 14/03/2023) because S. moschata was formerly a subspecies of S. exarata.
The incomplete isolation taxa shown here is challenging to reconcile taxonomy with evolutionary theory. It challenges also our understanding of speciation and how species maintain their genetic integrity. Almost a century ago, Lotsy (1925) illustrated the syngameon concept with Saxifraga and Betula case studies. This concept has recently enjoyed renewed interest (Cannon and Petit 2020) because it emphasizes the role of adaptive introgression at ecological margins (Buck et al. 2022). The question of whether the syngameon is a relevant issue for S. exarata and its relatives remains to be answered. In parallel with this theoretical aspect, our results suggest that S. exarata probably encompass unknown diverging lineages that need to be studied more thoroughly. We confirmed the presence of at least two different forms of S. exarata in the Queyras, which were already suggested by Villars (1779). The population from the Traversette Pass is morphologically differentiated from S. exarata. According to RADseq based phylogeny, it forms a new lineage close to S. delphinensis and S. cebennensis but not completely isolated from S. exarata as shown by its shared co-ancestry or genetic admixtures with other S. exarata.
Last but not least, we must acknowledge that Ravaud proposed a close relationship of S. delphinensis with Pyrenean taxa based on morphological similarities (Ravaud 1885). At first glance, this hypothesis is not supported by RADseq phylogeny or ITS network, but we cannot exclude an early influence of taxa from the Pyrenees or north Iberian mountains in the history of S. delphinensis. First, the plastome phylogeny (Smyčka et al. 2022) suggests a close relationship with the Iberian endemic S. vayredana, which is confirmed by ABBA-BABA tests and f-branch analysis of RADseq data. Second, S. delphinensis forms a well-supported clade with S. cebennensis and with the S. exarata population from the Traversette Pass and the morphological characteristics of this clade (three to five leaf tips, strong leaf pilosity and complete leaf furrow) are also observed in certain Iberian taxa such as S. pubescens or S. vayredana. Such influence is quite consistent with the hypothesis of Ravaud (1885). Future research will aim to establish the sequence of divergence and hybridization events that shaped this story during Quaternary range shifts through several mountain ranges in Europe.
Data availability
Molecular data are available online (NCBI). Morphometrical and occurrence data are available in supplementary material.
References
Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJ, Bierne N et al (2013) Hybridization and speciation. J Evol Biol 26:229–246
Adams DC, Otárola-Castillo E (2013) geomorph: an R package for the collection and analysis of geometric morphometric shape data. Methods Ecol Evol 4:393–399
Aeschimann D, Lauber K, Moser DM, Theurillat JP (2004) Flora alpina: atlas des 4500 plantes vasculaires des Alpes. Belin
Aeschimann D, Rasolofo N, Theurillat JP (2011) Analyse de la flore des Alpes. 2: biodiversité et chorologie. Candollea 66:225–253
Bouchard J (1947) Saxifrages nouveaux. Bull Soc Bot Fr 93:214–219
Bouckaert R, Heled J, Kühnert D et al (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537
Buck R, Ortega-Del Vecchyo D, Gehring C, Michelson R, Flores-Rentería D, Klei B, Whipple AV, Flores-Rentería L (2022) Sequential hybridization may have facilitated ecological transitions in the Southwestern pinyon pine syngameon. New Phytol 237:2435–2449
Cannon CH, Petit RJ (2020) The oak syngameon: more than the sum of its parts. New Phytol 226:978–983
Carnicero P, Kröll J, Schönswetter P (2023) Homoploid hybrids are common but evolutionary dead ends, whereas polyploidy is not linked to hybridization in a group of Pyrenean saxifrages. Mol Phylogenet Evol 180:107703
Chifman J, Kubatko L (2014) Quartet inference from SNP data under the coalescent model. Bioinformatics 30:3317–3324
Christie K, Fraser LS, Lowry DB (2022) The strength of reproductive isolating barriers in seed plants: Insights from studies quantifying premating and postmating reproductive barriers over the past 15 years. Evolution 76:2228–2243
Cuénot L (1917) Sepia officinalis est une espèce en voie de dissociation. Arch Zool Exp Gén 56:315–346
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, 1000 Genomes Project Analysis Group (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
de Queiroz K (2020) An updated concept of subspecies resolves a dispute about the taxonomy of incompletely separated lineages. Herpetol Rev 51:459–461
Dray S, Dufour AB (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22:1–20
Eaton DA, Overcast I (2020) ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 36:2592–2594
Ebersbach J, Muellner-Riehl AN, Michalak I, Tkach N, Hoffmann MH, Röser M, Sun H, Favre A (2017) In and out of the Qinghai-Tibet Plateau: divergence time estimation and historical biogeography of the large arctic-alpine genus Saxifraga L. J Biogeogr 44:900–910
Ebersbach J, Tkach N, Röser M, Favre A (2020) The role of hybridisation in the making of the species-rich arctic-alpine genus Saxifraga (Saxifragaceae). Diversity 12:440
Edelman NB, Mallet J (2021) Prevalence and adaptive impact of introgression. Annu Rev Genet 55:265–283
Feliner GN (2014) Patterns and processes in plant phylogeography in the Mediterranean Basin. A review. Perspect Plant Ecol Evol Syst 16:265–278
Folk RA, Stubbs RL, Mort ME, Cellinese N, Allen JM, Soltis PS, Soltis D, Guralnick RP (2019) Rates of niche and phenotype evolution lag behind diversification in a temperate radiation. PNAS 116:10874–10882
Frichot E, François O (2015) LEA: An R package for landscape and ecological association studies. Methods Ecol Evol 6:925–929
García-Camacho R, Totland Ø (2009) Pollen limitation in the alpine: a meta-analysis. Arct Antarct Alp Res 41:103–111
Garraud L (2004) Flore de la Drôme. Atlas écologique et floristique. Conservatoire Botanique National Alpin de Gap-Charance
Grant V (1981) Plant speciation. Columbia University Press, Columbia
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071
Kalyaanamoorthy S, Minh BQ, Wong TK, Von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Karbstein K, Tomasello S, Hodač L, Dunkel FG, Daubert M, Hörandl E (2020) Phylogenomics supported by geometric morphometrics reveals delimitation of sexual species within the polyploid apomictic Ranunculus auricomus complex (Ranunculaceae). Taxon 69:1191–1220
Kerguélen M (1993) Index synonymique de la flore de France. Muséum national d’histoire naturelle, Paris
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547
Lotsy JP (1925) Species or linneon. Genetica 7:487–506
Luizet D (1913) Contribution à l’étude des Saxifrages du groupe des Dactyloides Tausch. Bull Soc Bot Fr 60:58–64
Malinsky M, Trucchi E, Lawson DJ, Falush D (2018a) RADpainter and fineRADstructure: population inference from RADseq data. Mol Biol Evol 35:1284–1290
Malinsky M, Svardal H, Tyers AM, Miska EA, Gener MJ, Turner GF, Durbin R (2018b) Whole-genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nat Ecol Evol 457:830
Malinsky M, Matschiner M, Svardal H (2021) Dsuite-Fast D-statistics and related admixture evidence from VCF files. Mol Ecol Res 21:584–595
Mallet J, Besansky N, Hahn MW (2016) How reticulated are species? BioEssays 38:140–149
Mas De Xaxars G, García-Fernández A, Barnola P, Martín J, Mercadé A, Vallès J, Vargas P, Vigo J, Garnatje T (2015) Phylogenetic and cytogenetic studies reveal hybrid speciation in Saxifraga subsect. Triplinervium (Saxifragaceae). J Syst Evol 53:53–62
Mouly A, Ferrez Y (2020) D’une saxifrage endémique jurassienne (France), Saxifraga giziana (Saxifragaceae): historique et publication de nom. Nouv Arch Flore Jurass 17:133–137
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
Ortiz EM (2019) Vcf2phylip. GitHub. https://github.com/edgardomortiz/vcf2phylip
Paradis E (2010) Pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26:419–420
Paradis E, Schliep K (2019) ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35:526–528
Rambaut A (2009) FigTree v1. 3.1. http://tree.bio.ed.ac.uk/software/figtree/
Ravaud LC (1885) Guide du botaniste dans le Dauphiné. Excursions bryologiques et lichénologiques (6me excursion). Le Grand-Veymont et le Diois. Les forêts du Vercors. Drevet Editeur, Grenoble
Rieseberg LH, Wood TE, Baack EJ (2006) The nature of plant species. Nature 440:524–527
Seguinot J, Ivy-Ochs S, Jouvet G, Huss M, Funk M, Preusser F (2018) Modelling last glacial cycle ice dynamics in the Alps. Cryosphere 12:3265–3285
Smyčka J, Roquet C, Boleda M et al (2022) Tempo and drivers of plant diversification in the European mountain system. Nat Commun 13:1–13
Suarez-Gonzalez A, Lexer C, Cronk QC (2018) Adaptive introgression: a plant perspective. Biol Lett 14:20170688
Swofford DL (2018). PAUP*(* Phylogenetic Analysis Using PAUP). Version 4a161
Tison JM, de Foucault B (2014) Flora gallica: flore de France. Biotope, New York
Tkach N, Röser M, Miehe G, Muellner-Rieh AN, Ebersbach J, Favre A, Hoffmann MH (2015) Molecular phylogenetics, morphology and a revised classification of the complex genus Saxifraga (Saxifragaceae). Taxon 64:1159–1187
Vargas P (2000) A phylogenetic study of Saxifraga sect. Saxifraga (Saxifragaceae) based on nrDNA ITS sequences. Plant Syst Evol 223:59–70
Villars D (1779) Histoire des Plantes de Dauphiné tome 3:673–674
Villesen P (2007) FaBox: an online toolbox for fasta sequences. Mol Ecol Notes 7:965–968
Voisin C, Dentant C, Rioux D, Boucher FC (2023) Introgression of an isolated Primula lineage suggests the existence of a glacial refugium in the Écrins range (Southwestern French Alps). Alp Bot 133:21–33
Acknowledgements
Data used in this study were produced in part in the technical facilities of the SCBM (IMBE, Marseille). All bioinformatics were done on the High-Performance Computing Cluster from the Pytheas IT facility (OSU Institut Pytheas Aix Marseille Univ, INSU-CNRS UMS 3470). Our grateful thanks to the following people for their help in obtaining the authorization to collect plant or for field sampling: Camille Thomas (PNR Volcan d’Auvergne), Thierry LEROY (Réserve naturelle nationale de Chastreix-Sancy), Pierre-Mari Le Hénaff (Conservatoire Botanique National Massif Central), Mathias Pires (Conservatoire Botanique National Méditerranéen), Nadia Noblet (PNR Vercors). Our grateful thanks to Matthieu Lefebvre (GRM curator, Museum Grenoble City) for providing precious information on saxifrage herbarium specimens, Régnier Camille (IMBE) for her help scanning the plant leaves, Baptiste Pierre (IMBE) for his help drawing the sampling map and to the IMBE administrative staff for their support. Professional English editing was performed by Thomas Smith.
Funding
Devoluy municipality, CNRS UMR 7263.
Author information
Authors and Affiliations
Contributions
AB, CR, LG, SA, CD, AM, MV and MC contributed to the sampling and to design of the study. AB performed the bioinformatics of RADseq and morphometric analyses. AB, KC, CM, JS and LA performed molecular data analyses. AB wrote the manuscript with the help of CR, SL, LG, SA, CD, AM and MV and all authors approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest to disclose. Funding for this study was provided by Dévoluy municipality (Le Dévoluy FR-05250) and by the IMBE.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Baumel, A., Roquet, C., Lavergne, S. et al. Evolutionary distinctiveness with incomplete isolation of the narrow endemic alpine plant Saxifraga delphinensis Ravaud. Alp Botany 133, 85–99 (2023). https://doi.org/10.1007/s00035-023-00297-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00035-023-00297-2