
Abstract
Nicotinic acetylcholine receptors (nAChRs) are pentameric ion channels expressed in the central nervous systems. nAChRs containing the α4, β2 and α5 subunits are specifically involved in addictive processes, but their functional architecture is poorly understood due to the intricacy of assembly of these subunits. Here we constrained the subunit assembly by designing fully concatenated human α4β2 and α4β2α5 receptors and characterized their properties by two-electrodes voltage–clamp electrophysiology in Xenopus oocytes. We found that α5-containing nAChRs are irreversibly blocked by methanethiosulfonate (MTS) reagents through a covalent reaction with a cysteine present only in α5. MTS-block experiments establish that the concatemers are expressed in intact form at the oocyte surface, but that reconstitution of nAChRs from loose subunits show inefficient and highly variable assembly of α5 with α4 and β2. Mutational analysis shows that the concatemers assemble both in clockwise and anticlockwise orientations, and that α5 does not contribute to ACh binding from its principal (+) site. Reinvestigation of suspected α5-ligands such as galantamine show no specific effect on α5-containing concatemers. Analysis of the α5-D398N mutation that is linked to smoking and lung cancer shows no significant effect on the electrophysiological function, suggesting that its effect might arise from alteration of other cellular processes. The concatemeric strategy provides a well-characterized platform for mechanistic analysis and screening of human α5-specific ligands.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nicotinic receptors form a family of acetylcholine (ACh) gated ion channels that are widely expressed in the nervous system. They result from the assembly of 5 subunits taken from a repertoire of 16 subunit genes in the human genome. In the brain, a single subunit α7 can form homopentamers, while the others contain both principal (α2, 3, 4, 6) and complementary (β2, 4) subunits, together or not with an accessory (α5, β3) subunit. The combinatorial association of various subunits thus generates a large repertoire of receptors. Genomic studies associate the α5 gene to smoking and lung cancer through the existence of a polymorphism that produces a protein variant of α5 (D398N) [1]. In mice, α4−/− and β2−/− genotypes fail to self-administer nicotine, while α5−/− genotype displays a unique altered behavior towards nicotine, with partly reduced nicotine-related behaviors [2,3,4] and also towards ethanol, whose effects are partly enhanced [5], together with anxiety-related behaviors [6]. In rats, a recent study also highlighted a phenotype linked to nicotine and alcohol relapse processes [7, 8]. In the brain, α5 incorporates in α3β4α5 and α4β2α5 nAChRs, and both nAChRs may contribute to these effects. Among them, α4β2α5 nAChRs in the dorsal striatum were found critical to regulate dopamine transmission and release [1, 9, 10]. Likewise, deletion of α5 in ventral tegmental dopaminergic neurons, which also express α4 and β2, markedly shifts to higher concentrations nicotine-elicited increase in neuron firing as well as nicotine self-administration. These effects are reversed, either totally or partially, by the re-expression of wild-type versus D398N α5 subunit [3, 11]. These observations indicate a key contribution of the α4β2α5 combination in addictive processes.
Despite their therapeutic potential in physiology and translation, α4β2α5 nAChRs are poorly understood. This is due to the intricate assembly of the subunits that associate in various stoichiometries. First, co-expression in recombinant systems of α4 and β2 generates two populations of functional receptors: 1/(α4)2(β2)3, carrying two ACh binding sites at the interface between the “principal” or (+) face of α4 and the “complementary” or (−) face of β2 [named α4(+)–β2(−) sites], and 2/(α4)3(β2)2, carrying two α4(+)–β2(−) sites and one α4(+)–α4(−) site, this latter displaying low ACh affinity [12]. Additional co-expression of the α5 subunit is thought to mostly yield a receptor with a (α4)2(β2)2α5 stoichiometry, with unknown number of ACh binding sites since it is not clear whether α5 can participate to one, and since α5 can potentially occupy any position within the pentamer.
Detailed functional and pharmacological study of these receptors requires overcoming the “stoichiometry issue”. Two main strategies have been developed: (1) by varying with the ratios of transfected/injected DNAs of the distinct subunits, which is assumed to favor the proportion of a particular subunit in the pentamer, but this technique cannot certify that a single stoichiometry is being expressed; (2) another approach is to fuse the subunits into concatemeric nAChRs whose subunits are covalently connected by linkers of various sizes. However, di-, tri-, tetramers were found not to definitely solve the stoichiometry issue [13], and the current approach is to generate fully concatenated pentameric receptors that are thought to unambiguously constrain the stoichiometry, providing that the protein is not cleaved within the cell.
Several pentameric concatemers of heteromeric receptors have been described incorporating the α4β2 and α3β4 subunits [11, 14,15,16,17,18,19,20,21,22], as well as the α3β4α5 subunits [11, 19]. In contrast, a single study described the α4β2α5 combination, with an arrangement (β2α4α5α4α4/β2) where only one α4β2 pair (and therefore one canonical α4/β2 binding site) is present in the pentamer and that are endowed with weak ACh-gated currents (less than 50 nA in oocytes [19]). In the present paper, we describe α4β2α5 containing concatemers where α5 is assembled with two α4β2 pairs and that generate substantial (in the 0.1–1 µA range) ACh-gated currents. This enables unambiguous expression of defined nAChRs stoichiometries, allowing us to revisit the functional and pharmacological properties of these key receptors involved in addiction.
Methods
Molecular biology
The design of nAChR concatemers used in this study followed the same strategy as a previous concatemer design for GABAA receptors [23]. Human α4, β2 and α5 nAChR subunits were first subcloned into the pRK5 vector. To enhance expression in Xenopus oocytes, a 37 bp 5′UTR region of Xenopus β-globin mRNA was added upstream of the open reading frame in pRK5 and the Kozak sequence was optimized. To facilitate the cloning, the endogenous HindIII and NheI sites from the β2 subunit were removed by site directed mutagenesis.
Inverse PCR was further used to add 20 primer-encoded glutamines to the C-terminal end of the α4 subunits and 15 to the end of the α5 and β2 subunits—except for subunits used as the last subunit in the concatemers—and the individual subunits were cloned to be flanked by unique restriction sites.
The five subunits were then assembled into a single open reading frame between ClaI and HindIII sites, with the first subunit containing a signal peptide (α4 signal peptide for the 3(α4), 3(β2) and α5last concatemers; α5 signal peptide for the α5first concatemer). All the other subunits were composed only of their mature proteins as described in the Uniprot.
The final arrangements are: for the 3(α4) concatemer ClaI-Kozak-SP(α4)–α4-Q20-AgeI–β2-15Q-SalI–α4-20Q-NheI–β2-15Q-XhoI–α4-Stop-HindIII, for the 3(β2) concatemer ClaI-Kozak-SP(α4)–α4-20Q-AgeI–β2-15Q-SalI–α4-20Q-NheI–β2-15Q-XhoI–β2-Stop-HindIII, for the α5last concatemer ClaI-Kozak-SP(α4)–α4-20Q-AgeI–β2-15Q-SalI–α4-20Q-NheI–β2-15Q-XhoI–α5-Stop-HindIII and for the α5first concatemer ClaI-Kozak-SP(α5)–α5-15Q-AgeI–β2-15Q-SalI–α4-20Q-NheI–β2-15Q-XhoI–α4-Stop-HindIII.
Concatemers containing selected point mutations in particular subunits were constructed by mutating the corresponding single subunits, which were then cloned back into the concatemer.
When specified, we used GFP-fusion concatemers, inspired from Nashmi et al. [24]: the coding sequence of the eGFP was inserted in the intracellular domain of α4 between Lys423 and Ser424 (according to the Uniprot P43681 numbering) to give the following arrangement […PGPSCK-eGFP-SPSDQL…].
HEK cells expression and Western blotting
HEK293 cells were cultured in 100 mm dishes in DMEM containing fetal bovine serum (10% v/v) and antibiotics (penicillin/streptomycin, 10 U/ml and 10 µg/ml respectively), and 10 µg DNA was transfected using JetPrime reagent (Polyplus) according to manufacturer instructions. After 48 h expression at 37 °C, cells were resuspended in lysis buffer containing 150 mM Tris pH 7.6, 200 mM NaCl and 1% Triton, and used for Western blotting. Western blotting was performed using mouse anti-GFP antibody (Invitrogen) and goat anti-mouse HRP-coupled antibody (Vectorlabs), and revealed with SuperSignal West Pico ECL kit (ThermoPierce).
Two-electrode voltage–clamp electrophysiology
Xenopus laevis oocytes were obtained from Centre de Ressources Biologiques-Rennes, France or from EcoCyte Bioscience, Germany, and maintained in modified Barth’s medium (88 mM NaCl, 1 mM KCl, 1 mM MgSO4, 2.5 mM NaHCO3, 0.7 mM CaCl2, 5 mM HEPES pH 7.3). Defolliculated oocytes were submitted to intranucleus injection of 2–5 ng of cDNA and kept at 18 °C for 2–5 days before recording.
For DNA ratio injections, cDNA sequences of human α4, β2 and α5 inserted in pRK5 vectors were mixed to the desired ratio while keeping a constant total DNA concentration of 0.06 ng/nl, and were recorded 1–2 days after injection.
Recordings were performed with a Digidata 1550A (Molecular Devices) digitizer, an Oocyte Clamp OC-725C (Warner Instruments) amplifier and using the pClamp 10.5 software. Oocytes were perfused with Ringer’s buffer (100 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES pH 7.3) using a gravity-driven system. Drugs were applied after dilution in Ringer’s buffer. MTSEA solutions were prepared immediately before recording. All currents were measured at − 60 or − 80 mV.
Data analysis
Recordings were analyzed using ClampFit, AxoGraph X, Plot and GraphPad Prism. Measurements were performed at the peak of the response. Except when stated otherwise, for concentrations response curves, points were fitted to a mono- or bi-phasic Hill equation for each cell before averaging EC50 and Hill slopes. For the statistical comparisons using GraphPad Prism, we performed Student’s t tests when comparing two groups and one-way ANOVA when comparing more than two groups (with Dunnett’s or Tukey’s post hoc tests, depending on whether the data are compared to a control group or not). Figures representing published X-ray structures were prepared using PyMol. Figures were prepared using GraphPad Prism and Inkscape.
Results
Concatemer design and characterization
Concatemer design
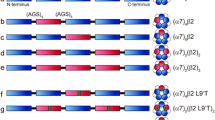
The design we adopted is “Signal Peptide (SP)–α–β–α–β–α/β”, giving for the α4β2 constructs SP(α4)–α4–β2–α4–β2–α4, called the 3(α4) concatemer, and SP(α4)–α4–β2–α4–β2–β2, called the 3(β2) concatemer (Fig. 1a). For α4β2α5 concatemers, we generated two constructs, one where α5 and its SP are inserted as the first subunit, SP(α5)–α5–β2–α4–β2–α4, called the α5first concatemer, and another construct where α5 is inserted as a last subunit, SP(α4)–α4–β2–α4–β2–α5, called α5last. We chose the linkers based on sequences alignment and the α4β2 X-ray structures [25]. It consists of 15 or 20 glutamine residues plus 2 residues corresponding to unique restriction sites used for cloning (Thr-Gly, Val-Asp, Ala-Ser and Leu-Glu in this order), a strategy already used to generate pentameric GABAA concatemers [23]. To compensate the fact that, as compared to α5 and β2, the C-terminus of α4 (post-M4 helix) is shorter by 12 and 15 residues respectively, the linkers are 15 glutamines long in the α4–β2 connection and 20 glutamines long for the α5–β2, β2–α4 and β2–α5 ones.

A robust concatemer design. a Schematic of the concatemers’ arrangement. Each subunit is depicted by an oval, with the first subunit in the N-terminal having its signal peptide as a circle. Linkers are depicted with a black line. b Anti-GFP Western blotting of whole extracts of HEK cells expressing the designated concatemer. c Sample traces recorded on oocytes expressing the 3(α4) (blue), 3(β2) (red), α5first (dark green) and α5last (light green) concatemers. For the 3(α4), ACh concentration was 100 µM, and for the 3(β2) and the α5, ACh concentration was 10 µM. d ACh concentration–response curves of the currents recorded for the four concatemers. Points are mean ± SD, with n ≥ 7
Concatemers metabolic stability
To assess the metabolic stability of the concatemers, we selected a representative subset of constructs. We inserted the GFP coding sequence in the 3(α4), 3(β2) and α5last concatemers. GFP was inserted within the intracellular domain of α4 similarly to previous work on the mouse α4 [24], and in the first subunit of the concatemer to visualize all sub-products if any. Constructs were transfected in HEK cells and whole cell extracts were subjected to Western blot analysis against GFP. All constructs show a single band at a high molecular weight >190 kDa), consistent with a pentameric concatemer, showing that the protein undergoes little, if any, degradation (Fig. 1b).
Concatemers ACh concentration–response curves
Concatemers were expressed in Xenopus oocytes and acetylcholine-evoked currents were recorded by two-electrode voltage–clamp electrophysiology. The 3(α4) and 3(β2) concatemers generate robust currents in the 1–10 µA range, while α5 concatemers generate lower, yet reasonable, currents in the 0.1–1 µA range, with some cells displaying maximal currents above 1 µA (Fig. 1c). ACh concentration–response curve of the 3(β2) and the α5 concatemers are similar with EC50s of 1.45, 0.75 and 1.17 µM for 3(β2), α5first and α5last respectively (Fig. 1d, Supplementary Table 1). The 3(α4) concatemer, known to carry an additional low affinity α4–α4 site, yields a biphasic curve as expected. Constraining the high-affinity EC50 to 1 µM yields a second component with an EC50 of 28.9 µM. All values are consistent with the literature on α4β2 receptors, both on loose subunits [17, 26] and on pentameric concatemeric α4β2 constructs [17].
Rotational direction of concatemers
In 2018, Ahring and colleagues showed that concatemers of nicotinic receptors often assemble readily in both the clockwise and the counterclockwise orientations, yielding heterogeneity of subunit arrangements within the pentamer even for fully concatenated constructs (Supplementary Fig. 1). Their approach was based on the use of the positive modulator NS9283 [13]. NS9283 was shown by X-ray crystallography to occupy the orthosteric site of the acetylcholine binding protein [27]. On the α4β2 nAChR, NS9283 potentiation is disrupted by three-point mutations introducing a VFL β2-specific motif on the complementary face of the α4 subunit [13] (Supplementary Fig. 2), while introducing the HQT α4-specific motif on the complementary face of the β2 subunit converts it into an agonist. Combined data strongly support that NS9283 binds specifically at the α4–α4 interface [27]. Single channel measurements also showed that NS9283 potentiation was sensitive to mutations performed on the principal face of the β2 subunit [22].
Considering the 3(α4) concatemer, a clockwise orientation would place the first α4 subunit contributing to the (−) face of the α4–α4 site, and conversely an anticlockwise orientation would place the 5th subunit in this position. NS9283 binding specifically at the α4–α4 interface, a fixed orientation of the concatemer should thus yield an alteration of its binding when mutating only one of the two terminal subunits (Fig. 2a). We thus introduced the mutation triplets in the α4 subunits of the 3(α4) concatemer, one subunit at a time (calling the resulting constructs SU1, SU3 and SU5). We also constructed a concatemer in which both the 1st and the 5th subunit harbor the VFL triplet (SU1 + SU5). ACh concentration response shows marked gain of function effect of the mutations on the four constructs (Fig. 2b and Supplementary Table 1), showing an increase of the ACh apparent affinity as compared to the WT. Thus, to reliably evaluate the potentiation by NS9283, we used an ACh test concentration of 0.1 µM which is below the EC20 of the four constructs. On the WT 3(α4) concatemer, when we co-applied ACh at 0.1 µM with 0.1, 1, 3, 10, 30 and 100 µM of NS9283, we observed a dose-dependent potentiation of the currents as previously described (Fig. 2c, d). We controlled that NS9283 does not significantly potentiate the ACh-gated currents of the SU1 + SU5 construct, in agreement with its binding at the α4–α4 interface [13] (Fig. 2c, d). Strikingly, all single-subunit mutations, SU1, SU3 and SU5 constructs, display robust potentiation of the currents by NS9283 (Fig. 2c,d). We conclude that the 3(α4) concatemer produces a mixture of both orientations at the cell surface (Fig. 2a), both participating substantially to the electrophysiological response.

Rotation direction of the 3(α4) concatemer. a Potential integrity of the NS9283 binding site depending on the position of the VFL triple-mutation. In the WT, NS9283 binds at the α4–α4 interface. In the triple-mutation VFL situation on all three α4 subunits, the NS9283 binding site is impaired and no potentiation is detected (Ahring 2017). When the VFL mutation is introduced in the middle α4 subunit (“SU3”) the NS9283 binding site is predicted to be intact. When it is introduced in the first (“SU1”) or the last (“SU5”) α4 subunit, pentamers that adopt an orientation that makes the VFL residues pointing in the NS9283 binding site should not be potentiated anymore. b ACh concentration–response curves of WT, SU1, SU3, SU5 and SU1 + SU5 constructs show that all mutants display gain-of-function phenotypes towards ACh, as previously described (Ahring 2017). Points are mean ± SD, with n ≥ 3. Unlike the original study, we choose a test ACh concentration that would allow to observe clear potentiation for the 3 constructs: 0.1 µM which is around the EC10 of SU3, SU5 and SU1 + SU5, and EC05 of SU1. c Example traces of NS9283 potentiation of the WT and the VFL mutants. Vertical bar is 100 nA, horizontal bar is 10 s. d Concentration–response curves of the potentiation of 0.1 µM ACh currents by NS9283. A dash line is drawn to figure 100%. Values measured for 100 µM NS9283 were not included in the curve fit because they were reproducibly lower than the 30 µM values, possibly due to a channel block effect, but they are still displayed as empty circles. Points are mean ± SD with n ≥ 3. * for SU1 + SU5, a Student’s t test found those two values to be non-statistically different
Investigating α5-nAChR pharmacology
For the sake of simplicity, and if not stated otherwise, we show here data related to the α5first concatemer, first because it displays similar properties to those of the α5last, and second because, as shown above, the mixture of orientations likely results in the same mixture of orientations for both constructs (Supplementary Fig. 1). In the search for α5 ligands, we investigated compounds that were described in the literature to bind to the α5 subunit from work on loose or partially concatenated subunits.
ACh and Sazetidine-A
Since co-expression of α5 and β2 without α4 does not generate functional channels, it is believed that α5 does not contribute to the principal component of the ACh site, but this idea has been challenged in a recent study [28]. In α4β2 nAChRs, the canonical ACh binding site is well characterized, as illustrated by the X-ray structures of α42β23 in complex with nicotine [25] (Fig. 3a). The site is composed of three regions called loops from the principal α4 component, each of them contributing aromatic residues to bind the positive ammonium moiety of the ligand, and four regions of the complementary β2 component. The principal component is only partially conserved in α5, questioning its ability to bind ACh from of its (+) face (Supplementary Figs. 1 and 2). For instance, some critical aromatic residues in the (+) face of α4 are present in α5 (Trp in Loop B, the key binding aromatic residue stabilizing the ligand through cation-pi interaction [29], as well as Tyr C2), while some are absent (Tyr in Loop A and Loop C1).

The orthosteric site. a The nicotine binding pocket in the α4β2 X-ray structure visualized using PyMol (Morales-Perez 2018, pdb 5KXI). α4 subunit is shown in blue cartoon representation, β2 in red cartoon representation. Loop B and Loop C are labeled, Trp 156 is shown in orange stick representation and nicotine is depicted in yellow stick representation. b ACh concentration–response curves for the α5 concatemers WT and W169A mutant. Points are mean ± SD with n ≥ 3. c Sample traces of the effect of saturating concentrations of ACh and Sazetidine-A (Saz-A) on a single oocyte for the three concatemers. d Concentration–response curves for Saz-A of the three concatemers. Currents were normalized with the maximal current recorded with ACh for each cell. Points are mean ± SD with n ≥ 6
If α5(+) contributes to ACh binding, its mutation should alter the ACh EC50 as recorded by TEVC. To challenge this idea, we mutated the α5 Loop B Trp in α5first and in α5last into alanine (α5-W169A). Concentration–response curves of the α5-W169A concatemers are indistinguishable from the WT with EC50 of 0.87 and 1.00 µM respectively, and nH of 1.00 and 1.08 (Fig. 3b, Supplementary Table 1). This suggests that the (+) face of α5 does not bind ACh. This idea is fully consistent with our observation that the α5 and the 3(β2) concatemers (which carry an α5 or a β2 subunit at the “accessory” position) have highly similar EC50 for ACh.
However, in the course of our pharmacological investigations, we observed that the α5first and the 3(β2) concatemers have different concentration–response curves for sazetidine. Sazetidine-A (Saz-A) is a full agonist of α42β23 nAChRs but a partial agonist of α43β22 since it does not bind to the α4–α4 interface [30, 31]. On the 3(α4) concatemers, Saz-A generates small currents that do not exceed 6.0 ± 2% of the maximal response to ACh (Fig. 3c and Supplementary Table 1). In contrast, we found that Saz-A is a full agonist on both α5first and 3(β2) concatemers, but it displays higher EC50 and lower cooperativity for α5first (22 nM and nH = 0.86 as compared to 3(β2) 6.6 nM and nH = 1.58, Fig. 3d and Supplementary Table 1). Interestingly, looking at the concatemer assemblies in both rotational directions allows proposing a simple interpretation of this discrepancy. While the clockwise and anticlockwise assemblies of the 3(β2) concatemer both carry two α4(+)–β2(−) sites, the clockwise assembly of α5first carries two α4(+)–β2(−) sites but the anticlockwise assembly of α5first carries one α4(+)–β2(−) site and one α4(+)–α5(−) site. The lower cooperativity of α5first suggests the contribution of a heterogeneous class of binding sites, and we infer that, in the anticlockwise assembly, the α4(+)–α5(−) site might display an affinity similar to that of the α4(+)–β2(−) for ACh, but a lower affinity for sazetidine.
A lower affinity of sazetidine for the α4(+)–α5(−) site would also be in line with its stronger sensitivity toward the amino-acid composition of the complementary component of the site, which explains its inactivity on the α4(+)–α4(−) site. An illustration of this are the binding modes of Sazetidine-A derivatives on ACh binding proteins (AChBP) that shows that while the ammonium moiety interacts with the aromatic box of the principal component, the rest of the compound lies on the complementary component and contacts more residues than smaller agonists, notably one hydrophobic residue from Loop E aligning with α5-Thr139 (Supplementary Fig. 2b, d).
Overall, our data support the idea that the α5(+) face does not contribute to ACh activation in the concatemers, but they indirectly suggest that the α4(+)–α5(−) interface might also bind and be activated by ACh and sazetidine, displaying respectively similar and lower affinity than the canonical α4(+)–β2(−) site.
Investigating other α5-ligand candidates
Galantamine is an inhibitor of acetylcholine esterase and is used for treatment for Alzheimer’s disease with debated efficacy. It was shown to potentiate ACh responses of the neuronal α4β2 and α7 receptors by no more than 2-folds [32]. It was further described as potentiating only α4β2α5 receptors as opposed to α4β2 and α4β2β3 assemblies in calcium flux measurements of α4β2 stable cell lines transfected with α5[33]. More recently, another study using loose subunits surprisingly described the absence of potentiation on both α4β2 and α7 receptors [34], spreading confusion on the validity of galantamine effect. Herein, we applied galantamine for 3 s or for 20 s before ACh pulses at its EC20, as well as in co-application. We could not observe any significant potentiation, nor inhibition, of any of the concatemers by galantamine (Fig. 4a, b), neither α5first nor 3(β2) or 3(α4).

Galantamine and progesterone fail to selectively modulate α5-containing concatemers. a Sample traces of Galantamine effect on the 3(α4) concatemer. Three protocols are displayed for the same cell: a 3-s pre-application, a 20-s pre-application and a co-application of galantamine with a sub-saturating ACh concentrations, showing no effect on the peak current. b Normalized peak currents obtained with the three protocols for the three concatemers. The apparent potentiation of 3(α4) is not significant as compared with the 3(β2). Displayed are mean ± SD with n ≥ 3. “n.s.” indicates that a one-way ANOVA with a Tukey’s post hoc test was performed and found the values not statistically different from each other. c Sample traces of progesterone inhibition on the three concatemers. For each cell, a control application of 17 mM of Ethanol was perform to account for the ethanol concentration used for progesterone solubilization. d Percentage inhibition of 30 µM ACh-evoked currents by 3.14 mM progesterone. Data on the right are corrected by the ethanol effect for each cell at 17 mM. The corrected values for progesterone inhibition at 3.14 µM are similar for the three concatemers with n ≥ 3: 63.0 ± 12% for 3(α4), 71.7 ± 12% for 3(β2) and 61.7 ± 12% for α5first. “n.s.” indicates that a one-way ANOVA with a Tukey’s post hoc test was performed and found the values not statistically different from each other
Finally, we tested the neurosteroid progesterone because (1) its action on neuronal nAChRs was not investigated further than in the original study [35], and (2) it was linked to α5-nAChR modulation in α5−/− mice. Progesterone is soluble at 1 mg/ml in ethanol, we thus systematically applied a control solution with the ethanol concentration used in the progesterone solution. We found that 3.14 µM progesterone inhibits ACh-evoked currents on the three concatemers to similar extents (Fig. 4c, d). Thus, we conclude that progesterone is a non-selective inhibitor of α43β22, α42β23 and α42β22α5 nAChRs, having similar potencies for the various assemblies.
Exploring the D398N α5 mutation
The α5 D398N single nucleotide polymorphism (SNP) was linked to nicotine addiction and lung cancer by genome-wide association studies in humans [1]. The mutation lies in the ICD, far away from the ACh/nicotine binding sites, the channel lumen or from any known allosteric modulators’ sites. However, several studies have proposed various effects of the D398N on the function of recombinant α4β2α5 and α3β4α5 nAChRs, pointing at desensitization [36], calcium permeability [36, 37], orthosteric ligands apparent affinities [37] or maximal currents [11], effects that are rather moderate and not consistent from study to study, while one study found no effect [38].
We thus introduced the D398N mutation in the α5 concatemers. We first compared the metabolic stability of the WT and D398N α5last constructs in HEK cells, by Western blotting after insertion of GFP in the first subunit. As with the WT, we did not observe degradation of the concatemer after expression (Fig. 5a). We then injected WT and D398N α5first concatemers in Xenopus oocytes to investigate potential electrophysiological differences. ACh elicited robust currents from the D398N mutated concatemer, with amplitudes similar to those of the wild-type α5 concatemer (Fig. 5b). Concentration–response curves for ACh are similar, with an EC50 of 1.08 µM and nH of 1.17 for α5-D398N concatemer (Fig. 5c, Supplementary Table 1). To assess a potential effect of the α5-D398N mutation on the receptor’s desensitization, we analyzed the decaying phase of agonist-evoked responses during prolonged ACh application. We superposed currents traces generated at saturating and sub-saturating ACh concentration for the wild-type and D398N concatemers from oocytes coming from the same animal and observed no significant differences (Fig. 5d). After 5 s of a 100 µM ACh treatment, the current remaining corresponds to 61.3 ± 6.6% of the initial peak of the wild-type α5 and 55.9 ± 4.4% for the D398N mutant (Fig. 5e). It is noteworthy that, in oocyte, the ACh-gated currents increase intracellular calcium, thereby activating endogenous calcium-activated chloride channels. The measurements performed herein are thus only semi-quantitative, but overall, we could not detect any obvious difference between our WT and α5-D398N concatemers.

Apparent lack of functional effects of the α5-D398N mutation. a Anti-GFP western blot of whole cell extracts of HEK cells expressing the WT and D398N α5 concatemers. b Scattered dot-plot of maximal currents recorded in oocytes expressing the WT and D398N mutant α5 concatemers. Measurements were performed on injections series where the WT and mutant constructs were injected on the same oocytes batches and recorded the same day. Each dot corresponds to a single cell with n ≥ 10. “n.s.” indicates that a Student’s t test was performed and found the values not statistically different from each other. c ACh concentration–response curves for the α5 concatemers WT and D398N mutant. Points are mean ± SD with n ≥ 4. d Sample traces of responses to sub-saturating and saturating ACh concentrations for the WT (green) and D398N (purple) α5 concatemers. Traces were normalized to the peak to facilitate comparison. Oocytes from the same animal were compared. e Mean residual current observed after 5 s application of saturating ACh (10 µM) for the WT and D398N α5 concatemers. Oocytes from the same animal were compared with n ≥ 3. Displayed are mean ± SD. “n.s.” indicates that a Student’s t test was performed and found the values not statistically different from each other
Identification of an α5 specific irreversible channel-block reaction
In the course of SCAM (substituted cysteine accessibility method) experiments on the α5 concatemers, we applied the commonly used cysteine-modifying reagent MTSEA. When co-applied with ACh for 20 s, 200 µM MTSEA inhibited currents of all concatemers (Fig. 6). This inhibition is moderate and reversible upon washing for the 3(α4) and the 3(β2) concatemers, suggesting a pore-blocking effect. However, on the α5 concatemer, this inhibition is stronger and is not reversible upon washing, with still 65 ± 13% of the current silenced after a 5-min wash (Fig. 6b, it is noteworthy that inhibition is nearly complete (higher than 90%) when using 1 mM MTSEA, Fig. 7). In the α5 sequence, we noticed the presence of a cysteine residue in the second transmembrane segment (2′ position Cys261) which faces the ion channel lumen (Fig. 6c, d). This cysteine is not present in α4 and β2, and is thus a good candidate for α5-specific covalent reaction with MTSEA. We thus mutated the cysteine to serine (α5-C261S mutant), which fully abolished the irreversible component of MTSEA inhibition (Fig. 6e), demonstrating that the observed effect is due to the covalent modification of this residue in the channel lumen. In addition, we applied on the α5 concatemer another cysteine-modifying reagent used in SCAM studies, MTSET (Supplementary Fig. 3). We observed that MTSET is also able to irreversibly inhibit the α5 concatemer (40% inhibition after 1 min at 200 µM), likely through the reaction with the same Cys261.

Irreversible channel-block of α5-nAChR by MTSEA. a, b Sample trace of the effect of MTSEA (200 µM) on the 3(α4) and α5 concatemers during a 20 s co-application with ACh and after 5 min wash. c Sequence alignment of the M2 transmembrane segment for α4, β2 and α5. Arrows indicate the cysteine residue in 2′ mutated in α5. d View from the top of the pore of the α4β2 nAChR X-ray structure visualized using PyMol (Morales-Perez 2018 pdb 5KXI). The α-carbon of each of the 2′ residues is depicted as yellow spheres. e Mean irreversible inhibition after a 20 s MTSEA (200 µM) + ACh(3 µM) application for the 3(α4), 3(β2), α5 and α5-C261S concatemers. Displayed are mean ± SD with n ≥ 4. *** Statistically different from α5first (one-way ANOVA with Dunnett’s test using α5 as control group, p < 0.001)

Evaluation of α5 content in pentamers from loose subunits. a, b Sample traces of the effect of MTSEA on the 3(α4) and α5 concatemers during a 30 s 1 mM co-application with ACh and after 5 min wash. c Scattered dot-plot presenting the permanent inhibition measured for each cell injected with the indicated DNA ratio and with n ≥ 4. One-way ANOVA with Dunnett’s test using 1:1:0 as control group found those values not different from the 1:1:0 ration (n.s.) or statistically different with p < 0.001 (***)
In conclusion, we identified reagents that irreversibly inhibit human α5-containing nAChRs while leaving α4β2 receptors unaffected.
MTSEA as a tool to estimate the α5 content in α4β2 nAChRs
To study α4β2α5 nAChRs, it is common practice to transfect cells with an excess of α5-encoding cDNA, a 1:1:10 ratio for α4:β2:α5 being in most cases considered as sufficient to generate a majority of α5-containing receptors population. We challenged this assumption using the specificity of the MTSEA irreversible blockade of α5. We co-injected α4 and β2 cDNAs with increasing amounts of α5 cDNAs. We evaluated the irreversible inhibition of ACh-elicited currents by applying 1 mM MTSEA for 30 s in presence of ACh, followed by a 5-min wash, conditions for which we reach > 90% inhibition of the α5 concatemer (Fig. 7a, b), incidentally demonstrating that blockade by MTSEA can reach completion by increasing its concentration.
In the absence of α5 (1:1:0), we detect a moderate short-term current inhibition and no irreversible inhibition (Fig. 7c). This is consistent with what we observed for the 3(α4) and the 3(β2) concatemers. At 1:1:1 ratio, we observed no significant irreversible inhibition suggesting that little or no α5 is incorporated. At 1:1:2, half of the cells are not irreversibly inhibited by MTSEA, while the other half display partial irreversible inhibition. At 1:1:6, 1:1:10 and 1:1:12 ratios, the extent of irreversible inhibition progressively increases, but this is associated with a large variability from cell to cell, both between and within oocytes injection batches. At the 1:1:12 ratio, none of the oocytes display the nearly full inhibition of α5first. At higher ratios, such as 1:1:20, we could not detect currents most of the time, probably because overloading cells with α5 alters the assembly and/or export and/or function of receptors. Overall, even at the highest cDNA ratio of 1:1:12, MTSEA inhibition was highly variable, demonstrating that the population of receptors is still highly heterogenous with, for most cells, the significant contribution of α5-less receptors.
Altogether, these results show that for ratios higher than 1:1:6, incorporation of α5 does happen, sometimes in the majority on the pentamers, but is highly variable from cell to cell, and is never complete. This demonstrates that, even in excess of α5 cDNA, injection of loose subunits is not efficient to generate a homogenous population of α4β2α5 nAChRs.
Discussion
Design of concatemers unambiguously and quantitatively expressing α5-nAChRs
The study of α5-nAChRs is difficult in recombinant systems that express loose subunits, because the resulting population is heterogenous and hardly controlled by the experimenter. Here, we directly illustrate this issue by using our finding that MTSEA irreversibly inhibits only α5-containing receptor. In our system, even at the highest cDNA 1:1:12 ratio for α4:β2:α5, we show that oocytes express a heterogenous population of receptors with the significant contribution of α5-less assemblies, in a proportion highly variable from cell to cell. Interestingly, in the seminal paper of Ramirez-Latorre et al. [39], the authors used substituted-cysteine accessibility method at several positions in the ion channel of the mouse α5, including the 2′ (since mouse α5 carries an endogenous serine at position 2′), to demonstrate the incorporation of α5 into α4β2 nAChRs. Their study, following RNA injection of loose subunits in oocyte, is consistent with ours with a partial (30%) irreversible inhibition of the currents by 1 mM MTSET for the α4:β2:α5(S2′C) at a 1:1:10 ratio, as compared to the (40%) irreversible inhibition by 200 μM MTSET for the α5first concatemer. However, our results contrast with those of Marotta et al. [40] which show that, for a 1:1:10 ratio for α4:β2:α5(V9′S) expressed by RNA injection in the oocyte, a nearly homogenous population containing the α5(V9′S) subunit, which specifically shows fast-reversible mecamylamine inhibition, is expressed. Since we show here a high variability in α5 incorporation, it is possible that this discrepancy comes from different oocyte sources or RNA versus DNA injection. We also cannot exclude species differences since they used mouse nicotinic subunits for their experiments. Alternatively, it can come from the presence of a mutation in the channel pore in the Marotta et al. study (V9′S), since single mutations in the transmembrane domain have been reported to strongly alter subunit assemblies in GABAA receptors [41].
In the present study, we developed a convenient concatemeric system to force the subunit stoichiometry, which display currents large enough for characterization. We conducted biochemical validation to ensure the integrity of the final protein products after expression, highlighting the absence of any cleavage. Moreover, for the α5-containing concatemers, we uncovered the irreversible inhibition by MTSEA due to the cysteine in the α5 pore region, thereby fully assigning the electrophysiological currents to α5-containing receptors. However, we provide evidence that the concatemers assemble according to two orientations, clockwise and anticlockwise, a feature that must be considered when interpreting pharmacological data. In the literature, we found another study describing two concatemeric α4β2α5 nAChRs. They differ from ours from the linker used and the subunit order [19], which in this case inserts α5 between two α4s (β2–α4–α5–α4–α4/β2). The resulting constructs displayed low current amplitudes, with maximal currents around 30 nA, precluding their use for systematic functional and pharmacological investigation.
On the effect of SNP mutant D398N on nAChR signaling
We found no significant effect of the D398N mutation on the concatemeric function, a feature also reported by other groups [11, 38] and which contrasts with its strong effect in the human population, as well as in animal models. We thus speculate that D398N might rather alter other features of the receptor, such as its biosynthesis, cellular trafficking, plasma membrane diffusion and/or internalization processes. In this line, it is noteworthy that the mutation is located in the ICD, which is involved in interaction with cytoplasmic proteins and allosteric regulation by phosphorylation. When looking at the recent cryo-EM structure of the α3β4 nAChR [42] for which part of the ICD is solved, the residues of α3 and β4 that are homologous to α5Asp398 have their side chain accessible to the solvent. This position could thus even be directly involved in interaction with cytoplasmic proteins. Unfortunately, those processes cannot be studied in recombinant systems, especially Xenopus oocytes, as they are far from recapitulating the neuronal conditions.
Our MTSEA assay suggests that α5 incorporation into α4β2 receptors is rather inefficient, since even a large excess of this subunit cDNA does not drive quantitatively the assembly to α5-containing receptors. It is thus likely that α5 displays a low propensity of assembly with the α4 and β2 subunits in the course of maturation within the endoplasmic reticulum, an observation that is in line with the smaller currents recorded on α5-containing concatemers. This contrasts with the case of α3β4 receptors, where α5 is shown to efficiently integrate into pentamers, and for which pentameric concatemers containing the α5 subunit at the fifth position display stronger currents than α5-less concatemers [11].
The α5 subunit contribution to ligand binding
Our data support that the principal face of α5 does not contribute to ACh binding, since the 3(β2) and α5-containing concatemers are identical in terms of ACh dose–response curve, which is not altered when the canonical Trp from the α5 principal site is mutated. This idea is fully consistent with a previous work by Marotta et al. [40], where mutation of loops A, B and C of α5 did not alter the ACh dose–response curves as well. In contrast, Jain et al. studied α4β2 concatemeric dimers co-expressed with loose α5 subunits [28]. Their data suggest that ACh does bind to the α5–α4 and/or α5–β2 interfaces, yet with a lower affinity to that of α4–β2 interface, yielding biphasic ACh dose response curves. Their data also suggest that Saz-A is a partial agonist of α5-nAChRs yielding an efficacy of approximately 30% relative to Ach [28]. Herein, we did not replicate the Saz-A effect, since applying it on α5first elicited robust currents matching ACh-evoked currents (95.9 ± 6%), pointing to a full agonistic activity. We provisionally propose that such experimental discrepancies might be due to the use of dimeric concatemers, which do not strictly exclude the contribution of nAChRs devoid of α5 subunits, notably the α43β22 combination, to the whole-cell currents. Indeed, the MTSEA inhibition of our α5-containing concatemers, which we also observed with MTSET and which we extended to α5-containing nAChRs assembled from free subunits, hampers the use of MTS compounds for SCAM studies when using human α5-containing nAChRs. The absence of MTSET inhibition at human α5-containing nAChRs in the Jain et al. study might thus reflect a substantial contribution of the α43β22 combination.
Interestingly, we found that sazetidine displays different dose response curves on α5-containing concatemers as compared to the 3(β2). Our data indirectly suggest that it may bind to the α4(+)–α5(−) site with a lower affinity than to the classical α4(+)–β2(−) site. The idea that the α4(+)–α5(−) site contributes to ACh binding was already documented [19], but we suggest here that sazetidine displays significant selectivity for α4(+)–β2(−) site, opening for further pharmacological investigation.
In light of previous difficulties in studying α5-containing nAChRs, we also chose to reinvestigate their pharmacology, focusing on ligands that have been proposed to show some selectivity towards these receptors, such as galantamine and progesterone. While the former has no effect on any of our concatemers, the latter appears to be a non-selective inhibitor, modulating α4β2 and α4β2α5 nAChRs with similar potencies.
In conclusion, our concatemeric designs, which force the incorporation of the α5 subunit, offer a well-characterized platform to screen for allosteric modulators of α5-containing nAChRs, and decipher their mechanisms of action. These concatemers will also allow for a precise dissection of the gating mechanisms, since they enable to introduce mutations one subunit at a time in the entire pentamer to assess the interactions between subunits, as well as the long-range allosteric communication between distant binding sites, similarly to previous work on GABAA receptors [23, 43, 44].
Abbreviations
- ACh:
-
Acetylcholine
- GABA:
-
γ-Amino butyric acid
- GFP:
-
Green fluorescent protein
- MTS:
-
Methanethiosulfonate
- nAChR:
-
Nicotinic acetylcholine receptor
- SCAM:
-
Substituted cysteine accessibility method
References
Wen L, Jiang K, Yuan W et al (2014) Contribution of variants in CHRNA5/A3/B4 gene cluster on chromosome 15 to tobacco smoking: from genetic association to mechanism. Mol Neurobiol 53:472–484. https://doi.org/10.1124/jpet.107.132977
Salas R, Orr-Urtreger A, Broide RS et al (2003) The nicotinic acetylcholine receptor subunit alpha 5 mediates short-term effects of nicotine in vivo. Mol Pharmacol 63:1059–1066. https://doi.org/10.1124/mol.63.5.1059
Morel C, Fattore L, Pons S et al (2014) Nicotine consumption is regulated by a human polymorphism in dopamine neurons. Mol Psychiatry 19:930–936. https://doi.org/10.1038/mp.2013.158
Fowler CD, Lu Q, Johnson PM et al (2011) Habenular α5 nicotinic receptor subunit signalling controls nicotine intake. Nature 471:597–601. https://doi.org/10.1038/nature09797
Dawson A, Wolstenholme JT, Roni MA et al (2018) Knockout of alpha 5 nicotinic acetylcholine receptors subunit alters ethanol-mediated behavioral effects and reward in mice. Neuropharmacology 138:341–348. https://doi.org/10.1016/j.neuropharm.2018.06.031
Besson M, Guiducci S, Granon S et al (2016) Alterations in alpha5* nicotinic acetylcholine receptors result in midbrain- and hippocampus-dependent behavioural and neural impairments. Psychopharmacology. https://doi.org/10.1007/s00213-016-4362-2
Forget B, Scholze P, Langa F et al (2018) A human polymorphism in CHRNA5 is linked to relapse to nicotine seeking in transgenic rats. Curr Biol 28:3244–3253.e7. https://doi.org/10.1016/j.cub.2018.08.044
Besson M, Forget BXT, Correia C et al (2019) Profound alteration in reward processing due to a human polymorphism in. Neuropsychopharmacology. https://doi.org/10.1038/s41386-019-0462-0
Exley R, McIntosh JM, Marks MJ et al (2012) Striatal 5 nicotinic receptor subunit regulates dopamine transmission in dorsal striatum. J Neurosci 32:2352–2356. https://doi.org/10.1523/JNEUROSCI.4985-11.2012
Grady SR, Salminen O, McIntosh JM et al (2009) Mouse striatal dopamine nerve terminals express α4α5β2 and two stoichiometric forms of α4β2*-nicotinic acetylcholine receptors. J Mol Neurosci 40:91–95. https://doi.org/10.1124/mol.54.6.1124
George AA, Lucero LM, Damaj MI et al (2012) Function of human α3β4α5 nicotinic acetylcholine receptors is reduced by the α5(D398N) variant. J Biol Chem 287:25151–25162. https://doi.org/10.1124/mol.63.5.1059
Mazzaferro S, Benallegue N, Carbone A et al (2011) Additional acetylcholine (ACh) binding site at alpha4/alpha4 interface of (alpha4beta2)2alpha4 nicotinic receptor influences agonist sensitivity. J Biol Chem 286:31043–31054. https://doi.org/10.1074/jbc.M111.262014
Ahring PK, Liao VWY, Balle T (2018) Concatenated nicotinic acetylcholine receptors: a gift or a curse? J Gen Physiol 150:453–473. https://doi.org/10.1124/mol.54.6.1124
Groot-Kormelink PJ, Broadbent S, Beato M, Sivilotti LG (2006) Constraining the expression of nicotinic acetylcholine receptors by using pentameric constructs. Mol Pharmacol 69:558–563. https://doi.org/10.1124/mol.105.019356
Carbone A-L, Moroni M, Groot-Kormelink P-J, Bermudez I (2009) Pentameric concatenated (alpha4)(2)(beta2)(3) and (alpha4)(3)(beta2)(2) nicotinic acetylcholine receptors: subunit arrangement determines functional expression. Br J Pharmacol 156:970–981. https://doi.org/10.1111/j.1476-5381.2008.00104.x
Mazzaferro S, Benallegue N, Carbone A et al (2011) Additional acetylcholine (ACh) binding site at α4/α4 interface of (α4β2) 2α4 nicotinic receptor influences agonist sensitivity. J Biol Chem 286:31043–31054. https://doi.org/10.1523/JNEUROSCI.0627-09.2009
Benallegue N, Mazzaferro S, Alcaino C, Bermudez I (2013) The additional ACh binding site at the α4(+)/α4(−) interface of the (α4β2) 2α4 nicotinic ACh receptor contributes to desensitization. Br J Pharmacol 170:304–316. https://doi.org/10.1074/jbc.M111.221754
Eaton JB, Lucero LM, Stratton H et al (2013) The unique α4(+)/(−) α4 agonist binding site in (α4)3(β2)2 subtype nicotinic acetylcholine receptors permits differential agonist desensitization pharmacology versus the (α4)2(β2)3 subtype. J Pharmacol Exp Ther 348:46–58. https://doi.org/10.1124/mol.54.6.1124
Jin X, Bermudez I, Steinbach JH (2014) The nicotinic α5 subunit can replace either an acetylcholine-binding or nonbinding subunit in the α4β2* neuronal nicotinic receptor. Mol Pharmacol 85:11–17. https://doi.org/10.1124/mol.113.089979
Lucero LM, Weltzin MM, Eaton JB et al (2016) Differential α4(+)/(−)β2 agonist-binding site contributions to α4β2 nicotinic acetylcholine receptor function within and between isoforms. J Biol Chem 291:2444–2459. https://doi.org/10.1124/mol.115.098061
George AA, Bloy A, Miwa JM et al (2016) Isoform-specific mechanisms of a3b4*-nicotinic acetylcholine receptor modulation by the prototoxin lynx1. FASEB J 31:1398–1420. https://doi.org/10.1124/mol.115.098061
Mazzaferro S, Bermudez I, Sine SM (2018) Potentiation of a neuronal nicotinic receptor via pseudo-agonist site. Cell Mol Life Sci. https://doi.org/10.1007/s00018-018-2993-7
Gielen MC, Lumb MJ, Smart TG (2012) Benzodiazepines modulate GABAA receptors by regulating the preactivation step after GABA binding. J Neurosci 32:5707–5715. https://doi.org/10.1523/JNEUROSCI.5663-11.2012
Nashmi R, Dickinson ME, McKinney S et al (2003) Assembly of alpha4beta2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J Neurosci 23:11554–11567
Morales-Perez CL, Noviello CM, Hibbs RE (2016) Manipulation of subunit stoichiometry in heteromeric membrane proteins. Structure 24:797–805. https://doi.org/10.1016/j.str.2016.03.004
Hamouda AK, Deba F, Wang Z-J, Cohen JB (2016) Photolabeling a nicotinic acetylcholine receptor (nAChR) with an ( α4) 3( β2) 2nAChR-selective positive allosteric modulator. Mol Pharmacol 89:575–584. https://doi.org/10.1124/mol.54.6.1124
Olsen JA, Ahring PK, Kastrup JS et al (2014) Structural and functional studies of the modulator NS9283 reveal agonist-like mechanism of action at α4β2 nicotinic acetylcholine receptors. J Biol Chem 289:24911–24921. https://doi.org/10.1074/jbc.M114.568097
Jain A, Kuryatov A, Wang J et al (2016) Unorthodox acetylcholine binding sites formed by α5 and β3 accessory subunits in α4β2* nicotinic acetylcholine receptors. J Biol Chem 291:23452–23463. https://doi.org/10.1016/j.ymeth.2010.01.012
Xiu X, Puskar NL, Shanata JAP et al (2009) Nicotine binding to brain receptors requires a strong cation. Nature 458:534–537. https://doi.org/10.1038/nature07768
Zwart R, Carbone AL, Moroni M et al (2008) Sazetidine-A is a potent and selective agonist at native and recombinant α4β2 nicotinic acetylcholine receptors. Mol Pharmacol 73:1838–1843. https://doi.org/10.1124/mol.54.6.1124
Mazzaferro S, Gasparri F, New K et al (2014) Non-equivalent ligand selectivity of agonist sites in (α4β2) 2α4 nicotinic acetylcholine receptors. J Biol Chem 289:21795–21806. https://doi.org/10.1113/jphysiol.1989.sp017717
Samochocki M, Höffle A, Fehrenbacher A et al (2003) Galantamine is an allosterically potentiating ligand of neuronal nicotinic but not of muscarinic acetylcholine receptors. J Pharmacol Exp Ther 305:1024–1036. https://doi.org/10.1046/j.1471-4159.2000.0752492.x
Kuryatov A, Onksen J, Lindstrom J (2008) Roles of accessory subunits in α4β2* nicotinic receptors. Mol Pharmacol 74:132–143. https://doi.org/10.1074/jbc.273.44.28721
Kowal NM, Ahring PK, Liao VWY et al (2017) Galantamine is not a positive allosteric modulator of human α4β2 or α7 nicotinic acetylcholine receptors. Br J Pharmacol 175:2911–2925. https://doi.org/10.1124/mol.54.6.1124
Valera S, Ballivet M, Bertrand D (1992) Progesterone modulates a neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci USA 89:9949–9953. https://doi.org/10.1073/pnas.89.20.9949
Kuryatov A, Berrettini W, Lindstrom J (2011) Acetylcholine receptor (AChR) α5 subunit variant associated with risk for nicotine dependence and lung cancer reduces (α4β2)2α5 AChR function. Mol Pharmacol 79:119–125. https://doi.org/10.1124/mol.110.066357
Tammimäki A, Herder P, Li P et al (2012) Impact of human D398N single nucleotide polymorphism on intracellular calcium response mediated by α3β4α5 nicotinic acetylcholine receptors. Neuropharmacology 63:1002–1011. https://doi.org/10.1016/j.neuropharm.2012.07.022
Li P, McCollum M, Bracamontes J et al (2011) Functional characterization of the α5(Asn398) variant associated with risk for nicotine dependence in the α3β4α5 nicotinic receptor. Mol Pharmacol 80:818–827. https://doi.org/10.1124/mol.111.073841
Ramirez-Latorre J, Yu CR, Qu X et al (1996) Functional contributions of alpha5 subunit to neuronal acetylcholine receptor channels. Nature 380:347–351. https://doi.org/10.1038/380347a0
Marotta CB, Dilworth CN, Lester HA, Dougherty DA (2014) Neuropharmacology. Neuropharmacology 77:342–349. https://doi.org/10.1016/j.neuropharm.2013.09.028
Hannan S, Smart TG (2018) Cell surface expression of homomeric GABA A receptors depends on single residues in subunit transmembrane domains. J Biol Chem 293:13427–13439. https://doi.org/10.1002/prot.22488
Gharpure A, Teng J, Zhuang Y et al (2019) Agonist selectivity and ion permeation in the α3β4 ganglionic nicotinic receptor. Neuron 104:501–511.e6. https://doi.org/10.1016/j.neuron.2019.07.030
Szabo A, Nourmahnad A, Halpin E, Forman SA (2019) Monod–Wyman–Changeux allosteric shift analysis in mutant α1 β3 γ2L GABA A receptors indicates selectivity and crosstalk among intersubunit transmembrane anesthetic sites. Mol Pharmacol 95:408–417. https://doi.org/10.1038/81800
Absalom NL, Ahring PK, Liao VW et al (2019) Functional genomics of epilepsy-associated mutations in the GABA A receptor subunits reveal that one mutation impairs function and two are catastrophic. J Biol Chem 294:6157–6171. https://doi.org/10.1371/journal.pone.0141359
Acknowledgements
This work was founded by the ERC Grant (No. 788974, Dynacotine), the ANR Grant (No. 17-CE11-0030 Nicofive), the “équipe FRM” (Fondation pour la Recherche Médicale) grant DEQ20140329497, a fellowship from the Fondation ARC to MSP and a European Commission Research Executive Agency individual fellowship to MG (Marie Sklodowska-Curie Action, Individual Fellowship 659371). The authors would like to thank Uwe Maskos, Stéphanie Pons, Morgane Besson, Akos Nemecz and Alexandre Mourot for critical reading of the manuscript.
Author information
Authors and Affiliations
Contributions
MSP, MG and P-JC designed the study. MSP, HB, NB and MG performed and analyzed the experiments. MSP, MG and P-JC wrote the manuscript with inputs of all authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Prevost, M.S., Bouchenaki, H., Barilone, N. et al. Concatemers to re-investigate the role of α5 in α4β2 nicotinic receptors. Cell. Mol. Life Sci. 78, 1051–1064 (2021). https://doi.org/10.1007/s00018-020-03558-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03558-z