
Abstract
Vascular smooth muscle cells (VSMCs) are involved in phenotypic switching in atherosclerosis. This switching is characterized by VSMC dedifferentiation, migration, and transdifferentiation into other cell types. VSMC phenotypic transitions have historically been considered bidirectional processes. Cells can adopt a physiological contraction phenotype or an alternative "synthetic" phenotype in response to injury. However, recent studies, including lineage tracing and single-cell sequencing studies, have shown that VSMCs downregulate contraction markers during atherosclerosis while adopting other phenotypes, including macrophage-like, foam cell, mesenchymal stem-like, myofibroblast-like, and osteochondral-like phenotypes. However, the molecular mechanism and processes regulating the switching of VSMCs at the onset of atherosclerosis are still unclear. This systematic review aims to review the critical outstanding challenges and issues that need further investigation and summarize the current knowledge in this field.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mature VSMCs modulate the diameter of blood vessels and hence regulate blood flow distribution and blood pressure. In the blood vessels in adults, VSMCs exhibit low synthetic activity and proliferation rates and possess special signalling molecules, ion channels, and proteins that regulate the contraction of blood vessels. Differentiated VSMCs show a high level of plasticity, allowing them to flexibly change phenotypes depending on the local environmental stimuli [1]. However, most studies have traditionally focused on the conversion of VSMCs from contractile to secretory types, whereas the current research focuses on subsets of smooth muscle cells (SMCs) and their transformation to different cell types. Contractile VSMCs often alter their phenotypes and proliferation and migration patterns to participate in tissue repair. Once the reparative process ends, VSMCs regain their nonproliferating contractile phenotype. In atherosclerosis, a fibrous cap forms at the injury site. The restoration of VSMCs is dysregulated in certain contexts, leading to the transformation of VSMCs into other cell types, e.g., fibroblast-like cells [2, 3], macrophage-like cells [3,4,5,6,7,8,9], adipocyte-like cells [10, 11], and chondrocyte-like cells [2, 3, 12, 13]. In addition, accumulating evidence suggests that these transitions to an abnormal phenotype may contribute to vascular diseases, e.g., atherosclerosis. Understanding the mechanisms and signalling cascades that lead to a specific phenotype could uncover important avenues for designing effective interventions for vascular diseases.
Phenotype of VSMCs
Contractile vs synthetic VSMC phenotype in atherosclerosis
The transition of VSMCs from a basal state contractile phenotype to a so-called synthetic phenotype was first described in the early 1980s [14]. In healthy arteries, medial arterial contractile VSMCs are spindle-shaped and function as arterial elastic constrictors and produce extracellular matrix (ECM). SMC-specific differentiation proteins are mainly important components of the contractile machinery and cell anchoring of smooth muscle cells (e.g., α-smooth muscle actin (α-SMA), smooth muscle 22α (SM22α), myosin heavy chain 11 (MYH11), calmodulin, and h-calmodulin). In disease states such as atherosclerosis and vascular injury, VSMCs switch to a secretory phenotype characterized by reduced expression of SMC-specific markers and a change in cell morphology from elongated/spindle-shaped to rhombic/epithelial-like [15]. It is critical to note that the attributes listed above refer to the "averaged" or "ideal" dedifferentiation state. Within an injured or sick vasculature, populations of dedifferentiated VSMCs can be extremely variable, both in terms of dedifferentiation grade and phenotypic divergence caused by exposure to different environmental signals.
VSMCs-derived phenotype in atherosclerosis
VSMCs play a role in atherosclerosis by remodelling the arterial wall to sustain blood flow in damaged arteries due to atherosclerotic alterations. VSMCs have been proven in vitro and in vivo to be capable of transdifferentiating into other cell types, such as chondrocyte-like cells, macrophage-like cells and foam cells. As early as 1968, Wissler proposed the idea that VSMCs formed at least part of the foam cells in atherosclerotic plaques [16]. Later, Andreeva et al. found colocalization of the macrophage marker CD68 and the smooth muscle cell marker α-SMA in atherosclerotic human aortic cells [17]. In addition to forming macrophage-like cells and foam cells, VSMCs can also transdifferentiate into osteoblastic/chondrocyte-like cells [12, 13, 18]. During the VSMC phenotypic transition, VSMC-specific marker expression changes, such as the loss of α-SMA, SM22α, and MYH11. At the same time, VSMCs gain the ability to express markers of other cell types such as pluripotent vascular stem cells, macrophages, adipocytes and fibroblasts, which posses a significant challenge for VSMC identification.
Detection methods for VSMCs phenotypes
Traditional methods for detection of VSMC phenotype
Traditional methods for detecting the phenotype of VSMCs generally include observation of cell morphology under light microscopy, transmission electron microscopy, phase-contrast microscopy, and immunohistochemical blotting and immunofluorescence methods to detect SMC-specific differentiation markers [19,20,21]; however, these methods may misidentify cell types, and therefore, the use of advanced gene fate targeting technology is required.
New approaches for detection of VSMCs phenotype
The advent of lineage tracing and single-cell sequencing in recent years has not only confirmed the previous view that VSMCs transdifferentiate into macrophage-like cells and chondrocyte-like cells but has also revealed the transdifferentiation of VSMCs to various cell phenotypes. The differentiation of SMCs into 'inflammatory' macrophage-like cells may contribute to the instability of atherosclerotic lesions, whereas the transition of SMCs to 'synthetic' fibrotic SMCs may stabilize lesions by increasing the thickness of the protective fibrous cap. These unique cells derived from VSMCs drive the molecular events that regulate atherosclerotic plaque stability, including fibrous cap formation and rupture, lipid retention, inflammation, calcification, and extracellular matrix composition. Whether the phenotypic shift in SMCs is primarily protective or detrimental to atherosclerosis and whether both effects depend on the microenvironment remain uncertain; however, it is possible that VSMC-derived cells play a dual role such that they not only enhance plaque stability but also exacerbate plaque rupture.
Lineage tracing of VSMCs in atherosclerosis
Traditional pedigree tracing methods include those that rely on dye injection, fluorescent proteins, or marker tracing. With the development of genetic recombination and sequencing technologies, related barcoding techniques relying on the Sleeping Beauty transposase, Cre-loxP and CRISPR-Cas9 systems have gradually been developed and are widely used [22]. Several studies based on lineage tracing advanced our understanding of the functional consequences of clonality, plasticity and eventually the fate of VSMCs within plaques, indicating that they play a more significant and complex role in atherosclerosis than previously believed [6, 23,24,25,26,27,28]. In mice, Feil et al. used lineage tracing to demonstrate that VSMCs can clonally expand and transform into macrophage-like cells, which constitute a major component of advanced atherosclerotic lesions [5]. Shankman et al. showed that > 80% of smooth muscle cells in advanced atherosclerotic plaques lose classical contractile markers, such as ACTA2, 30% of such cells have an activated state of lgals3, and a small proportion of these cells express mesenchymal stem cell (MSC) markers or myofibroblast-like markers, such as SCA1 and ACTA2/PDGFRβ [6]. Notably, the knockout of Krüppel-like factor 4 (KLF4), a transcription factor required for VSMC de-differentiation, led to a significant decrease in the number of macrophage-like cells and the extent of atherosclerotic lesions [6]. Dobnikar et al. also identified VSMC-derived cells expressing the MSC marker SCA1 in healthy arterial media and atherosclerotic plaques [29]. These cells may represent a highly flexible intermediate population that is highly inflammatory-susceptible and capable of generating VSMCs that can contract or phenotypically switch. Several other studies have shown that fibrotic caps include most αSMA-positive cells [30,31,32], rejecting earlier assumptions that αSMA-positive cells are generated by bone marrow-derived cells [33, 34]. The rainbow confetti system combined with the genetic lineage tracing strategy of VSMCs has demonstrated, surprisingly, that the origin of VSMCs in mouse atherosclerotic plaques arises from the oligoclonal expansion of a few cells in the vessel wall [5, 26,27,28]. Chappell et al. used multicolour labelling to demonstrate that VSMCs in atherosclerotic plaques are oligoclonal, originating from a small subpopulation of VSMCs [26]. Additionally, lineage tracing revealed that the progeny of individual VSMCs were involved in Mac-3-expressing macrophage-like core cells in plaques in addition to constituting αSMA-positive fibrous caps. Jacobsen et al. reached a similar conclusion based on an analysis of the clonal structure of SMCs in chimeric mice and mice with a randomly recombinant fluorescent transgene [27]. The above studies suggest that the contribution of SMCs to disease arises from the overproliferation of a few pre-existing medial cells and that plaque cell expression from VSMCs is traditionally associated with macrophages (CD68, LGALS3/MAC2 and LAMP2/MAC3) [5, 23, 26, 28], mesenchymal stem cells (SCA1) [6, 29], myofibroblasts (ACTA2/PDGFRβ) [6, 28] and chondrocyte-like cells (BMP2, RUNX2, OPN, ALP and BGLAP) [13, 35,36,37,38]. Genetic fate mapping of VSMCs in atherosclerosis is presented in Table 1.
Single-cell RNA sequencing of VSMCs in atherosclerosis
Single-cell RNA-seq (scRNA-seq) analyses examine the gene expression of each cell and assemble cells into distinct subpopulations [39]. This approach has shown that cells express a whole set of genes that are known to participate in the conversion process, and although these cells do not necessarily show the characteristics of fully converted cells, they can be found during the conversion process in a way that was not possible before.
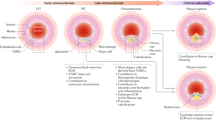
Recently, single-cell RNA sequencing was employed to gain a better understanding of the heterogeneity of atherosclerotic plaques, including heterogeneity in specific cell types. In particular, even in healthy arteries, VSMCs show variation in morphology and gene expression, including the discovery of rare, atypical SCA1-positive VSMCs and encoding genes associated with phenotypic behavioural transitions [29, 40]. Alencar et al. constructed a model of thoracic aortic atherosclerosis and then collected samples for RNA-seq and chromatin immunoprecipitation sequencing of KLF4 and octamer-binding transcription factor-4 (OCT4). These authors found that SMC-specific KLF4 gene deletions exhibit genomic features almost opposite to those of Oct4 knockouts and that their putative target genes play an important role in regulating SMC phenotypic changes [41]. The authors also found that Lgals3-positive vascular smooth muscle-derived cells appear to be an earlier transitional state that subsequently gives rise to at least three other SMC phenotypes in advanced lesions, including a KLF4-dependent osteogenic phenotype that may lead to plaque calcification and plaque instability [41]. Similarly, Pan et al. combined scRNA-seq and SMC fate maps of human and mouse atherosclerotic plaques to identify an intermediate cell state of SMC origin [3]. SMC-derived intermediate cells are referred to as 'SEM' cells with pluripotent potential due to their ability to differentiate into macrophage-like cells and fibrochondrocyte-like cells and revert to an SMC-like phenotype. Furthermore, in symptomatic human atherosclerosis, all-trans retinoic acid (RA) prevented the translocation of SMCs to SEM cells by activating RA signalling, reducing the atherosclerotic burden and promoting fibrous cap stabilization. Subsequently, the authors identified several retinol-responsive genes with downregulated expression in unstable human atherosclerotic lesions. Notably, these genes were closely associated with coronary heart disease (CAD)-related risk variants. However, these associations require further investigation. Wirka et al. analysed human and mouse atherosclerotic lesion samples by scRNA-seq and found that the transcriptional phenotype of SMCs in the samples was regulated and that these cells acquired distinct fibroblast-like cell phenotypes instead of the typical macrophage-like phenotype [2]. However, this finding contradicts the results obtained in other studies investigating macrophage-like SMCs in mouse and human lesions [4,5,6,7]. This discrepancy may be explained as follows: previous studies were performed in the aortic root, which contains numerous mesenchymal and epicardial cells, thereby severely limiting the sensitivity of detecting VSMC phenotypic shifts in the lesion. This discrepancy has prompted researchers to pay attention to the heterogeneity of spectral tracer markers, objective factors, such as the dose of tamoxifen, and the need to combine lipid staining with spectral tracers or molecular markers of cellular status when analysing macrophage states in atherosclerosis. Dobnikar et al. found significant differences in gene expression in seven clusters that segregate Myh11-expressing SMCs into VSMCs within the abdominal aorta (AA) and thoracic aortic (TA) [29]. Several of these genes are involved in the regulation of VSMC phenotype, inflammation, and cardiovascular disease, suggesting that VSMCs perform diverse functions. These findings elucidated the clonality of SMC invasion and growth in atherosclerotic plaques. Wong et al. completed the eQTL mapping of human coronary artery smooth muscle cells (HCASMCs) by analyzing RNA-seq data and whole genomes of unrelated multiethnic donors, and the results indicated that certain SMC genes (e.g., SMAD3, PDGFRA, and SIPA1) may increase the risk of CAD, while other SMC genes (e.g., TCF21 and FES) may exert antiatherosclerotic effects. Transcription factor 21 (TCF21) is a CAD gene associated with the disease. Specific knockdown of TCF21 in SMCs significantly inhibits phenotypic regulation of SMCs in mice, leading to a reduction in fibroblasts and protective fibrous caps at damaged sites. Furthermore, in patients' coronary arteries, TCF21 expression was highly connected with SMC phenotypic regulation, whereas increased TCF21 expression was associated with a decreased risk of developing CAD in human CAD-associated tissues [2, 42]. These results suggest that the conversion of VSMCs to fibroblasts has atheroprotective effects. Single-cell transcriptome technology also identified a small number of VSMCs that may be Sca1 + progenitor cells that 'overreact' to inflammatory stimuli [43]. Tang et al. performed scRNA-seq and cell fate mapping and found that indeterminate Sca1 + PDGFRa + cells in the arterial wall transmigrated into the medial layer after severe injury, where they formed SMCs, which is similar to previous research showing that the contribution of SMCs to disease arises from the overproliferation of a few pre-existing medial cells [44]. Further studies found that SMCs formed under the control of yes-associated protein (YAP) had a higher proliferative capacity in vascular repair than pre-existing SMCs [44]. These findings concerning heterogeneity offer new possibilities to study different smooth muscle cell subpopulations and their function in atherosclerosis (Fig. 1).

Overview of vascular smooth muscle cells (VSMC) phenotypic transition within media and atherosclerotic lesions in mice. Lineage tracing and scRNA-seq studies have shown that partially contractile VSMCs transform into transitional pluripotent cells (e.g. lgals3 + VSMCs, SEM cells and MSC-like SMCs) in response to environmental stimuli. SEM cells have the potential to differentiate into macrophage-like SMCs and fibrochondrocyte-like SMCs and even revert to their contractile phenotype. ATRA can inhibit the conversion of SMCs to SEM cells. KLF4-dependent Lgals3-activated VSMCs subsequently exhibited a shift to a variety of SMC phenotypes, including inflammatory cells, ECM-rich cells, osteogenic phenotypes and macrophage-like cells. Whether MSC-like SMCs can differentiate into other SMC phenotypes is currently unclear. There are two possible sources of SMC foam cells in atherosclerotic plaques. One is foam cells derived directly from SMCs and the other is from macrophage-like SMCs. It is unclear whether SMCs need to acquire a macrophage-like phenotype to become foam cells, or whether they become foam cells before expressing macrophage markers. OCT4 and SMC-specific knockout of TCF21 inhibit SMC phenotypic modulation in mice. ATRA indicates all-trans retinoic acid; KLF4, krüppel-like factor 4; TCF21, transcription factor 21; OCT4, octamer-binding transcription factor 4
Factors and signalling pathways influencing phenotypic switching of VSMCs in atherosclerosis
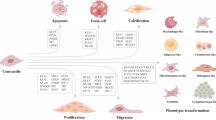
The phenotypic transformation of VSMCs in the body is often affected by a series of molecules and the integration of environmental factors, which mainly include lipids, retinoids, inflammatory mediators, growth factors, oxidative stress, blood flow shear stress, and cell-to-cell interactions. Then, VSMCs are transdifferentiated into various types of cells in the disease state, such as macrophage-like cells, foam cells, osteochondral-like cells, mesenchymal stem-like cells, and myofibroblast-like cells in disease states (Fig. 2). In recent decades, researchers have identified multiple signalling pathways that regulate the VSMC phenotypic transition process under the induction of various risk factors (Fig. 3).

Influencing factors and transcription factors/co-factors of the phenotypic switching of VSMCs. The phenotypic switching of VSMCs in vivo is often influenced by the integration of a range of extracellular signals, signalling pathways, and transcription factors. The main extracellular signals (e.g. lipids, retinoic acid, inflammatory mediators, growth factors, reactive oxygen species) activate signalling pathways that converge on transcription factors (e.g. KLF4, NF-κB, SP-1, and TCF21) to regulate the transdifferentiation of VSMCs into various cell types such as macrophage-like cells, foam cells, osteochondrocytes, mesenchymal stem cell-like cells and myofibroblast-like cells in disease states. MYOCD is a cofactor of SRF that binds to the CArG-box element within the promoter of contraction-related genes to promote VSMC contractile gene expression. Many transcription factors (e.g. KLF4, NF-κB, SP-1, and TCF21) repress the expression of contraction-related genes by inhibiting SRF binding to CArG-boxes. TGFβ, transforming growth factor-β; IGF, insulin-like growth factor; ECM, extracellular matrix; Ang II, angiotensin II; ROS, reactive oxygen species; MYOCD, myocardin; SRF, serum response factor; KLF4, krüppel-like factor 4; NF-κB, nuclear factor-κB; SP-1, specificity protein-1; Elk1, Ets-like protein 1; Runx2, runt-related transcription factor 2; TCF21, transcription factor 21

Signals mediating the phenotypic switching of VSMCs. The TGF-β signalling pathway is involved in the differentiation of SMC towards a contractile phenotype. Some growth factors (e.g., PDGF) exert their role in promoting VSMC dedifferentiation mainly through the Ras/Raf/MEK/ERK, PI3K/Akt, NF-κB, and JAK2/STAT3 pathways. Activation of Wnt/β-catenin protein signalling may promote the osteogenic transdifferentiation of VSMC. ECM binding to integrins causes activation of FAK. Phosphorylated FAK recruits Grb2-SoS complex, which then activates its downstream Ras and PI3K pathways. H2O2 is released from NOX4, which activates the PI3K and NF-κB pathways and mediates the VSMC phenotypic switch. In the nucleus, RA acts as a ligand for the RA receptor (RAR) and binds to the retinoic acid X receptor (RXR) near the target gene as a heterodimer with the RA response element (RARE) and thus regulates VSMC phenotype switching. Abbreviations: TGF-β, transforming growth factor β; SARA, Smad anchor for receptor activation; Grb2, growth factor receptor-bound protein 2; SoS, son of seven-less; Ras, rat sarcoma; Raf, Raf protein kinase; MEK, mitogen-activated ERK-regulating kinase; ERK, extracellular signal-regulated kinase; Elk1, Ets-like protein 1; PI3K, phosphatidylinositol 3-kinase; Akt/PKB, protein kinase B; mTOR, mammalian target of Rapamycin; FOXO4, forkhead box protein O; JAK, The Janus kinases; H2O2, hydrogen peroxide; NOX4, NADPH oxidase-4; ROS, reactive oxygen species; IKK, I kappa B kinase; NF-κB, nuclear factor kappa-B; ECM, extracellular matrix; FAK, focal adhesion kinase; RA, retinoic acid
VSMCs transdifferentiate from the contractile type to synthetic-like type
Transforming growth factor-β (TGF-β) is a multifunctional cytokine [45]. Typically, TGF-β signalling induces the differentiation of VSMCs and regulates the interaction of cells with the extracellular matrix [46]. KLF4 inhibits this effect of TGF-β and promotes the dedifferentiation of VSMCs [47]. Glycoprotein M6B (GPM6B) influences SMC differentiation by activating TGF-β-Smad2/3 signal transduction [48]. It is generally accepted that both insulin-like growth factor 1 (IGF-1) and TGF-β inhibit the phenotypic transformation of VSMCs [49, 50].
Platelet-derived growth factor-BB (PDGF-BB) was among the first factors identified to promote the phenotypic transformation of VSMCs and has been widely used in in vitro experiments as a model for generating dedifferentiated cells from VSMCs. PDGF-BB exerts its pro-SMC dedifferentiation effects mainly through the Ras/Raf/MEK/ERK [51], PI3K/Akt [52], NF-κB [53] and JAK2/STAT3 [54, 55] pathways. This leads to transcription of dedifferentiated genes under the control of SRF, as well as suppression of SMC-specific markers. In addition, extracellular signal-regulated kinase (ERK) phosphorylates myocardin-related transcription factor (MRTF) in the cytoplasm, preventing its nuclear translocation. Most importantly, PDGF-BB induces KLF4, which controls the transcriptional program of SMC-specific genes by preventing the SRF/MYOCD complex from binding to the promoters of pro-differentiation genes. PDGF-BB intervention activates Rho-associated coiled-coil-forming kinases (ROCKs) levels in VSMCs from human aortas [56]. ROCKs mediate Rho-induced changes in the actin cytoskeleton, mainly through phosphorylation of the myosin light chain (MLC), thereby mediating the contraction of VSMCs [56, 57]. Other growth factors, such as basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF), are widely considered factors that induce the maturation and differentiation of VSMCs [58].
The ECM (collagen IV, laminin, and perlecan) drives VSMC phenotypic transformation by binding integrin receptors on the VSMC surface and triggering a series of intracellular signal transduction pathways [59]. Generally, the basal lamina surrounds SMCs, and laminin and collagen IV in the basement membrane is crucial for maintaining VSMCs in a quiescent phenotype [60]. However, some abnormal components of the basement membrane appear under the disease state, such as fibronectin and syndecan-4. Collagen I can promote the transformation of VSMCs to a synthetic phenotype from a contractile phenotype and this conversion is blocked by an integrin beta-1 antibody [61]. Shi et al. showed that when VSMCs are cultured in vitro, fibronectin polymerization can promote the transformation of VSMCs to a synthetic phenotype from a contractile phenotype via a Rac1-dependent pathway [62]. Syndecan-4 is a transmembrane acetyl heparan sulfate proteoglycan. Ikesue et al. showed that in mice, deletion of syndecan-4 resulted in the restricted formation of neointima after vascular injury and reduced proliferation of vascular smooth muscle cells [63]. A recent study found that the basement membrane protein Nidogen-2 enhanced the interaction between Jagged1 and Notch3 and amplified downstream signalling activation, thereby maintaining the contractile phenotype of VSMCs and inhibiting subsequent neointimal formation following vascular injury [64].
NOX4-ROS signalling is involved in the phenotypic switching of VSMCs in the vascular system [65]. Reactive oxygen species (ROS) are generated mainly from NADPH oxidase (NOX) [66]. The mammalian NOX family mainly has seven members: NOX1-5 and DUOX1/2 [66]. In rodent and human aortic SMCs, the main ROS-producing NOXs are NOX1 and NOX4 [67,68,69]. NOX1 is associated with the de-differentiation of VSMC [70]. Moreover, NOX1 plays a key role in VSMC migration, proliferation, and extracellular matrix production following vascular injury [71, 72]. In contrast to NOX1, NOX4 is required for the maintenance of the differentiated VSMC phenotype [70]. In addition, NOX4 also mediates VSMC migration [73]. It is exactly an active area of investigation how NOX1 and NOX4 regulate such diverse functions in VSMC. A previous study found that the NOX4-mediated modulation of the levels of VSMC-specific genes is involved in p38 MAPK/NF-κB activation [74]. The JAK/STAT cascade modifies NOX1 and NOX4 in human aortic SMCs [75]. In addition, studies have shown that E2F transcription factors are positive regulators of NOX4 transcription in VSMCs [76] and that mechanical tension stimulation increases the production of NOX1 through myocyte enhancer factor 2 (MEF2), resulting in the conversion of contractile VSMCs to a synthetic phenotype [77]. In addition to the previously listed components, haemodynamic factors [78] and intercellular interactions [79] both contribute to the phenotypic alteration of VSMCs.
VSMCs transdifferentiate to a macrophage-like state and foam cells
It has long been thought that VSMCs can take up and accumulate lipids to form foam cells, thereby influencing the progression of atherosclerosis [16]. Human arteries exhibit diffuse intimal thickening (DIT) before the development of atherosclerosis, and lipids occur in the deeper layers of the SMC- and extracellular matrix-rich DIT [80]. Notably, SMC and SMC foam cells are not as pronounced in mouse models of atherosclerosis as they are in human lesions [81]. In mouse models, lipids appear to be deposited in narrow subendothelial spaces with limited SMCs and ECM [80]. Hypercholesterolaemia and dyslipidaemia promote the accumulation of low-density lipoprotein (LDL) within the vascular wall, where LDL is modified and transformed into oxidized low-density lipoprotein (oxLDL) in the presence of increased ROS. Moreover, VSMCs migrate in the subendothelial space, phagocytose oxidized LDL via scavenger receptors (e.g., LOX-1, SR-A, CD36, SR-BI and CXCL16/SR-PSOX) and differentiate into lipid-containing foam cells [82]. SMCs with a synthetic phenotype may be converted to foam cells through a different lipid metabolism than contractile SMCs and partly due to reduced expression of cholesterol esterase and the cholesterol efflux transporter ABCA1 (ATP-binding cassette transporter). Higashimori M et al. found that SMC cholesterol enrichment in vitro stimulated the expression of cholesterol acyltransferase-1 mRNA and that cytoplasmic cholesteryl ester accumulation was expressed in a Toll-like receptor 4 (TLR4)-dependent manner [83]. Yin et al. discovered that oxLDL activated the TLR4/MyD88/NF-B inflammatory signalling pathway in VSMCs, which, in turn, increased ACAT1 expression and promoted VSMC foam cell formation [84]. Another study showed that TLR4 is involved in the formation of VSMC foam cells induced by oxidized LDL via the Src and Sirt1/3 pathways [85]. The above results suggest that inflammatory signalling mediated by TLR4 may be involved in the formation of VSMC foam cells induced by oxLDL. The original observations by Rong XJ et al. demonstrated that after cholesterol loading, mouse aortic smooth muscle cells transdifferentiated to a macrophage-like state in vitro, accompanied by the loss of VSMC markers (α-SMA, SM22α, MYH11, and CNN1) and the appearance of macrophage-like markers (CD68 and Mac-2) that accelerate atherosclerosis progression [4]. Further studies by Dr. Fisher's laboratory revealed that cholesterol loading induced the conversion of VSMCs to a macrophage-like phenotype through the downregulation of the miR-143/145–myocardin axis [8]. However, to date, the exact mechanism of VSMC foam cell formation has not been elucidated. The question of whether SMCs need to acquire a macrophage-like phenotype to become foam cells or whether they become foam cells and then express macrophage markers remains unanswered. SMC foam cells have the full characteristics of foam cells and constitute most foam cells in atherosclerotic plaques in both humans [7] and mice [86], whereas the properties and functions of macrophage-like SMCs are still not fully clear. The net effect of macrophage-like SMCs on the various stages of atherosclerotic plaque formation needs to be investigated in depth.
In addition to lipid metabolism, inflammatory factors, vasoactive substances, and oxidative stress are important factors influencing VSMC transdifferentiation. Macrophage migration inhibitory factor (MIF) is a critical pro-inflammatory mediator. Fan et al. found that MIF inhibited the p68 protein and reduced the expression of SRF, which controls the transcription of VSMC differentiation marker genes, thereby inducing VSMC dedifferentiation [87]. Interleukin-lβ (IL-1β) increases the uptake of unmodified LDL via LDL receptors and enhances the conversion of VSMCs to foam cells by increasing cholesterol esterification [88]. In addition, interleukin-l9 (IL-19) reduced LDL receptor adapter protein 1 (LRP1) expression and oxLDL uptake, thereby reducing lipid accumulation in VSMCs [89]. LRP1 is a lipoprotein receptor that actively converts VSMCs into lipid-rich foam cells. Sendra et al. showed that in human VSMCs, Ang II increased LRP1 expression and LRP1-mediated aggregated LDL uptake, thereby contributing to the formation of lipid-rich atherosclerotic lesions [90]. Secretory phospholipase A2 upregulates SR-A1 and LOX-1 protein expression and enhances LDL uptake in VSMCs, inducing a macrophage-like transition from smooth muscle foam cells [91]. The activity of Noxa1-dependent NADPH oxidase is critical for VSMC plasticity since it promotes KLF4-mediated transformation to macrophage-like cells and plaque inflammation and expansion [92]. Bao et al. discovered that late glycosylation end products induced the formation of foam cells from VSMCs and their transdifferentiation to a macrophage-like state [93]. Hyperglycaemia promotes VSMC-derived foam cell formation by increasing CD36-mediated modified LDL uptake and decreasing ABCG1-regulated cellular cholesterol efflux, an effect associated with increased oxidative stress and activation of NF-κB pathway signalling [94]. SMC Drebrin inhibits atherosclerosis by reducing the transdifferentiation of SMCs to macrophage-like foam cells in a Nox1- and KLF4-dependent manner [95].
VSMCs transdifferentiate to an osteoblastic-like state
VSMCs lose their ability to express smooth muscle-specific markers in response to multiple stimuli and phenotypically change from contractile to osteoblast-like cells, which may subsequently lead to atherosclerotic calcification in vivo [96,97,98]. The exposure of VSMCs to high concentrations of phosphate and calcium promotes this transformation, a process that activates bone morphogenetic proteins (BMPs) and the WNT signalling pathway by upregulating Runt-related transcription factor 2 (RUNX2) and msh homeobox 2 (MSX2) transcription factors [98,99,100]. This process can also be mediated by oxygen free radicals [101, 102]. After transformation, the cells can produce matrix proteins [98, 103]. Furthermore, Sirtuin 1 can delay the transformation of high phosphate-induced VSMCs into an osteogenic phenotype [104, 105]. Inhibition of the H2O2-activated AKT signalling pathway blocks osteogenic phenotypic transdifferentiation and Runx2 induction in VSMCs [102]. Chronic exposure to high glucose (HG) alone or in combination with oxLDL enhanced the expression of bone morphogenetic protein-2 (BMP2), secreted phosphoprotein 1 (SPP1), and alkaline phosphatase (ALPL) in VSMCs, implying a process of transdifferentiation towards osteoblast-like cells [106]. Calcium and osteoprotegerin inhibit vascular calcification by regulating IGF1R expression [107]. Kanno et al. found that NO inhibited the differentiation of VSMCs towards osteoblasts and that cyclic guanosine phosphate (cGMP)-dependent protein kinase inhibitors restored the inhibitory effect on osteoblast differentiation of VSMCs [108]. Further studies revealed that NO inhibited TGF-β signalling via a CGMP-dependent pathway, thereby preventing the differentiation of VSMCs towards osteoblasts [108]. Uraemic toxin [109], AGEs [101], and oestrogen [110] all lead to phenotypic changes in VSMCs towards osteoblasts. RANKL is expressed in VSMCs and is thought to stimulate the osteogenic differentiation of VSMCs [111]. In addition, secreted Frizzled Related Protein 5 (SFRP5) inhibits VSMC transformation to osteoblast-like cells and is involved in regulating the wnt3a-mediated noncanonical signalling pathway by inhibiting the Rho/ROCK/JNK signalling pathway [112]. In addition to VSMC conversion to foam cells, cholesterol loading is involved in VSMC osteogenic transdifferentiation, altered bone-related protein gene expression, and calcification [113,114,115,116]. Further studies revealed that the NFAT signalling pathway regulated the oxLDL-induced transition of VSMCs to an osteoblast-like phenotype [114]. Parhami et al. showed that HDL-C in vitro inhibited the osteogenic differentiation of VSMCs [117]. More recently, Neven et al. suggested that midlayer calcification is a phenotypic transformation of VSMCs to chondrocytes rather than osteoblasts [118]. Still, the mechanism of vascular calcification to the osteoblast or chondrocyte phenotype needs further investigation.
VSMCs transdifferentiate to a mesenchymal stem cell-like state and a myofibroblast-like state
Chen et al. found that the specific deletion of SMC TGF-β signalling was associated with the transformation of contractile SMCs into MSC-like intermediates that can develop into osteoblast-like cells, chondrocyte-like cells, adipocyte-like cells, and macrophage-like cells in Apoe −/− mice fed a high-cholesterol diet [119]. Recent studies have found that stem cell antigen-1 (Sca 1) is expressed in VSMC-derived cells within healthy vessels and atherosclerotic lesions [6, 29]. However, although a subset of SMC-derived cells in atherosclerotic lesions express multiple markers of MSCs, there is no evidence that they are pluripotent and therefore do not appear to be capable of MSCs. The SMC-derived SEM cells identified by Pan et al. have stem cell properties and have the potential to differentiate into fibrochondrocyte-like cells. The proportion of ACTA2 + cells was significantly increased after induction of SEM cells with TGFβ1. Notably, all-trans retinoic acid (ATRA)-activated retinoic acid (RA) signalling inhibited the expression of SEM cell markers in vitro and suppressed SMC-to-SEM cell transformation. It is suggested that the RA signal plays an important role in regulating the SMC phenotypic transition. RA, an active metabolite of vitamin A, regulates cell proliferation and differentiation by binding the following two subfamilies of nuclear receptors: retinoid X receptors (RXRγ, RXRβ, and RXRα) and RA receptors (RARγ, RARβ, and RARα) [120]. Previous studies have demonstrated that RA can regulate VSMC phenotypes by binding RA-reactive elements located in target genes through nuclear receptors [121]. The RA receptor-specific agonist Am80 inhibits the activity of the transcription factor KLF5 to attenuate the phenotypic modulation of SMCs [122]. Smoothelin, a marker of late vascular smooth muscle cell differentiation, is not expressed in differentiated myofibroblasts and can, therefore, be used to differentiate differentiated myofibroblasts from mature VSMCs [123, 124]. During the evolution of vascular diseases, such as atherosclerosis or restenosis, VSMCs may lose specific differentiation markers, such as smoothelin markers, and become similar to myofibroblasts [125].
Transcriptional factors/co-factors mediating phenotypic switching of VSMCs in atherosclerosis
The expression of SMC-specific contractile proteins is dependent on the regulation of a network of transcription factors/cofactors in the promoter regions of their genes. Three transcription factors, namely, myocardin (MYOCD), serum response factor (SRF), and Krüppel-like factor 4 (KLF4), are thought to be key components of this network [6, 126, 127]. MYOCD is a critical regulator of SMC contractility [128]. MYOCD is highly expressed in VSMCs and effectively activates CArG-dependent [129] and CArG-independent [130] target gene expression. MYOCD cannot attach directly to the target gene's CArG box due to the absence of a DNA binding structural domain. In contrast, MYOCD can interact with SRF's N-terminal MADS-box via a Q-rich structural domain and bind the target gene's CArG box, forming an MYOCD-SRF-CArG box complex that activates the gene responsible for smooth muscle cell contraction, thereby maintaining the differentiation state and contractile phenotype of VSMCs [127, 131, 132]. MYOCD-related transcription factors (MRTFs)-A and B have also been identified as coactivators of SRF. SRF is a stimulus-responsive transcription factor that is a member of the Mcm1-Agamous-Deficiens-SRF-structural domain family of transcriptional regulators. SRF contains the following two functional domains: the N-terminus is a DNA-binding structural domain that associates with the serum response element or CArG-box and contains the MADS structural domain that facilitates DNA binding and homodimerization, and the C-terminus is thought to be a structural domain that regulates cofactor binding [126]. Almost all smooth muscle contraction marker genes have one or more SRF binding sites (CArG-box sequences) in the promoter or first intron. As transcription factors, SRFs are not transcriptionally active on their own and, therefore, need to interact with many SRF cofactors to coregulate the expression of target genes [133]. Under basal conditions, KLF4 expression in VSMCs is very low and is rapidly induced after injury to VSMCs; it is generally believed that KLF4 may inhibit the expression of specific genes encoding contractile proteins by competing with SRF for binding to CArG [134]. Other transcription factors acting on the MYOD/SRF complex have been demonstrated to play a critical role in SMC differentiation/dedifferentiation. By interacting with SRF and MYOCD, phosphatase and tensin homologues (PTEN) [135] have been demonstrated to maintain the contractile phenotype of SMCs. In contrast, phosphorylated ETS domain-containing protein-1 (pELK-1) [136], Yin Yang 1 (YY1) [137] and forkhead box protein O (FOXO4) [138] induce SMC dedifferentiation through their interaction with the MYOCD/SRF complex. By interacting with the KLF4 promoter, specificity protein-1 (Sp-1) [139] regulates SMC phenotypic alterations. Dedicator of cytokinesis 2 (DOCK2) [140], a rac activator, was recently identified as a novel regulator of the SMC phenotype. It competes with MYOCD for interaction with SRF, which is required for SMC dedifferentiation, and DOCK2 and KLF4 synergistically suppress MYOCD/SRF interactions. Transcription factor 21 (TCF21) knockdown experiments in SMCs demonstrate that TCF21 promotes phenotypic regulation in vivo [2]. Further studies established that TCF21 may interact directly with MYOCD to disrupt its functional association with SRF and transcriptional regulation of SMC genes [141].
Epigenetic mechanisms regulating the phenotypic switching of VSMCs in atherosclerosis
The epigenetic state is coordinated by numerous converging and reinforcing signals, including DNA methylation, histone modifications and non-coding RNAs [142]. In recent years, extensive progress has been made in our understanding of the complex epigenetic mechanisms underlying the phenotypic modulation of VSMCs in atherosclerosis (Fig. 4).

Epigenetic mechanisms regulating the phenotypic switching of VSMCs in response to vascular injury or atherosclerotic disease. The epigenetic processes include DNA methylation, histone modifications, non-coding RNA expression. During the phenotypic regulation of SMCs cultured in vitro, overall hypomethylation occurs. In the basal state, SMC contractile genes (e.g. SMα-Actin) are post-translationally modified through alterations such as acetylation of histones 3 and 4 (H3Ac and H4Ac) and dimethylation of H3K4 (H3K4diMe). These modifications are thought to remodel chromatin structure, allowing the SRF-Mycardin complex to bind to CArG-box elements and drive SMC-selective gene expression. In contrast, loss of previously activated histone modifications (e.g. H3/H4 Ac and H3K4diMe) and reduced ability of the SRF-Mycardin complex to bind to the CArG-box suppressed the expression of SMC marker genes. Non-coding RNAs (mainly microRNAs, long non-coding RNAs and circular RNAs) also play a key role in the VSMC phenotypic switch. The arrows and T-shaped ends represent promoting and inhibiting phenotype switch, respectively
DNA methylation
In mammalian cells, DNA methylation is a process through which methylation is selectively added to cytosine by the DNA methyltransferases DNMT1, DNMT3A, and DNMT3B, and this process mediates gene silencing [143]. Early studies involving further analysis of genome-wide methylation in normal and atherosclerotic arteries by high-pressure liquid chromatography have shown that the content of 5-methylcytosine (5-MC) tends to decrease in atherosclerotic arteries compared with that in normal arteries [144]. Liu et al. discovered that ten-eleven translocation-2 (TET2) and 5-hydroxymethylcytosine (5hmC) were abundant in a differentiated VSMC experimental model but were dramatically reduced in a dedifferentiated VSMC experimental model and human atherosclerosis [145]. Additionally, silencing TET2 reduced the expression of contractile genes in VSMCs while significantly increasing the expression of synthetic phenotypic markers. In ApoE−/− mice fed a high-fat diet, the DNA methyltransferase inhibitor 5-aza-2-deoxycytidine (5-Aza) significantly reduced atherosclerotic lesions and inhibited the activity of DNA methyltransferase while reducing the overall 5-methylcytosine content in atherosclerotic lesions [146]. In addition, during the phenotypic regulation of SMCs cultured in vitro, overall hypomethylation occurs, which is accompanied by a decrease in DNA methyltransferase activity [147, 148].
Several studies have shown that DNA methylation regulates certain SMC genes, thereby affecting the regulation of the SMC phenotype and the development of vascular diseases. The methylation-dependent combination of recombination signal-binding protein for the immunoglobulin J region (RBPJ) and a GC repressor has been shown to negatively regulate the activity of smooth muscle myosin heavy chain (SM MHC) promoters and RBPJ has been shown to inhibit the expression of SMC markers in phenotypically regulated SMCs [149]. Interestingly, LPS or a high-fat diet downregulates miR-152, a DNMT1 repressor, resulting in the hypermethylation of the oestrogen receptor-α gene in human or rat aortic SMCs [150]. MIR-1298 is methylation-dependent and affects the proliferation and migration of VSMCs by targeting connexin 43 [151]. DNMT1, a negative regulator of arterial stiffening, maintains the contractile phenotype of VSMCs. A previous study demonstrated that DNMT1 plays a vital role in the PDGF-induced phenotypic transformation of rat airway smooth muscle cells [152]. Recently, a report demonstrated that DNMT suppression with 5-Aza enhances mineralization in human aortic SMCs in vitro by demethylating the promoter of alkaline phosphatase [153].
The ECM influences SMC phenotypes and DNA methylation [148]. During SMC proliferation, the collagen type XV alpha 1 gene (COL15A1) is hypomethylated, leading to the elevation of gene expression involved in atherosclerosis development and the SMC phenotype [154, 155]. DNA methylation levels are altered in rat visceral SMCs grown on denatured versus native collagen [148]. Plating-damaged collagen increases SMC proliferation, and this increase is reversed by 5-Aza [148]. The matrix precisely regulates the localization and expression of DNMT3A and affects the differentiation of SMCs exposed to denatured matrix ± hypoxia [156]. In addition, the study further revealed that the expression of nuclear DNMT3 in SMCs on damaged collagen was increased. Substrate stiffening in VSMCs induces the phenotypic transition of VSMCs by downregulating DNMT1 expression. Through the extracellular matrix, altered DNA methylation may affect the SMC phenotype, resulting in the transdifferentiation of VSMCs to an osteoblast-like phenotype. These studies demonstrate that DNA methylation occurs during the phenotypic transition of SMCs, but additional evidence is required to determine whether DNA methylation is causally related to this process and, if so, the underlying mechanisms.
Histone modifications
Histone acetylation and deacetylation are strictly regulated by histone acetyltransferase (HAT) and histone deacetylase (HDAC) and are closely related to eukaryotic gene transcription. In the early SMC differentiation A404 cell model induced by RA, SRF enrichment and SMC CARG inclusion region histone H3 and H4 hyperacetylation were observed [157]. It has been shown that stimulation of the coactivator CREB binding protein (CBP) to the SM22 promoter depends on HAT activity. HDAC overexpression inhibits SM22 promoter activity, whereas the HDAC inhibitor trichostatin A stimulates SM22 promoter activity in a CArG box-dependent manner and induces endogenous SM22 gene expression [158, 159]. Myocardin is a coactivator of SRF transcription specifically expressed in the heart and smooth muscle and can induce the histone acetylation of nucleosomes surrounding SRF-binding sites near the control region of the smooth muscle gene. Other studies have found that myocardin increases the association of SRF with methylated histones and CArG box chromatin during the activation of the SMC gene. McDonald et al. observed that the deacetylation of histone H4 and the loss of SRF binding inhibit SMC differentiation after vascular injury. In contrast, KLF4 recruits histone H4 deacetylase activity into the SMC gene and prevents SRF from binding methylated histones and CArG box chromatin, thereby inhibiting SMC gene expression [160]. The relationship between histone acetylation and the pathway affects the phenotypic transformation of SMCs. The overexpression of HAT proteins (P300 and CBP) has been shown to enhance TGFβ1-induced SM22 promoter activity [161]. In comparison, the overexpression of HAT inhibitors, such as Twist1, but not Twist2/Dermo-1 and E1A, inhibits TGFβ1's effect. Studies involving chromatin precipitation measurements have revealed that PDGF-BB induces the dissociation of the MKL factor from the CArG-containing region of SMC marker genes. This type of dissociation is mediated by the MKL factor and the phosphorylation of Elk-1 at early stages but is later mediated by the histone acetyl enzymes HDAC2, HDAC4, and HDAC5, which are induced by low levels of the promoter regions of acetylated histone H4 [162]. HDAC4 regulates the proliferation and migration of SMCs induced by PDGF-BB by generating ROS via Ca2 + /calmodulin-dependent protein kinase and activating p38 mitogen-activated protein kinase/heat shock protein 27 signals [163]. Additionally, the inhibition of SMC differentiation marker genes by poVPC is associated with a low level of histone H4 acetylation at the SM-actin promoter, which is mediated by the recruitment of HDAC2 and HDAC5 [164]. During phenotypic switching, the sequence combination of pELK-1 and KLF4 in G/C repressor regions largely silenced SMC marker genes. HDAC2 is recruited by the pELK-1-KLF4 complex, which results in reduced histone acetylation and epigenetic silencing [136]. A recent study performed H3K4me2 demethylation in smooth muscle-specific genomes by constructing a specific epigenetic editing system (Myocd-LSD1) and found that stable histone modification of H3K4me2 mediated DNA demethylation by acting as a TET2 binding site and that H3K4me2-demethylated smooth muscle cells lost their cell identity and muscle function and enhanced smooth muscle cell phenotypic plasticity [165]. This study sheds new light on the mechanism of "epigenetic memory" in smooth muscle cells and provides new clues for vascular smooth muscle-mediated prevention and treatment of cardiovascular disease. Christian L et al. found that mutations that affect both TGF-β signalling and the VSMC cytoskeleton lead to the formation of the HDAC9-MALAT1-BRG1 tricompound histone, which binds chromatin and inhibits contractile protein gene expression while causing increased trimethylated modifications of histone H3-lysine 27 [166].
MicroRNAs
MicroRNAs (miRNAs) are involved in various regulatory pathways, including the phenotypic switching of VSMCs. MiRNAs also modulate VSMC phenotypes by interacting with transcription factors (SRF, myocardin, MRTFs, and KLF) and the cytoskeleton, affecting the differentiation of VSMCs. Albinsson S et al. used SM22Cre-Dicerflox mice to reveal that the knockout of Dicer triggers embryonic lethality on embryonic day 16.5, which was characterized by decreased SMC marker proteins and proteins, impaired contractile function and disarrangement of aortic elastic lamellae [167]. It was demonstrated that in contrast to cells in a normal vascular wall, proliferating VSMCs have reduced miR-143/145 levels [168]. Cordes et al. discovered that miR-145 modulates the levels of VSMC differentiation transcription factors, e.g., CaMKIIδ, myocardin, and KLF4 [169]. Evidence indicates that miR-145 elevates myocardin levels but downregulates CaMKIIδ and KLF4 levels, which is consistent with the fact that miR-145 promotes a contractile phenotype in VSMCs. Farina FM et al. discovered that miR-128-3p is a new phenotypic switch regulator of VSMCs, and KLF4 is a direct target of miR-128 capable of regulating the methylation status of the key VSMC gene MYH11 [170]. MiR-22 is also a novel phenotypic switch regulator in VSMCs and targets ecotropic viral integration site 1 (EVI1) and methyl CpG binding protein 2 (MECP2), which regulate H3K9me3 enrichment in the promoter regions of SMC-specific genes [171]. MiR-31-5p significantly inhibits the myocardin levels and aggravates the pathological VSMC phenotypic switch and aortic aneurysm/dissection (AAD), while the inhibition of aldehyde dehydrogenase 2 (ALDH2) can attenuate this effect [172]. Platelet-derived miR-223 seems to promote the phenotypic switch of VSMCs in arterial injury repair by directly targeting PDGFRβ [173].
LncRNAs
LncRNAs are RNAs that regulate gene expression and usually do not encode proteins [174]. Lung adenocarcinoma metastasis-associated transcript 1 (MALAT1) can be combined with the immunoprecipitate of histone deacetylase 9 (HDAC9) and the chromatin remodelling enzyme Brahma-related gene 1 (BRG1), and silencing MALAT1 expression can inhibit immunoprecipitate stability, downregulate the levels of proliferating cell nuclear antigen, cyclin D1, and osteopontin genes, and promote the switch of SMCs from a synthetic phenotype to a contractile phenotype [166, 175]. MYOSLID, a VSMC-selective and SRF/CArG-dependent lncRNA that promotes the assembly of fibronectin through the nuclear transfer of myocardin-related transcription factor A, transcriptionally activates the VSMC contraction gene and its downstream signals and thus stabilizes the VSMC contraction phenotype [176]. Ahmed et al. further found that nuclear paraspeckle assembly transcript 1 (NEAT1) can block the interaction between histone methyltransferase 1 and WD repeat-containing protein 5 (WDR5) by binding the epigenetic activator WDR5 protein, and this blockade regulates histone methyltransferase 1 catalytic function, reduces the combination of serum response factors and the CArG box in the smooth muscle-specific gene promoter, and inhibits the expression of smooth muscle contraction-related proteins [177]. Bell et al. performed RNA sequencing of VSMCs in coronary arteries from humans and found that the new lncRNA SENR (smooth muscle and endothelial cell-rich migration/differentiation-associated) exhibited cytoplasmic localization, and its downregulated expression stabilized the contractile phenotypes of SMCs [178]. Studies have shown that lncRNA-GAS5 can regulate VSMC function through the Wnt/β-catenin signalling pathway [179]. In addition, GAS5 can use multiple Smad-binding elements to competitively bind the Smad3 protein as a molecular decoy to negatively regulate the TGF-β/Smad3 signalling pathway and thus inhibit TGF-β-induced VSMC differentiation. A recent study found that the ECM gene was downregulated and that the contraction gene was upregulated in the lncRNA Mymsl gene-knockout mouse aorta, indicating that the lncRNA Mymsl regulates the phenotypic transition of VSMCs [180]. LncRNA ANRIL modulates the phenotypic switch of HASMCs by acting as a molecular scaffold to promote the combination of HDAC3 and WDR5 to form an HDAC3 and WDR5 complex, regulating the expression of target genes, such as NOX1, through histone modification and upregulating the level of ROS [181]. In conclusion, lncRNAs constitute a new class of cellular phenotype transition regulators that may also be used as therapeutic targets for atherosclerosis and related cardiovascular diseases.
CircRNAs
CircRNAs constitute a class of non-coding RNAs with a covalently closed-loop structure. In contrast to conventional linear RNAs, circRNAs are formed by reverse splicing and do not have a 5′ end cap or a 3′ end poly(A) tail structure and, therefore, have features and functions that linear RNAs do not have [182]. CircRNAs could act as microRNA sponges to regulate VSMC phenotypes. Lasse S Kristensen et al. showed that circ_Lrp6, a circular RNA enriched in VSMCs with multiple putative binding sites for miR-145, blocked miR-145-mediated regulation of VSMC differentiation [183]. Another study used circRNA sequencing to identify circMAP3K5 in human coronary artery SMCs, which play a major role in the dedifferentiated phenotype of VSMCs, and demonstrated at the cellular and animal levels that circMAP3K5 acts as a competitive endogenous RNA to close miR-22-3p and that miR-22-3p deletion indeed fails to inhibit the TET2 pathway, thereby maintaining VSMC differentiation [184]. Xu et al. also found that circDiaph3 regulates the differentiation of rat VSMCs. circDiaph3 expression upregulation inhibits the function of miR-148a-5p, thereby downregulating Diaph3 protein levels and converting mature contractile VSMCs to dedifferentiated immature synthetic VSMCs by reducing the synthesis of contractile SMC markers and increasing the synthesis of type I collagen and elastin fibres [185]. The overexpression of circ-SATB2 was found to inhibit SM22α expression [186]. Yang et al. found that circCHFR is also involved in the regulation of phenotypic changes in VSMCs [187]. An increasing number of researches suggest that circRNAs play significant roles in the pathogenesis of atherosclerosis induced by VSMCs phenotypic switching. However, the lack of a comprehensive understanding of the precise biogenesis and regulatory mechanisms of circRNAs has hindered progress in this area of research and delayed the use of circRNAs in clinical diagnosis and therapy.
Conclusion and future perspective
For many years, VSMCs were undervalued, mischaracterized, and commonly labelled cells that promote atherosclerosis and/or stable plaques. Recent progress in VSMC lineage tracing and new transcriptomic technologies in mouse models of atherosclerosis further validate the idea that VSMCs have broad plasticity, reveal transitional cell types derived from previously constricted VSMCs with distinct molecular characteristics with different phenotypes playing distinct roles in lesion development, and suggest that different stimuli trigger behavioural changes in these cells. New research has also confirmed the theory from decades ago that the clonal expansion of a few VSMC-derived cells occurred in atherosclerosis. In addition to confirming previous ideas and theories, these new techniques revealed the existence of transcriptomic heterogeneity in VSMCs in atherosclerosis and led to the identification of additional cell subpopulations and VSMC-associated genes. However, there are still some problems, such as the heterogeneity factor of spectral tracer markers, the dose of tamoxifen, and objective factors, such as vascular tissue analysis techniques, that can affect the reliability of the results.
To develop effective therapeutic strategies to limit cardiovascular risk, it is necessary to address several questions. First, does the limited number of VSMCs have a predetermined route to migrate from the mid-membrane and undergo a fate transition in the endothelium to the SEM cell state? Second, do VSMCs have a predetermined route to migrate towards the plaque fibrous cap or necrotic core? Finally, are there specific factors that induce the differentiation of SEM cells into multiple VSMC-derived cells and their relative importance in the pathogenesis of atherosclerosis? Understanding the mechanisms of VSMC plasticity is necessary for achieving easy targeting of vascular smooth muscle phenotypic therapy to inhibit atherosclerotic plaque progression. scRNA-seq analyses of normal and diseased human coronary arteries are expected to identify CAD candidate genes coexpressed in cells expressing SMC markers and define distinct SMC subpopulations in diseased coronary arteries that could elucidate the pathogenesis of the disease. The next step is to integrate VSMC-specific fate maps and single-cell transcriptomics with human genetics to find novel regulatory mechanisms and candidate targets for the therapeutic modulation of VSMC phenotype switching. Advances in imaging and nanotechnology can also be used to detect inducers of clinically unstable plaque lesions.
Theoretically, the observation of an increase in the number of transformed functional cells in healthy vessels should be alarming. Again, knowledge of the molecular characteristics of these cells may help selectively target these cells with specific drugs, and treatment should specifically target a harmful subpopulation of VSMCs, reversing their phenotype or reprogramming the cells into atheroprotective VSMCs. However, such prospects are still in the early stages. Recent studies have been performed in mice, allowing access to large numbers of vascular smooth muscle cells and modifying their genomes for genealogical labelling. Further studies are needed to translate the available results first into human cells and then into the clinical detection of an increase in the number of functional cells converted into healthy vasculature.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
Consent for publication
Not applicable.
Consent to participate
Not applicable.
References
Owens GK, Kumar MS, Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84:767–801
Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL et al (2019) Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med 25:1280–1289
Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J et al (2020) Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation 142:2060–2075
Rong JX, Shapiro M, Trogan E, Fisher EA (2003) Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A 100:13531–13536
Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M et al (2014) Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res 115:662–667
Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM et al (2015) KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 21:628–637
Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA (2014) Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 129:1551–1559
Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO et al (2015) Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol 35:535–546
Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K et al (2018) Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res 122:1661–1674
Berry DC, Jiang Y, Graff JM (2016) Mouse strains to study cold-inducible beige progenitors and beige adipocyte formation and function. Nat Commun 7:10184
Long JZ, Svensson KJ, Tsai L, Zeng X, Roh HC, Kong X et al (2014) A smooth muscle-like origin for beige adipocytes. Cell Metab 19:810–820
Bobryshev YV (2005) Transdifferentiation of smooth muscle cells into chondrocytes in atherosclerotic arteries in situ: implications for diffuse intimal calcification. J Pathol 205:641–650
Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL et al (2009) Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res 104:733–741
Chamley-Campbell J, Campbell GR, Ross R (1979) The smooth muscle cell in culture. Physiol Rev 59:1–61
Bochaton-Piallat ML, Ropraz P, Gabbiani F, Gabbiani G (1996) Phenotypic heterogeneity of rat arterial smooth muscle cell clones. Implications for the development of experimental intimal thickening. Arterioscler Thromb Vasc Biol 16:815–820
Wissler RW (1967) The arterial medial cell, smooth muscle, or multifunctional mesenchyme? Circulation 36:1–4
Andreeva ER, Pugach IM, Orekhov AN (1997) Subendothelial smooth muscle cells of human aorta express macrophage antigen in situ and in vitro. Atherosclerosis 135:19–27
Bobryshev YV, Killingsworth MC, Lord RS (2008) Spatial distribution of osteoblast-specific transcription factor Cbfa1 and bone formation in atherosclerotic arteries. Cell Tissue Res 333:225–235
Jiang H, Lun Y, Wu X, Xia Q, Zhang X, Xin S et al (2014) Association between the hypomethylation of osteopontin and integrin beta3 promoters and vascular smooth muscle cell phenotype switching in great saphenous varicose veins. Int J Mol Sci 15:18747–18761
Chamley JH, Groschel-Stewart U, Campbell GR, Burnstock G (1977) Distinction between smooth muscle, fibroblasts and endothelial cells in culture by the use of fluoresceinated antibodies against smooth muscle actin. Cell Tissue Res 177:445–457
Skalli O, Ropraz P, Trzeciak A, Benzonana G, Gillessen D, Gabbiani G (1986) A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol 103:2787–2796
Wu SS, Lee JH, Koo BK (2019) Lineage tracing: computational reconstruction goes beyond the limit of imaging. Mol Cells 42:104–112
Feil S, Hofmann F, Feil R (2004) SM22alpha modulates vascular smooth muscle cell phenotype during atherogenesis. Circ Res 94:863–865
Gomez D, Shankman LS, Nguyen AT, Owens GK (2013) Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods 10:171–177
Albarrán-Juárez J, Kaur H, Grimm M, Offermanns S, Wettschureck N (2016) Lineage tracing of cells involved in atherosclerosis. Atherosclerosis 251:445–453
Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD et al (2016) Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res 119:1313–1323
Jacobsen K, Lund MB, Shim J, Gunnersen S, Füchtbauer EM, Kjolby M et al (2017) Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight. https://doi.org/10.1172/jci.insight.95890
Misra A, Feng Z, Chandran RR, Kabir I, Rotllan N, Aryal B et al (2018) Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat Commun 9:2073
Dobnikar L, Taylor AL, Chappell J, Oldach P, Harman JL, Oerton E et al (2018) Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat Commun 9:4567
Bentzon JF, Sondergaard CS, Kassem M, Falk E (2007) Smooth muscle cells healing atherosclerotic plaque disruptions are of local, not blood, origin in apolipoprotein E knockout mice. Circulation 116:2053–2061
Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E (2006) Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol 26:2696–2702
Yu H, Stoneman V, Clarke M, Figg N, Xin HB, Kotlikoff M et al (2011) Bone marrow-derived smooth muscle-like cells are infrequent in advanced primary atherosclerotic plaques but promote atherosclerosis. Arterioscler Thromb Vasc Biol 31:1291–1299
Sata M, Saiura A, Kunisato A, Tojo A, Okada S, Tokuhisa T et al (2002) Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med 8:403–409
Caplice NM, Bunch TJ, Stalboerger PG, Wang S, Simper D, Miller DV et al (2003) Smooth muscle cells in human coronary atherosclerosis can originate from cells administered at marrow transplantation. Proc Natl Acad Sci U S A 100:4754–4759
Naik V, Leaf EM, Hu JH, Yang HY, Nguyen NB, Giachelli CM et al (2012) Sources of cells that contribute to atherosclerotic intimal calcification: an in vivo genetic fate mapping study. Cardiovasc Res 94:545–554
Speer MY, Li X, Hiremath PG, Giachelli CM (2010) Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J Cell Biochem 110:935–947
Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM (2018) Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res 114:590–600
Kim JB, Zhao Q, Nguyen T, Pjanic M, Cheng P, Wirka R et al (2020) Environment-sensing aryl hydrocarbon receptor inhibits the chondrogenic fate of modulated smooth muscle cells in atherosclerotic lesions. Circulation 142:575–590
Saliba AE, Westermann AJ, Gorski SA, Vogel J (2014) Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res 42:8845–8860
Kaur H, Carvalho J, Looso M, Singh P, Chennupati R, Preussner J et al (2017) Single-cell profiling reveals heterogeneity and functional patterning of GPCR expression in the vascular system. Nat Commun 8:15700
Alencar GF, Owsiany KM, Karnewar S, Sukhavasi K, Mocci G, Nguyen AT et al (2020) Stem cell pluripotency genes Klf4 and Oct4 regulate complex SMC phenotypic changes critical in late-stage atherosclerotic lesion pathogenesis. Circulation 142:2045–2059
Xie Y, Martin KA (2020) TCF21: flipping the phenotypic switch in SMC. Circ Res 126:530–532
Kayashima Y, Maeda-Smithies N (2020) Atherosclerosis in different vascular locations unbiasedly approached with mouse genetics. Genes 11:1427
Tang J, Wang H, Huang X, Li F, Zhu H, Li Y et al (2020) Arterial Sca1(+) vascular stem cells generate De Novo smooth muscle for artery repair and regeneration. Cell Stem Cell 26:81–96
Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C et al (2017) Force interacts with macromolecular structure in activation of TGF-beta. Nature 542:55–59
Wang X, Hu G, Betts C, Harmon EY, Keller RS, Van De Water L, et al (2011) Transforming growth factor-beta1-induced transcript 1 protein, a novel marker for smooth muscle contractile phenotype, is regulated by serum response factor/myocardin protein. J Biol Chem 286:4189–4199
Li HX, Han M, Bernier M, Zheng B, Sun SG, Su M et al (2010) Kruppel-like factor 4 promotes differentiation by transforming growth factor-beta receptor-mediated Smad and p38 MAPK signaling in vascular smooth muscle cells. J Biol Chem 285:17846–17856
Zhang X, Xie H, Chang P, Zhao H, Xia Y, Zhang L et al (2019) Glycoprotein M6B interacts with TbetaRI to activate TGF-beta-Smad2/3 signaling and promote smooth muscle cell differentiation. Stem Cells 37:190–201
Wang L, Han Y, Shen Y, Yan ZQ, Zhang P, Yao QP et al (2014) Endothelial insulin-like growth factor-1 modulates proliferation and phenotype of smooth muscle cells induced by low shear stress. Ann Biomed Eng 42:776–786
Martin KA, Merenick BL, Ding M, Fetalvero KM, Rzucidlo EM, Kozul CD et al (2007) Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem 282:36112–36120
Lee CK, Lee HM, Kim HJ, Park HJ, Won KJ, Roh HY et al (2007) Syk contributes to PDGF-BB-mediated migration of rat aortic smooth muscle cells via MAPK pathways. Cardiovasc Res 74:159–168
Wang H, Zhong B, Geng Y, Hao J, Jin Q, Zhang Y et al (2021) TIPE2 inhibits PDGF-BB-induced phenotype switching in airway smooth muscle cells through the PI3K/Akt signaling pathway. Respir Res 22:238
Lu QB, Wan MY, Wang PY, Zhang CX, Xu DY, Liao X et al (2018) Chicoric acid prevents PDGF-BB-induced VSMC dedifferentiation, proliferation and migration by suppressing ROS/NFkappaB/mTOR/P70S6K signaling cascade. Redox Biol 14:656–668
Yellaturu CR, Rao GN (2003) Cytosolic phospholipase A2 is an effector of Jak/STAT signaling and is involved in platelet-derived growth factor BB-induced growth in vascular smooth muscle cells. J Biol Chem 278:9986–9992
Neeli I, Liu Z, Dronadula N, Ma ZA, Rao GN (2004) An essential role of the Jak-2/STAT-3/cytosolic phospholipase A(2) axis in platelet-derived growth factor BB-induced vascular smooth muscle cell motility. J Biol Chem 279:46122–46128
Tang L, Dai F, Liu Y, Yu X, Huang C, Wang Y et al (2018) RhoA/ROCK signaling regulates smooth muscle phenotypic modulation and vascular remodeling via the JNK pathway and vimentin cytoskeleton. Pharmacol Res 133:201–212
Qi Y, Liang X, Dai F, Guan H, Sun J, Yao W (2020) RhoA/ROCK pathway activation is regulated by AT1 receptor and participates in smooth muscle migration and dedifferentiation via promoting actin cytoskeleton polymerization. Int J Mol Sci 21:5398
Chen PY, Qin L, Li G, Tellides G, Simons M (2016) Fibroblast growth factor (FGF) signaling regulates transforming growth factor beta (TGFβ)-dependent smooth muscle cell phenotype modulation. Sci Rep 6:33407
Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687
Orr AW, Lee MY, Lemmon JA, Yurdagul AJ, Gomez MF, Bortz PD et al (2009) Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol 29:225–231
Graf K, Kappert K, Stawowy P, Bokemeyer J, Blaschke F, Schmidt G et al (2003) Statins regulate alpha2beta1-integrin expression and collagen I-dependent functions in human vascular smooth muscle cells. J Cardiovasc Pharmacol 41:89–96
Shi F, Long X, Hendershot A, Miano JM, Sottile J (2014) Fibronectin matrix polymerization regulates smooth muscle cell phenotype through a Rac1 dependent mechanism. PLoS ONE 9:e94988
Ikesue M, Matsui Y, Ohta D, Danzaki K, Ito K, Kanayama M et al (2011) Syndecan-4 deficiency limits neointimal formation after vascular injury by regulating vascular smooth muscle cell proliferation and vascular progenitor cell mobilization. Arterioscler Thromb Vasc Biol 31:1066–1074
Mao C, Ma Z, Jia Y, Li W, Xie N, Zhao G et al (2021) Nidogen-2 maintains the contractile phenotype of vascular smooth muscle cells and prevents neointima formation via bridging Jagged1-Notch3 signaling. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.120.053361
Obradovic M, Essack M, Zafirovic S, Sudar-Milovanovic E, Bajic VP, Van Neste C et al (2020) Redox control of vascular biology. BioFactors 46:246–262
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Clempus RE, Griendling KK (2006) Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res 71:216–225
Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y et al (2001) Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res 88:888–894
Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK (2004) Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 24:677–683
Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP et al (2007) Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol 27:42–48
Lee MY, San MA, Mehta PK, Dikalova AE, Garrido AM, Datla SR et al (2009) Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol 29:480–487
Schroder K, Helmcke I, Palfi K, Krause KH, Busse R, Brandes RP (2007) Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 27:1736–1743
Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P et al (2009) Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res 105:249–259
Lu X, Murphy TC, Nanes MS, Hart CM (2010) PPAR{gamma} regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-{kappa}B. Am J Physiol Lung Cell Mol Physiol 299:L559–L566
Manea A, Tanase LI, Raicu M, Simionescu M (2010) Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol 30:105–112
Zhang L, Sheppard OR, Shah AM, Brewer AC (2008) Positive regulation of the NADPH oxidase NOX4 promoter in vascular smooth muscle cells by E2F. Free Radic Biol Med 45:679–685
Rodríguez AI, Csányi G, Ranayhossaini DJ, Feck DM, Blose KJ, Assatourian L et al (2015) MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 35:430–438
Shi ZD, Tarbell JM (2011) Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Ann Biomed Eng 39:1608–1619
Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM et al (2012) Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol 14:249–256
Nakashima Y, Wight TN, Sueishi K (2008) Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res 79:14–23
Nakashima Y, Fujii H, Sumiyoshi S, Wight TN, Sueishi K (2007) Early human atherosclerosis: accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler Thromb Vasc Biol 27:1159–1165
Wagsater D, Olofsson PS, Norgren L, Stenberg B, Sirsjo A (2004) The chemokine and scavenger receptor CXCL16/SR-PSOX is expressed in human vascular smooth muscle cells and is induced by interferon gamma. Biochem Biophys Res Commun 325:1187–1193
Higashimori M, Tatro JB, Moore KJ, Mendelsohn ME, Galper JB, Beasley D (2011) Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 31:50–57
Yin YW, Liao SQ, Zhang MJ, Liu Y, Li BH, Zhou Y et al (2014) TLR4-mediated inflammation promotes foam cell formation of vascular smooth muscle cell by upregulating ACAT1 expression. Cell Death Dis 5:e1574
Chen Z, Xue Q, Cao L, Wang Y, Chen Y, Zhang X et al (2021) Toll-like receptor 4 mediated oxidized low-density lipoprotein-induced foam cell formation in vascular smooth muscle cells via Src and Sirt1/3 pathway. Mediators Inflamm 2021:6639252
Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE et al (2019) Smooth muscle cells contribute the majority of foam cells in ApoE (Apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol 39:876–887
Fan Y, Zhang J, Chen CY, Xiao YB, Asico LD, Jose PA et al (2017) Macrophage migration inhibitory factor triggers vascular smooth muscle cell dedifferentiation by a p68-serum response factor axis. Cardiovasc Res 113:519–530
Ruan XZ, Moorhead JF, Tao JL, Ma KL, Wheeler DC, Powis SH et al (2006) Mechanisms of dysregulation of low-density lipoprotein receptor expression in vascular smooth muscle cells by inflammatory cytokines. Arterioscler Thromb Vasc Biol 26:1150–1155
Gabunia K, Herman AB, Ray M, Kelemen SE, England RN, DeLa CR et al (2017) Induction of MiR133a expression by IL-19 targets LDLRAP1 and reduces oxLDL uptake in VSMC. J Mol Cell Cardiol 105:38–48
Sendra J, Llorente-Cortes V, Costales P, Huesca-Gomez C, Badimon L (2008) Angiotensin II upregulates LDL receptor-related protein (LRP1) expression in the vascular wall: a new pro-atherogenic mechanism of hypertension. Cardiovasc Res 78:581–589
Giannotti KC, Weinert S, Viana MN, Leiguez E, Araujo T, Laurindo F et al (2019) A secreted phospholipase A2 induces formation of smooth muscle foam cells which transdifferentiate to macrophage-like state. Molecules 24:3244
Vendrov AE, Sumida A, Canugovi C, Lozhkin A, Hayami T, Madamanchi NR et al (2019) NOXA1-dependent NADPH oxidase regulates redox signaling and phenotype of vascular smooth muscle cell during atherogenesis. Redox Biol 21:101063
Bao Z, Li L, Geng Y, Yan J, Dai Z, Shao C et al (2020) Advanced glycation end products induce vascular smooth muscle cell-derived foam cell formation and transdifferentiate to a macrophage-like state. Mediators Inflamm 2020:6850187
Xue JH, Yuan Z, Wu Y, Liu Y, Zhao Y, Zhang WP et al (2010) High glucose promotes intracellular lipid accumulation in vascular smooth muscle cells by impairing cholesterol influx and efflux balance. Cardiovasc Res 86:141–150
Wu JH, Zhang L, Nepliouev I, Brian L, Huang T, Snow KP et al (2021) Drebrin attenuates atherosclerosis by limiting smooth muscle cell transdifferentiation. Cardiovasc Res. https://doi.org/10.1093/cvr/cvab156
Moe SM, Chen NX (2008) Mechanisms of vascular calcification in chronic kidney disease. J Am Soc Nephrol 19:213–216
Shanahan CM (2005) Mechanisms of vascular calcification in renal disease. Clin Nephrol 63:146–157
Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM (2011) Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 109:697–711
Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89:747–754
Shroff RC, Shanahan CM (2007) The vascular biology of calcification. Semin Dial 20:103–109
Tada Y, Yano S, Yamaguchi T, Okazaki K, Ogawa N, Morita M et al (2013) Advanced glycation end products-induced vascular calcification is mediated by oxidative stress: functional roles of NAD(P)H-oxidase. Horm Metab Res 45:267–272
Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM et al (2008) Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem 283:15319–15327
Murshed M, McKee MD (2010) Molecular determinants of extracellular matrix mineralization in bone and blood vessels. Curr Opin Nephrol Hypertens 19:359–365
Takemura A, Iijima K, Ota H, Son BK, Ito Y, Ogawa S et al (2011) Sirtuin 1 retards hyperphosphatemia-induced calcification of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 31:2054–2062
Badi I, Mancinelli L, Polizzotto A, Ferri D, Zeni F, Burba I et al (2018) miR-34a promotes vascular smooth muscle cell calcification by downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arterioscler Thromb Vasc Biol 38:2079–2090
Navas-Madronal M, Castelblanco E, Camacho M, Consegal M, Ramirez-Morros A, Sarrias MR et al (2020) Role of the scavenger receptor CD36 in accelerated diabetic atherosclerosis. Int J Mol Sci 21:7360
Di Bartolo BA, Schoppet M, Mattar MZ, Rachner TD, Shanahan CM, Kavurma MM (2011) Calcium and osteoprotegerin regulate IGF1R expression to inhibit vascular calcification. Cardiovasc Res 91:537–545
Kanno Y, Into T, Lowenstein CJ, Matsushita K (2008) Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovasc Res 77:221–230
Adijiang A, Goto S, Uramoto S, Nishijima F, Niwa T (2008) Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol Dial Transplant 23:1892–1901
Peng YQ, Xiong D, Lin X, Cui RR, Xu F, Zhong JY et al (2017) Oestrogen inhibits arterial calcification by promoting autophagy. Sci Rep 7:3549
Collin-Osdoby P (2004) Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ Res 95:1046–1057
Oh YJ, Kim H, Kim AJ, Ro H, Chang JH, Lee HH et al (2020) Reduction of secreted frizzled-related protein 5 drives vascular calcification through Wnt3a-mediated Rho/ROCK/JNK signaling in chronic kidney disease. Int J Mol Sci 21:3539
Proudfoot D, Davies JD, Skepper JN, Weissberg PL, Shanahan CM (2002) Acetylated low-density lipoprotein stimulates human vascular smooth muscle cell calcification by promoting osteoblastic differentiation and inhibiting phagocytosis. Circulation 106:3044–3050
Goettsch C, Rauner M, Hamann C, Sinningen K, Hempel U, Bornstein SR et al (2011) Nuclear factor of activated T cells mediates oxidised LDL-induced calcification of vascular smooth muscle cells. Diabetologia 54:2690–2701
Cai Z, Ding Y, Zhang M, Lu Q, Wu S, Zhu H et al (2016) Ablation of adenosine monophosphate-activated protein kinase alpha1 in vascular smooth muscle cells promotes diet-induced atherosclerotic calcification in vivo. Circ Res 119:422–433
Schiffrin EL, Lipman ML, Mann JF (2007) Chronic kidney disease: effects on the cardiovascular system. Circulation 116:85–97
Parhami F, Basseri B, Hwang J, Tintut Y, Demer LL (2002) High-density lipoprotein regulates calcification of vascular cells. Circ Res 91:570–576
Neven E, De Schutter TM, De Broe ME, D’Haese PC (2011) Cell biological and physicochemical aspects of arterial calcification. Kidney Int 79:1166–1177
Chen PY, Qin L, Li G, Malagon-Lopez J, Wang Z, Bergaya S et al (2020) Smooth muscle cell reprogramming in aortic aneurysms. Cell Stem Cell 26:542–557
Gudas LJ, Wagner JA (2011) Retinoids regulate stem cell differentiation. J Cell Physiol 226:322–330
Streb JW, Long X, Lee TH, Sun Q, Kitchen CM, Georger MA et al (2011) Retinoid-induced expression and activity of an immediate early tumor suppressor gene in vascular smooth muscle cells. PLoS ONE 6:e18538
Fujiu K, Manabe I, Ishihara A, Oishi Y, Iwata H, Nishimura G et al (2005) Synthetic retinoid Am 80 suppresses smooth muscle phenotypic modulation and in-stent neointima formation by inhibiting KLF5. Circ Res 97:1132–1141
van der Loop FT, Gabbiani G, Kohnen G, Ramaekers FC, van Eys GJ (1997) Differentiation of smooth muscle cells in human blood vessels as defined by smoothelin, a novel marker for the contractile phenotype. Arterioscler Thromb Vasc Biol 17:665–671
van der Loop FT, Schaart G, Timmer ED, Ramaekers FC, van Eys GJ (1996) Smoothelin, a novel cytoskeletal protein specific for smooth muscle cells. J Cell Biol 134:401–411
Christen T, Verin V, Bochaton-Piallat M, Popowski Y, Ramaekers F, Debruyne P et al (2001) Mechanisms of neointima formation and remodeling in the porcine coronary artery. Circulation 103:882–888
Zhao Y, Samal E, Srivastava D (2005) Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 436:214–220
Ackers-Johnson M, Talasila A, Sage AP, Long X, Bot I, Morrell NW et al (2015) Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arterioscler Thromb Vasc Biol 35:817–828
Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E et al (2001) Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105:851–862
Chen J, Kitchen CM, Streb JW, Miano JM (2002) Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol 34:1345–1356
Qiu P, Ritchie RP, Fu Z, Cao D, Cumming J, Miano JM et al (2005) Myocardin enhances Smad3-mediated transforming growth factor-beta1 signaling in a CArG box-independent manner: Smad-binding element is an important cis element for SM22alpha transcription in vivo. Circ Res 97:983–991
Wang Z, Wang DZ, Pipes GC, Olson EN (2003) Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci U S A 100:7129–7134
Xia XD, Zhou Z, Yu XH, Zheng XL, Tang CK (2017) Myocardin: a novel player in atherosclerosis. Atherosclerosis 257:266–278
Onuh JO, Qiu H (2021) Serum response factor-cofactor interactions and their implications in disease. FEBS J 288:3120–3134
Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK (2005) Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem 280:9719–9727
Horita H, Wysoczynski CL, Walker LA, Moulton KS, Li M, Ostriker A et al (2016) Nuclear PTEN functions as an essential regulator of SRF-dependent transcription to control smooth muscle differentiation. Nat Commun 7:10830
Salmon M, Gomez D, Greene E, Shankman L, Owens GK (2012) Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22α promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ Res 111:685–696
Zheng JP, He X, Liu F, Yin S, Wu S, Yang M et al (2020) YY1 directly interacts with myocardin to repress the triad myocardin/SRF/CArG box-mediated smooth muscle gene transcription during smooth muscle phenotypic modulation. Sci Rep 10:21781
Liu ZP, Wang Z, Yanagisawa H, Olson EN (2005) Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin. Dev Cell 9:261–270
Deaton RA, Gan Q, Owens GK (2009) Sp1-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle. Am J Physiol Heart Circ Physiol 296:H1027–H1037
Guo X, Shi N, Cui XB, Wang JN, Fukui Y, Chen SY (2015) Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling. Circ Res 116:e71–e80
Nagao M, Lyu Q, Zhao Q, Wirka RC, Bagga J, Nguyen T et al (2020) Coronary disease-associated gene TCF21 inhibits smooth muscle cell differentiation by blocking the myocardin-serum response factor pathway. Circ Res 126:517–529
Bonasio R, Tu S, Reinberg D (2010) Molecular signals of epigenetic states. Science 330:612–616
Jones PA, Takai D (2001) The role of DNA methylation in mammalian epigenetics. Science 293:1068–1070
Laukkanen MO, Mannermaa S, Hiltunen MO, Aittomäki S, Airenne K, Jänne J et al (1999) Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler Thromb Vasc Biol 19:2171–2178
Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G et al (2013) Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation 128:2047–2057
Zhuang J, Luan P, Li H, Wang K, Zhang P, Xu Y et al (2017) The Yin-Yang dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Arterioscler Thromb Vasc Biol 37:84–97
Hiltunen MO, Turunen MP, Häkkinen TP, Rutanen J, Hedman M, Mäkinen K et al (2002) DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc Med 7:5–11
Jiang JX, Aitken KJ, Sotiropoulos C, Kirwan T, Panchal T, Zhang N et al (2013) Phenotypic switching induced by damaged matrix is associated with DNA methyltransferase 3A (DNMT3A) activity and nuclear localization in smooth muscle cells (SMC). PLoS ONE 8:e69089
Rozenberg JM, Tesfu DB, Musunuri S, Taylor JM, Mack CP (2014) DNA methylation of a GC repressor element in the smooth muscle myosin heavy chain promoter facilitates binding of the Notch-associated transcription factor, RBPJ/CSL1. Arterioscler Thromb Vasc Biol 34:2624–2631
Wang YS, Chou WW, Chen KC, Cheng HY, Lin RT, Juo SH (2012) MicroRNA-152 mediates DNMT1-regulated DNA methylation in the estrogen receptor α gene. PLoS ONE 7:e30635
Hu W, Wang M, Yin H, Yao C, He Q, Yin L et al (2015) MicroRNA-1298 is regulated by DNA methylation and affects vascular smooth muscle cell function by targeting connexin 43. Cardiovasc Res 107:534–545
Ning Y, Huang H, Dong Y, Sun Q, Zhang W, Xu W et al (2013) 5-Aza-2’-deoxycytidine inhibited PDGF-induced rat airway smooth muscle cell phenotypic switching. Arch Toxicol 87:871–881
Azechi T, Sato F, Sudo R, Wachi H (2014) 5-aza-2’-Deoxycytidine, a DNA methyltransferase inhibitor, facilitates the inorganic phosphorus-induced mineralization of vascular smooth muscle cells. J Atheroscler Thromb 21:463–476
Raines EW (2000) The extracellular matrix can regulate vascular cell migration, proliferation, and survival: relationships to vascular disease. Int J Exp Pathol 81:173–182
Connelly JJ, Cherepanova OA, Doss JF, Karaoli T, Lillard TS, Markunas CA et al (2013) Epigenetic regulation of COL15A1 in smooth muscle cell replicative aging and atherosclerosis. Hum Mol Genet 22:5107–5120
Xie SA, Zhang T, Wang J, Zhao F, Zhang YP, Yao WJ et al (2018) Matrix stiffness determines the phenotype of vascular smooth muscle cell in vitro and in vivo: Role of DNA methyltransferase 1. Biomaterials 155:203–216
Manabe I, Owens GK (2001) Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system. Circ Res 88:1127–1134
Qiu P, Li L (2002) Histone acetylation and recruitment of serum responsive factor and CREB-binding protein onto SM22 promoter during SM22 gene expression. Circ Res 90:858–865
Kee HJ, Kwon JS, Shin S, Ahn Y, Jeong MH, Kook H (2011) Trichostatin A prevents neointimal hyperplasia via activation of Krüppel like factor 4. Vascul Pharmacol 55:127–134
McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK (2006) Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest 116:36–48
Qiu P, Ritchie RP, Gong XQ, Hamamori Y, Li L (2006) Dynamic changes in chromatin acetylation and the expression of histone acetyltransferases and histone deacetylases regulate the SM22alpha transcription in response to Smad3-mediated TGFbeta1 signaling. Biochem Biophys Res Commun 348:351–358
Yoshida T, Gan Q, Shang Y, Owens GK (2007) Platelet-derived growth factor-BB represses smooth muscle cell marker genes via changes in binding of MKL factors and histone deacetylases to their promoters. Am J Physiol Cell Physiol 292:C886–C895
Usui T, Morita T, Okada M, Yamawaki H (2014) Histone deacetylase 4 controls neointimal hyperplasia via stimulating proliferation and migration of vascular smooth muscle cells. Hypertension 63:397–403
Yoshida T, Gan Q, Owens GK (2008) Kruppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am J Physiol Cell Physiol 295:C1175–C1182
Liu M, Espinosa-Diez C, Mahan S, Du M, Nguyen AT, Hahn S et al (2021) H3K4 di-methylation governs smooth muscle lineage identity and promotes vascular homeostasis by restraining plasticity. Dev Cell 56:2765–2782
Lino CC, Kessinger CW, Cheng Y, MacDonald C, MacGillivray T, Ghoshhajra B et al (2018) An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat Commun 9:1009
Albinsson S, Suarez Y, Skoura A, Offermanns S, Miano JM, Sessa WC (2010) MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler Thromb Vasc Biol 30:1118–1126
Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q et al (2009) MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res 105:158–166
Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN et al (2009) miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460:705–710
Farina FM, Hall IF, Serio S, Zani S, Climent M, Salvarani N et al (2020) miR-128-3p is a novel regulator of vascular smooth muscle cell phenotypic switch and vascular diseases. Circ Res 126:e120–e135
Yang F, Chen Q, He S, Yang M, Maguire EM, An W et al (2018) miR-22 is a novel mediator of vascular smooth muscle cell phenotypic modulation and neointima formation. Circulation 137:1824–1841
Yang K, Ren J, Li X, Wang Z, Xue L, Cui S et al (2020) Prevention of aortic dissection and aneurysm via an ALDH2-mediated switch in vascular smooth muscle cell phenotype. Eur Heart J 41:2442–2453
Zeng Z, Xia L, Fan X, Ostriker AC, Yarovinsky T, Su M et al (2019) Platelet-derived miR-223 promotes a phenotypic switch in arterial injury repair. J Clin Invest 129:1372–1386
Quinn JJ, Chang HY (2016) Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17:47–62
Michalik KM, You X, Manavski Y, Doddaballapur A, Zörnig M, Braun T et al (2014) Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res 114:1389–1397
Zhao J, Zhang W, Lin M, Wu W, Jiang P, Tou E et al (2016) MYOSLID is a novel serum response factor-dependent long noncoding RNA that amplifies the vascular smooth muscle differentiation program. Arterioscler Thromb Vasc Biol 36:2088–2099
Ahmed A, Dong K, Liu J, Wen T, Yu L, Xu F et al (2018) Long noncoding RNA NEAT1 (nuclear paraspeckle assembly transcript 1) is critical for phenotypic switching of vascular smooth muscle cells. Proc Natl Acad Sci U S A 115:E8660–E8667
Bell RD, Long X, Lin M, Bergmann JH, Nanda V, Cowan SL et al (2014) Identification and initial functional characterization of a human vascular cell-enriched long noncoding RNA. Arterioscler Thromb Vasc Biol 34:1249–1259
Wang YN, Shan K, Yao MD, Yao J, Wang JJ, Li X et al (2016) Long noncoding RNA-GAS5: a novel regulator of hypertension-induced vascular remodeling. Hypertension 68:736–748
Choi M, Lu YW, Zhao J, Wu M, Zhang W, Long X (2020) Transcriptional control of a novel long noncoding RNA Mymsl in smooth muscle cells by a single Cis-element and its initial functional characterization in vessels. J Mol Cell Cardiol 138:147–157
Zhang C, Ge S, Gong W, Xu J, Guo Z, Liu Z et al (2020) LncRNA ANRIL acts as a modular scaffold of WDR5 and HDAC3 complexes and promotes alteration of the vascular smooth muscle cell phenotype. Cell Death Dis 11:435
Kristensen LS, Andersen MS, Stagsted L, Ebbesen KK, Hansen TB, Kjems J (2019) The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20:675–691
Hall IF, Climent M, Quintavalle M, Farina FM, Schorn T, Zani S et al (2019) Circ_Lrp6, a circular RNA enriched in vascular smooth muscle cells, acts as a sponge regulating miRNA-145 function. Circ Res 124:498–510
Zeng Z, Xia L, Fan S, Zheng J, Qin J, Fan X et al (2021) Circular RNA CircMAP3K5 Acts as a MicroRNA-22-3p sponge to promote resolution of intimal hyperplasia via TET2-mediated smooth muscle cell differentiation. Circulation 143:354–371
Xu JY, Chang NB, Rong ZH, Li T, Xiao L, Yao QP et al (2019) circDiaph3 regulates rat vascular smooth muscle cell differentiation, proliferation, and migration. FASEB J 33:2659–2668
Mao YY, Wang JQ, Guo XX, Bi Y, Wang CX (2018) Circ-SATB2 upregulates STIM1 expression and regulates vascular smooth muscle cell proliferation and differentiation through miR-939. Biochem Biophys Res Commun 505:119–125
Yang L, Yang F, Zhao H, Wang M, Zhang Y (2019) Circular RNA circCHFR facilitates the proliferation and migration of vascular smooth muscle via miR-370/FOXO1/Cyclin D1 pathway. Mol Ther Nucleic Acids 16:434–441
Funding
This work was supported by the National Natural Science Foundation of China (No. 81974182 and No. 82171325 to LM, No. 81771249 to YPX).
Author information
Authors and Affiliations
Contributions
FZ and XG wrote this manuscript. LM and YX designed the review and approved this manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, F., Guo, X., Xia, Y. et al. An update on the phenotypic switching of vascular smooth muscle cells in the pathogenesis of atherosclerosis. Cell. Mol. Life Sci. 79, 6 (2022). https://doi.org/10.1007/s00018-021-04079-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-021-04079-z