
Abstract
Alzheimer’s Disease (AD) is the sixth-leading cause of death in industrialized countries. Neurotoxic amyloid-β (Aβ) plaques are one of the pathological hallmarks in AD patient brains. Aβ accumulates in the brain upon sequential, proteolytic processing of the amyloid precursor protein (APP) by β- and γ-secretases. However, so far disease-modifying drugs targeting β- and γ-secretase pathways seeking a decrease in the production of toxic Aβ peptides have failed in clinics. It has been demonstrated that the metalloproteinase meprin β acts as an alternative β-secretase, capable of generating truncated Aβ2–x peptides that have been described to be increased in AD patients. This indicates an important β-site cleaving enzyme 1 (BACE-1)-independent contribution of the metalloprotease meprin β within the amyloidogenic pathway and may lead to novel drug targeting avenues. However, meprin β itself is embedded in a complex regulatory network. Remarkably, the anti-amyloidogenic α-secretase a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) is a direct competitor for APP at the cell surface, but also a sheddase of inactive pro-meprin β. Overall, we highlight the current cellular, molecular and structural understanding of meprin β as alternative β-secretase within the complex protease web, regulating APP processing in health and disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The metalloprotease meprin β in health and disease
Meprin β is a membrane-bound multi-domain metalloenzyme (Fig. 1) and exhibits a unique cleavage specificity amongst all extracellular proteases [1]. The protease belongs to the metzincin superfamily characterized by the typical zinc-binding motif HExxHxxGxxH/D and a so-called Met-turn, the latter containing a tyrosine residue that functions as zinc-ligand [2]. Based on structural data it became obvious that all members of the metzincin superfamily, including matrix metalloproteases (MMPs), ADAMs and astacins, share a common fold of the catalytic domain, but exhibit unique features within the active site cleft [3]. Meprin β belongs to the astacin family of metalloproteases and in mammals is closely related to meprin α, bone morphogenetic protein 1 (BMP-1), mammalian tolloid (mTld), tolloid-like 1 and 2 (tll-1/2), and ovastacin [4]. Although all astacin members exhibit a cleavage preference for aspartate and glutamate in P1ʹ, only meprin β is capable of hydrolyzing completely acidic peptides [1, 5]. Several biologically important substrates have been identified, which links meprin β activity to inflammation, connective tissue homeostasis and neurodegeneration [6, 7].

Domain composition and dimeric structure of the metalloprotease meprin β. Cartoon representation of dimeric meprin β domain model (left) and a membrane-bound ribbon structure model (right) based on the crystal structure of the ectodomain of human meprin β (PDB: 4GWN) Pro propeptide, CAT catalytic domain, MAM meprin A5 protein tyrosine phosphatase μ domain, TRAF tumour-necrosis-factor-receptor-associated factor domain, EGF epidermal growth factor-like domain, TM transmembrane region, C C-terminal part. The disulfide bridge between the MAM domains responsible for dimerization is indicated as yellow bar
Employing different mouse models of acute and chronic inflammation meprin β was found to be a rather pro-inflammatory enzyme [6]. The pro-inflammatory trans-signaling of interleukin 6 (IL-6) can be induced by meprin β through the shedding of the IL-6 receptor from granulocytes then acting on other cells by binding to its β-receptor gp130 and inducing signal transducer and activator of transcription 3 (STAT3) phosphorylation [8]. However, in a dextran sulfate sodium (DSS)-induced colitis model, IL-6 levels were increased in Mep1a/b−/− mice compared to wild-type animals [9]. This is further supported by a study showing that meprin β can directly cleave and inactivate IL-6 [10]. Several studies demonstrated that meprin β contributes to the onset and progression of nephritis and acute kidney failure [11, 12]. Interestingly, nanoparticle-based application of meprin β specific siRNA showed a clear benefit in a corresponding mouse model [13]. Another aspect of meprin β’s pro-inflammatory activity is reflected by its capacity to promote transendothelial migration (TEM) of immune cells [14, 15]. Here, the cell adhesion molecule CD99 is a possible substrate candidate, as its cleavage by meprin β induces TEM and cell proliferation in vitro and in vivo. Meprin β was also described to promote cell migration through cleavage of extracellular matrix proteins, such as fibronectin or nidogen [16]. However, other studies highlighted a rather opposite function, because meprin β was identified as a procollagen proteinase, cleaving off the N- and C-terminal pro-domains of collagen I + III, thereby inducing collagen fibril assembly [17, 18]. This is further supported by the observations that increased meprin β expression is associated with skin and lung fibrosis [18].
In the small intestine, meprin β is essential for the detachment of the mucus by cleaving mucine 2 (MUC2) [19]. This is crucial for the functionality of the mucus barrier to impede bacterial overgrowth and infection [20]. Interestingly, the pathogenic protease gingipain R (RgpB) from Porphyromonas gingivalis is able to cleave meprin β thereby preventing mucus detachment [21]. Of note, cleavage by RgpB leads to activation of membrane-bound meprin β, which precludes its shedding by ADAM proteases. Solubilization of meprin β is a prerequisite for the protease to get access to the cleavage site in MUC2 [19].
Activation and shedding of meprin β are mutually exclusive events [21]. Besides RgpB, this was also demonstrated using matriptase-2 (MT-2) as a potent activator of membrane-bound meprin β [22]. The molecular mechanism why cleavage of the pro-peptide completely blocks shedding by ADAM10/17 is still ambiguous. However, it demonstrates how strict meprin β activity and localization is regulated. This is of utmost importance for the β-secretase function of meprin β in the processing of the amyloid precursor protein (APP), which is involved in the generation of Alzheimer’s disease (AD) [23]. Hence, understanding the interplay of the α-secretase ADAM10 with the alternative β-secretase meprin β on molecular, cellular and disease level will help to further elucidate decisive steps in the onset and progression of AD.
β-secretases: BACE and alternatives
Neurotoxic amyloid-β (Aβ) plaques are one hallmark of AD [24, 25]. Aβ deposits in the brain are composed of peptides derived from APP and consist of up to 42 amino acids. Several publications support different molecular mechanisms for Aβ mediated synaptic dysfunction and neuronal cell death, such as membrane disruption, ion dysregulation or oxidative stress induction [26,27,28,29]. However, the Aβ biology is rather complex, mainly due to its great hydrophobic interacting potential [30]. Thus, the entire and exact role of Aβ remains elusive. Aβ peptides derive from APP by sequential cleavage at the β- and γ-secretase cleavage sites [31, 32]. Intramembranous γ-cleavage is accomplished by the aspartic peptidases presenilin 1 and 2 (PS1/2) within the γ-secretase complex at position 40 or 42 (numbering according to Aβ sequence) [32]. Aβ1–40 is the major species, whereas Aβ1–42 levels are low in healthy brains, however, strongly increase during the progression of AD [33]. Of note, conditional PS1/2 double knock-out mice exhibit significantly reduced Aβ levels [34]. Further, more than one hundred PS1 related mutations were identified that lead to increased Aβ levels suggesting PS1 as a susceptibility gene for AD [35]. However, β-secretase cleavage is rate limiting for the Aβ formation [36, 37]. The first identified β-secretase is the aspartic protease β-site cleaving enzyme 1 (BACE-1). It is predominantly expressed in acidic compartments and exhibits a low pH optimum. Thus, BACE-1 dependent APP cleavage occurs in endosomal/lysosomal compartments [38]. The major cleavage event by BACE-1 at the β-site of APP is at position 1 resulting in the dominant Aβ species Aβ1–40/42 [39]. Since the Aβ formation in BACE-1 knock-out mice is strongly reduced, BACE-1 is thought to be the major β-secretase [40]. Thus, BACE-1 is one of the most promising therapeutic targets for AD treatment. However, all clinical trials using specific BACE-1 inhibitors have failed so far and have not shown any cognitive benefits for the patients (https://www.alzforum.org/news/conference-coverage/bump-road-or-disaster-bace-inhibitors-worsen-cognition; 02.11.2018). Therefore, the investigation of alternative β-secretases as potential drug targets is of great interest.
Besides the BACE-1-generated Aβ1–x species N-terminal truncated Aβ forms came into focus of research. Already many years ago, Konrad Beyreuther and Colin Masters described N-terminal truncated Aβ peptides in the core of amyloid plaques of AD patients [41]. Other groups have shown an increase of Aβ peptides starting at position 2 (Aβ2–x) in the brains of AD patients compared to other dementias or non-demented subjects [42]. A number of N-terminally truncated Aβ variants starting at different other positions have been reported in the cerebrospinal fluid (CSF), blood and brain tissue of AD patients [42,43,44]. Since BACE-1 is incapable to generate such peptides the hunt for these enzymes was evident. For instance, cathepsin B, S and L as lysosomal proteases are discussed as alternative β-secretases generating various Aβ species [45,46,47]. Inhibitor studies in cells and mice indicate a direct involvement of these proteinases in Aβ generation [45]. Of note, cathepsin B is thought to be involved in Aβ3–x formation, which is the progenitor of highly neurotoxic pyroglutamate-modified Aβ3–x [46]. However, cathepsin D is involved in the clearance of Aβ [48]. The detailed role of cathepsins in this context is not revealed so far, however, their low cleavage specificity on APP suggests a Aβ degrading role [49, 50]. Other candidates generating N-terminally truncated Aβ peptides are the Aminopeptidases A and N (APA/APN). APA generates Aβ2–x from Aβ1–x, whereas APN is thought to convert Aβ2–x to Aβ3–x [51, 52]. A promising alternative β-secretase that directly cleaves at p2 within full-length APP is the metalloprotease meprin β [23]. We and others could show that mRNA and protein levels of meprin β are significantly increased in AD brain [23, 53], which is in line with increased Aβ2–42 [42]. Of note, Aβ2–40 peptides not only exhibit a greater aggregation potential than Aβ1–40 but additionally induce Aβ1–40 aggregation [54]. Mass spectrometry-based degradomics identified APP as a substrate for meprin β at three different sites in the N-terminal region between Ser124/Asp125, Glu380/Thr381, and Gly383/Asp384 [55]. After incubation with all APP isoforms and meprin β, two fragments of 20 and 11 kDa were identified either in vitro or in cell culture-based assays (Fig. 2). Interestingly, these fragments derived from APP processing were found in human and wild-type mice brain lysates, but not in the brain of Mep1b−/− mice [55], proving APP as a physiological target of meprin β. The major cleavage site of meprin β in APP695 is between Asp597 and Ala598 resulting in the formation of Aβ2–x, and to a minor extend between Met596 and Asp597 at the BACE-1 site [7, 23]. Meprin β knock-out mice brains show increased sAPPα levels which could indicate that the absence of meprin β leads to more available substrate to α-secretase. It has been shown that meprin β and APP co-localize at the cell surface and in the secretory pathway leading to APP processing by meprin β in these cellular compartments [54]. Therefore, meprin β may compete at the cell surface with ADAM10, the main α-secretase in the brain [56]. A recent publication indicates that meprin β may also act as dipeptidyl-peptidase being able to convert Aβ1–x to Aβ3–x, which is the progenitor of highly neurotoxic pyroglutamate-modified Aβ3–x [53]. However, this observation is based on in vitro cleavage using truncated Aβ-peptides. Hence, further cell-based assays are necessary to validate these findings.

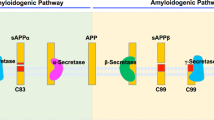
APP processing by BACE-1 and meprin β in the canonical and non-canonical pathway. BACE-1 and meprin β are shown to be involved in the generation of Aβ peptides in the canonical and non-canonical pathway. The so far well-described APP processing pathways by α-, β- and δ-secretases are described in green, orange and blue. Meprin β is involved in the generation of two APP N-terminal fragments of 20 and 11 kDa as well as the cleavage of APP at the β-secretase cleavage site (purple), providing a substrate for γ-secretase releasing Aβ peptides (red)
The role of meprin β-generated Aβ2–x has yet not been investigated in the common AD mouse models such as 5xFAD or tg2576. These and other mouse models contain the Swedish APP (APPswe) variant bearing two point mutations (KM595/N596L) N-terminal to the β-secretase cleavage site. These mutations originate from a rare genetic APP variant identified in two Swedish families [57, 58]. APPswe mutations are connected with an early loss of memory and dramatically increased the accumulation of Aβ1–x generated by BACE-1 [59, 60]. Of note, BACE-1 deficiency recovers the loss of memory and Aβ1–x accumulation [60]. However, the β-site of APPswe is not cleaved at position 2 by meprin β, resulting in complete reduction of N-terminal truncated Aβ peptides starting at p2 in mouse models carrying the APPswe mutation [7, 54]. This leads to the assumption that alterations of the amino acid composition close to the β-secretase cleavage site may inhibit meprin β activity on the generation of N-terminal truncated Aβ peptides. This could be further validated since the “protective” A673T mutation in APP, which results in reduced Aβ levels in patients, also prevents from meprin β cleavage at position p2 [54]. The cleavage sites of BACE-1 and meprin β on APPwt and APPswe are shown in Fig. 3.

Comparison of APPwt and APPswe processing by BACE-1 and meprin β. The local APP peptide sequence around the Aβ region (red boxes) is depicted. The Swedish APP mutation is highlighted by purple letters. The upper panel shows BACE-1-mediated and the lower panel meprin β-mediated APP processing at the β-site. The arrows indicate cleavage sites of BACE-1 or meprin β on APPwt in comparison to APPswe
Thus, it is not possible to address the biological relevance of meprin β-generated Aβ2–x in APPswe based mouse models. With regard to continuously failing BACE-1 inhibitor clinical trials, AD mouse models considering truncated Aβ species in an APP wild-type background are essential to promote AD research. A potential role of meprin β expression in AD may be reflected in genetic studies. Quantitative comparison of meprin β expression revealed significantly higher mRNA levels in brain tissue from AD patients versus controls [23]. Recently, Schlenzig and colleagues also detected a fivefold increase in expression levels of meprin β in postmortem tissue samples from AD patients compared to age-matched controls [53]. In addition, a more detailed histological analysis showed a prominent meprin β immunoreactivity in tissue sections from AD cases compared to controls. Interestingly, this work detected a noticeable immunoreactivity for meprin β in glial cells, more precisely in astrocytes of AD patients [53].
Using brain tissue from the Brains for Dementia Research (BDR) cohort, which was specifically created to address the shortages of high-quality brain tissue samples from healthy individuals as well as those with dementia, Patel and colleagues performed single variant and burden analysis on coding variants to identify significant associations with late-onset AD (LOAD). Using next-generation DNA sequencing (NGS) a synonymous mutation in MEP1B, the gene coding for meprin β, was identified to have greater frequency in AD cases than controls [61]. Although the sample size of this cohort is rather small and needs further validation, the result may provide more evidence to imply meprin β is in close association with AD.
β-secretase cleavage of APP is not only determined by the site preferences of the different enzymes. Cellular localization of APP/protease-interaction is also an important issue. BACE-1-dependent Aβ release occurs intracellularly due to its activity in late endosomes/lysosomes, whereas meprin β-dependent Aβ generation takes place at the cell surface or even in the late secretory pathway [54]. The α-secretase ADAM10 exhibits dual anti-amyloidogenic activity by cleaving APP within the Aβ-peptide sequence and by ectodomain shedding of meprin β thereby destroying its β-secretase activity [23, 62]. Hence, in contrast to BACE-1, ADAM10 and meprin β are direct competitors for the substrate APP at the cell surface.
Shedding of meprin β
The activity of meprin β is strictly regulated within the protease web (Fig. 4) [1, 21, 22]. During the secretory pathway, meprin β gets highly glycosylated and reaches the cell surface as zymogen. For maturation, the pro-peptide of soluble meprin β can be cleaved off by tryptic serine proteases, such as trypsin or human kallikrein-related peptidases (KLKs) [1, 63]. The membrane-tethered serine protease matriptase-2 (MT-2) has been identified as a potent activator of membrane-bound meprin β [22]. ADAM10 and ADAM17 were identified as sheddases of pro-meprin β, which is then maturated by soluble tryptic proteases [21, 63,64,65]. Importantly, shedding of meprin β by ADAM10/17 is completely abolished upon its activation by MT-2 at the cell surface [21]. The molecular mechanism why only the pro-form of meprin β can be shed is not understood.

Extracellular regulation of meprin β activity with respect to β-site cleavage of APP. Meprin β is expressed as zymogen at the cell surface. A disintegrin and metalloprotease 10 and 17 (ADAM10/17) act as sheddases of pro-meprin β. Shed meprin β can be activated by tryptic proteases. When activated as soluble protein, shed meprin β does not cleave at the β-site of APP. Alternatively, inactive meprin β can be maturated by the membrane-bound serine protease matriptase-2 (MT-2). Once activated at the cell surface membrane-bound meprin β cannot be shed any more and acts as β-secretase thus generating amyloid-β (Aβ)
Interestingly, there are subtle differences between the substrate pools of membrane-bound and soluble meprin β. The adhesion molecule CD99, essential for the transendothelial migration of leukocytes, is for example cleaved by both meprin β entities [14, 15]. For the meprin β substrates collagen-1 or the cytokines IL-6 and IL-18 it is still elusive if the membrane-bound and/or soluble form is involved in cleavage and requires further investigations [10, 17, 66]. In contrast, the receptor of the pro-inflammatory cytokine IL-6 is shed by membrane-bound meprin β only, and not by the soluble form [8]. As mentioned above, a specific substrate that is only cleaved by soluble meprin β is mucin-2 [19]. This cleavage event can be abrogated by the pathogenic protease RgpB that potently converts membrane-bound pro-meprin β into its active form thereby preventing its shedding by ADAM10/17 [21].
In terms of different substrate pools of soluble and membrane-bound meprin β, APP exhibits quite unique properties. Both meprin β forms were identified to cleave APP within its N-terminal region. Certain N-terminal APP fragments (N-APP) were discussed as neurotoxic factors through binding to the neuronal death receptor DR6 and inducing caspase-mediated cell death [67, 68]. However, meprin β generated N-APP fragments do not show neurotoxic properties at all [55]. A much more valid correlation of APP and neurodegeneration is based on the amyloid hypothesis. Sequential proteolytic cleavage of APP by β- and γ-secretase leads to the generation of aggregation-prone Aβ-peptides found in neurotoxic plaques in AD brains. We identified meprin β as an alternative β-secretase predominantly generating Aβ2–x peptides [7, 23]. This cleavage event requires membrane-bound meprin β and is prevented for the soluble shed protease. Thus, the meprin β sheddases ADAM10 and 17 may exhibit a dual function in preventing from amyloidation in AD. On the one hand, ADAM10 acts as α-secretase cleaving APP within the Aβ sequence and, thus, counteract against Aβ formation [62, 69]. On the other hand, ADAM10/17 prevent from amyloidation by shedding the β-secretase meprin β from the cell surface. However, whether ADAM proteases prefer APP over meprin β as shedding substrate or vice versa is completely unknown. Of note, meprin β itself was identified as an inducer of ADAM10 activity [64]. This finding complicates the protease network around ADAM10/17-mediated prevention of Aβ generation. Thus, further research on the exact mechanism of the dual protective role of ADAM 10 and 17 is required.
Structural properties of APP, ADAM10 and meprin β
As outlined above, the processing of APP is embedded in a quite complex network of different proteolytic checkpoints. In the last years, many investigations contributed to a better biochemical and cellular understanding of these complex regulatory mechanisms. Simultaneously, the results made apparent that a comprehensive knowledge of the molecular basis of APP processing is still missing. This is likewise reflected by the failure rate in developing successful inhibitory strategies for the treatment of AD. One essential bottleneck in that context is the so far only partly available structural information on APP itself and its processing proteases.
Structural information on APP is still enigmatic
APP is a single-span type-I multi-domain membrane protein belonging to the small gene family of APP-like proteins including also the amyloid precursor-like protein (APLP) 1 and 2. Overall, these three proteins and the existing isoforms share a highly similar domain organization and proteolytic processing, while only APP contains the Aβ-peptide sequence critical in the pathogenesis of AD [70,71,72,73,74,75]. APP proteins consist of three highly conserved regions: the extracellular E1 and E2 domains separated via a single transmembrane helix from the rather small C-terminal APP intracellular domain (AICD) (Fig. 5a). The E1 is composed of an N-terminal cysteine-rich growth factor-like subdomain (GFLD) with heparin binding properties (HBD) joined by a short linker with a zinc and copper-binding subdomain (CuBD) [76,77,78,79]. A structural flexible acidic domain (AcD), constituting of nearly 50% glutamate and aspartate residues, connects the E1 domain with the E2 domain in the neuronal isoform APP695 and ALPL1. The APP751 isoform consists of an additional Kunitz-type serine protease inhibitor domain (KPI) N-terminal to the E2 domain, which is in APP770 further followed by a 19 amino acid OX-2 domain, homologues to the immunoregulatory OX-2 antigen [80, 81]. The E2 domain, also known as central APP domain (CAPPD), is the largest of the conserved domains containing several substructures with interaction sites for binding partners like a second HBD, the RERMS pentapeptide motif [82,83,84] and a collagen binding domain [85,86,87] as well as two N-glycosylation sites [88, 89]. The natively unstructured juxtamembrane region (JMR) harbours an additional O-glycosylation site as well as the α- and β-secretase cleavage sites relevant for the shedding of APP. It connects the ectodomain with the TM-Helix, containing the γ-secretase cleavage site, which is followed by the intracellular AICD domain.

Extracellular regulation of meprin β activity with respect to β-site cleavage of APP. a Cartoon representation of a membrane-bound meprin β model (blue) based on the crystal structure of the ectodomain (PDB: 4GWN) in complex with part of APP (grey, Aβ peptide in red). Structures of additional N-terminal domains of APP695 are also shown as cartoons: E1 (PDB: 3KTM), E2 (PDB: 3NYL). Sequence stretches of unknown structure and the AICD domain are illustrated as dashed lines. The close up views in the right panel highlight determined meprin β cleavage sites, while P1 and P1ʹ residues are depicted as sticks. b Model of a membrane-bound ADAM10 based on the ectodomain (yellow, PDB: 6BE6) and pro-meprin β (blue). The pro-peptide of meprin β is shown in orange. The red arrow indicates the shedding site within pro-meprin β. E1/2 extracellular domains 1/2, Acd acidic domain, JMR juxtamembrane region, AICD APP intracellular domain
Even though in the last two decades many attempts have been made to structurally characterize APP, a structure with atomic resolution of full-length APP or its entire extracellular domain is still not available. Therefore, the current understanding is based on a set of substructure information. The GFLD and CuBD subdomains of the N-terminal E1 domain were initially described as individual folding units [76,77,78]. Further studies showed that the overall fold of both subdomains in isolation is very similar to their structures within the entire E1 domain, both comprising αβ topologies stabilized by disulfide bridges [79, 90]. The structural rigidity of E1 domain is determined by the interface interaction between GFLD and CuBD in a pH-dependent manner. An acidic pH leads to a tight interaction resulting in a compact structural conformation of the E1 entity [79, 90, 91]. Additionally, alterations of the pH regulate the self-dimerization of the isolated E1 domain as well as one of heparin-induced E1 dimers [79, 91]. However, size exclusion and light scattering experiments demonstrated the impact of the structural flexible linker connecting the E1 and E2 on dimerization of APP in solution [91]. In particular the presence of the acidic stretch AcD interfered with self- and the heparin-induced dimerization of the E1 domain, while the binding affinity for heparin was reduced [91]. Remarkably, for the entire extracellular domain and for the isolated E2 no short-chain heparin-induced dimerization and only a low self-dimerization potential at unphysiological high protein concentrations could be observed [91]. The E2 domain itself is an almost helical structure consisting of six α-helices arranged in two distinct coiled-coil substructures, which share a long continues central helix [88, 92,93,94]. In case of the E2 domain, metal binding correlates with the conformational flexibility, leading to increased rigidity as well as thermostability of the domain [92]. Also an X ray structure of an antiparallel APP E2 dimer was reported [88]. In contrast to this, limited proteolysis experiments and NMR analysis suggest that only parts of this domain are rigidly folded in solution [94]. As a third site of potential dimerization, the TM-helix was postulated. Several studies intended to understand the structure of various truncated transmembrane containing fragments under different solvent conditions [95,96,97,98,99]. The NMR analysis of the entire β-secretase cleavage product C99 offered for the first time a more comprehensive view on the helical nature of the transmembrane helix in presence of N- and C-terminal flanking APP regions [100]. In this structure, the β-cleavage site itself was found to be located within the unstructured juxtamembrane region. In contrast, the α-cleavage site is in close proximity to the outer membrane leaflet at the beginning of a short extracellular so-called N-helix. Further pulsed electron paramagnetic resonance (EPR) double electron–electron resonance (DEER) experiments confirmed a flexible, highly curved nature of the transmembrane helix, which is supposed to be well suited for its interaction with the γ-secretase. Interestingly, a second C-terminal helix, structurally uncoupled from the TM-helix, was shown to be likewise surface-associated. The transmembrane segment of APP features three consecutive glycine zipper motifs known to mediate dimerization in single-pass TM proteins [101,102,103,104]. Indeed, an involvement of these motifs in the dimerization of the transmembrane region with a regulatory impact on Aβ species generation was shown [99, 105,106,107,108]. While NMR studies structurally substantiate these observations by a dimeric transmembrane segment association under micellar conditions [109, 110], a monomeric state upon cholesterol binding was suggested [111]. Following molecular dynamic simulations support a determining effect of lipid composition, membrane thickness and membrane curvature influencing the C99 overall structure and the tendency to dimerize [112,113,114]. Finally, the C-terminal AICD domain that is intracellularly released after γ-secretase cleavage was shown to be intrinsically disordered by nuclear magnetic resonance (NMR) and circular dichroism (CD) experiments [115, 116]. However, it has been shown that this domain can adopt different conformations depending on its interaction partner as documented by the structures in complex with Dab1 and 2 [117], X11α [118] and the phosphotyrosine-binding Fe65-PTB domains [119].
Even though major progress has been made to understand the unique APP on the structural level, it still remains enigmatic to what extent the conformation of the individual subunits and their arrangement within the whole protein influence the processing by α, β or the γ-secretase complex. Especially the different derived models of cis- and trans-dimerization of APP need further investigations to conclusively clarify, how and if all proposed and analysed dimerization regions contribute to a membrane-bound dimer. In addition, it is possible that sAPP molecules interact differently with each other once cleaved by α- or β-secretases.
Structural properties of meprin β
The structure of the ectodomain of the alternative β-secretase meprin β revealed a compact disulfide-bridged dimer [120, 121]. Interestingly, both monomers interact in a nearly symmetric fashion between the catalytic domain of one monomer and the MAM domain of the other. By that, the two catalytic domains are accessible on opposite sites of the dimer and the active site clefts locate close to the plasma membrane (Figs. 1 and 5). Maturation of meprin β requires proteolytic processing of the N-terminal pro-peptide. Comparison of the available zymogen and mature meprin β structures indicates that the zymogen is already in a preformed conformation. Solely one-seventh of the protein is rearranged to gain catalytic activity. Given by the overall rigidity of meprin β, substrate engagement most likely requires structural flexible segments able to follow a “N-like” trajectory over the dimer to enter the active site cleft of one monomer. Overall, this mechanism would be compatible with most type-I transmembrane substrates. This is also nicely in line with the flexible, unstructured juxtamembrane region of APP, which contains the β-cleavage site (Fig. 5a). Taken this into account, it is hard to envision how APP cleavage of an e.g. TM-region associated dimer should be facilitated by meprin β. So far, N-APP cleavage by meprin β was only shown for soluble meprin β [55]. N-APP cleavage sites have been identified in the E1 and E2 domain (Fig. 2) [55]. Interestingly, so far no cleavage within the AcD domain was observed, even though the sequence stretches of alternating glutamate and aspartate residues represent ideal meprin β cleavage sites [5].
The α-secretase ADAM10 is a type-I transmembrane protein
ADAM10 is also a type-I transmembrane protein with a modular domain organization (Fig. 5b). In the extracellular ectodomain, the pro-domain is followed by a metalloprotease, disintegrin and cysteine-rich domain. The recently solved X ray structure of the ectodomain of ADAM10 gives first insights in activity regulatory mechanisms of the protease [122]. The structure adopts a compact fold resembling an arrowhead with the cysteine-rich domain partially occupying the active site (Fig. 5b). This suggests that the cysteine-rich domain has an autoinhibitory function to preclude unrestricted substrate access. Therefore, the authors suggested that a transient opening of ADAM10 is permissive for substrate capture. It is possible that the disintegrin and cysteine-rich region of ADAM10 directly contacts the substrate thereby stabilizing both proteins in their open conformations to promote cleavage. Further investigations are needed to explore this hypothesis in more detail. Nevertheless, deduced from the C99 [100] and the meprin β structure [120] it is very likely that cleavage of both proteins takes place in close proximity to the plasma membrane (Fig. 5b).
Taken together, understanding the structural basis of ADAM10-mediated APP cleavage at the α-site, and its shedding activity toward meprin β as competing β-secretase, would be a breakthrough to decipher homeostasis of APP processing in health and disease.
Conclusions and future directions/perspectives
Late-onset AD, the most common neurodegenerative disorder, is a progressive and to date incurable form of dementia that develops in the elderly population. In brains of AD patients, loss of neurons and synapses occur as a result of the accumulation of Aβ peptides. The aspartyl protease BACE-1 was identified as the major APP-cleaving β-secretase. However, certain AD associated N-terminally truncated Aβ peptides could not be assigned to BACE-1 activity, indicating the presence of additional β-secretases. We demonstrated that the metalloproteinase meprin β is capable of generating N-terminal truncated Aβ2–x peptides that have been described in AD patients [42], which may point to an important and BACE-independent contribution of the metalloprotease meprin β within the amyloidogenic pathway.
We could demonstrate physiological relevance of meprin β mediated APP cleavage, since we observed absence of N-APP fragments and increased endogenous sAPPα levels in the brains of meprin β knock-out mice [54, 55]. Moreover, we could show an interaction of APP and meprin β by coimmunoprecipitation and direct involvement of meprin β activity on the generation of Aβ2-x peptides in vitro. A recent study further supports the relevance of meprin β in AD, where a potential risk gene variant of meprin β (rs173032) for AD has been identified using whole-exome sequencing of the Brains for Dementia Research (BDR) cohort. Increased meprin β mRNA and protein expression specifically in AD cases has been observed previously [23].
Recently, the laboratory of Dennis Selkoe has shown, that meprin β co-fractionates with APP and PS1 in the same high molecular weight fraction in wild type mouse brains and that this fraction is responsible for the majority of Aβ generation [123]. Interestingly, ADAM10 is also present in these high molecular weight fractions. Hence, it is important to investigate how APP cleavage within these microdomains switches between competitive non-amyloidogenic (α-secretase) and amyloidogenic (β-secretase) processing in health and disease. Furthermore, the direct interaction with ADAM10 promotes shedding of meprin β, which in its soluble form loses its β-secretase activity [23]. Identification of regulatory elements responsible for the orchestration of substrates and proteases will be decisive for a better understanding of the molecular basis for AD pathology.
Abbreviations
- AD:
-
Alzheimer’s disease
- APP:
-
Amyloid precursor protein
- Aβ:
-
Amyloid-βADAM; a disintegrin and metalloproteinase domain-containing protein
- BACE-1:
-
β-Site cleaving enzyme 1
- PS1/2:
-
Presenilin 1 and 2
- MT-2:
-
Matriptase-2
References
Broder C, Becker-Pauly C (2013) The metalloproteases meprin alpha and meprin beta: unique enzymes in inflammation, neurodegeneration, cancer and fibrosis. Biochem J 450(2):253–264. https://doi.org/10.1042/BJ20121751
Bode W, Grams F, Reinemer P, Gomis-Ruth FX, Baumann U, McKay DB, Stocker W (1996) The metzincin-superfamily of zinc-peptidases. Adv Exp Med Biol 389:1–11
Gomis-Ruth FX (2003) Structural aspects of the metzincin clan of metalloendopeptidases. Mol Biotechnol 24(2):157–202. https://doi.org/10.1385/MB:24:2:157
Gomis-Ruth FX, Trillo-Muyo S, Stocker W (2012) Functional and structural insights into astacin metallopeptidases. Biol Chem 393(10):1027–1041. https://doi.org/10.1515/hsz-2012-0149
Becker-Pauly C, Barre O, Schilling O, Auf dem Keller U, Ohler A, Broder C, Schutte A, Kappelhoff R, Stocker W, Overall CM (2011) Proteomic analyses reveal an acidic prime side specificity for the astacin metalloprotease family reflected by physiological substrates. Mol Cell Proteomics 10(9):M111009233. https://doi.org/10.1074/mcp.m111.009233
Arnold P, Otte A (1864) Becker-Pauly C (2017) Meprin metalloproteases: molecular regulation and function in inflammation and fibrosis. Biochim Biophys Acta Mol Cell Res 11 Pt B:2096–2104. https://doi.org/10.1016/j.bbamcr.2017.05.011
Becker-Pauly C, Pietrzik CU (2016) The metalloprotease meprin beta is an alternative beta-secretase of APP. Front Mol Neurosci 9:159. https://doi.org/10.3389/fnmol.2016.00159
Arnold P, Boll I, Rothaug M, Schumacher N, Schmidt F, Wichert R, Schneppenheim J, Lokau J, Pickhinke U, Koudelka T, Tholey A, Rabe B, Scheller J, Lucius R, Garbers C, Rose-John S, Becker-Pauly C (2017) Meprin metalloproteases generate biologically active soluble interleukin-6 receptor to induce trans-signaling. Sci Rep 7:44053. https://doi.org/10.1038/srep44053
Banerjee S, Jin G, Bradley SG, Matters GL, Gailey RD, Crisman JM, Bond JS (2011) Balance of meprin A and B in mice affects the progression of experimental inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol 300(2):G273–G282. https://doi.org/10.1152/ajpgi.00504.2009
Keiffer TR, Bond JS (2014) Meprin metalloproteases inactivate interleukin 6. J Biol Chem 289(11):7580–7588. https://doi.org/10.1074/jbc.M113.546309
Oneda B, Lods N, Lottaz D, Becker-Pauly C, Stocker W, Pippin J, Huguenin M, Ambort D, Marti HP, Sterchi EE (2008) Metalloprotease meprin beta in rat kidney: glomerular localization and differential expression in glomerulonephritis. PLoS One 3(5):e2278. https://doi.org/10.1371/journal.pone.0002278
Bylander JE, Ahmed F, Conley SM, Mwiza JM, Ongeri EM (2017) Meprin metalloprotease deficiency associated with higher mortality rates and more severe diabetic kidney injury in mice with STZ-induced type 1 diabetes. J Diabetes Res 2017:9035038. https://doi.org/10.1155/2017/9035038
Alidori S, Akhavein N, Thorek DL, Behling K, Romin Y, Queen D, Beattie BJ, Manova-Todorova K, Bergkvist M, Scheinberg DA, McDevitt MR (2016) Targeted fibrillar nanocarbon RNAi treatment of acute kidney injury. Sci Transl Med 8(331):331ra339. https://doi.org/10.1126/scitranslmed.aac9647
Bedau T, Peters F, Prox J, Arnold P, Schmidt F, Finkernagel M, Kollmann S, Wichert R, Otte A, Ohler A, Stirnberg M, Lucius R, Koudelka T, Tholey A, Biasin V, Pietrzik CU, Kwapiszewska G, Becker-Pauly C (2017) Ectodomain shedding of CD99 within highly conserved regions is mediated by the metalloprotease meprin beta and promotes transendothelial cell migration. FASEB J 31(3):1226–1237. https://doi.org/10.1096/fj.201601113R
Bedau T, Schumacher N, Peters F, Prox J, Arnold P, Koudelka T, Helm O, Schmidt F, Rabe B, Jentzsch M, Rosenstiel P, Sebens S, Tholey A, Rose-John S, Becker-Pauly C (2017) Cancer-associated mutations in the canonical cleavage site do not influence CD99 shedding by the metalloprotease meprin beta but alter cell migration in vitro. Oncotarget 8(33):54873–54888. https://doi.org/10.18632/oncotarget.18966
Kruse MN, Becker C, Lottaz D, Kohler D, Yiallouros I, Krell HW, Sterchi EE, Stocker W (2004) Human meprin alpha and beta homo-oligomers: cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochem J 378(Pt 2):383–389. https://doi.org/10.1042/BJ20031163
Broder C, Arnold P, Vadon-Le Goff S, Konerding MA, Bahr K, Muller S, Overall CM, Bond JS, Koudelka T, Tholey A, Hulmes DJ, Moali C, Becker-Pauly C (2013) Metalloproteases meprin alpha and meprin beta are C- and N-procollagen proteinases important for collagen assembly and tensile strength. Proc Natl Acad Sci USA 110(35):14219–14224. https://doi.org/10.1073/pnas.1305464110
Kronenberg D, Bruns BC, Moali C, Vadon-Le Goff S, Sterchi EE, Traupe H, Bohm M, Hulmes DJ, Stocker W, Becker-Pauly C (2010) Processing of procollagen III by meprins: new players in extracellular matrix assembly? J Invest Dermatol 130(12):2727–2735. https://doi.org/10.1038/jid.2010.202
Schutte A, Ermund A, Becker-Pauly C, Johansson ME, Rodriguez-Pineiro AM, Backhed F, Muller S, Lottaz D, Bond JS, Hansson GC (2014) Microbial-induced meprin beta cleavage in MUC2 mucin and a functional CFTR channel are required to release anchored small intestinal mucus. Proc Natl Acad Sci USA 111(34):12396–12401. https://doi.org/10.1073/pnas.1407597111
Johansson ME, Hansson GC (2016) Immunological aspects of intestinal mucus and mucins. Nat Rev Immunol 16(10):639–649. https://doi.org/10.1038/nri.2016.88
Wichert R, Ermund A, Schmidt S, Schweinlin M, Ksiazek M, Arnold P, Knittler K, Wilkens F, Potempa B, Rabe B, Stirnberg M, Lucius R, Bartsch JW, Nikolaus S, Falk-Paulsen M, Rosenstiel P, Metzger M, Rose-John S, Potempa J, Hansson GC, Dempsey PJ, Becker-Pauly C (2017) Mucus detachment by host metalloprotease meprin beta requires shedding of its inactive Pro-form, which is abrogated by the pathogenic protease RgpB. Cell Rep 21(8):2090–2103. https://doi.org/10.1016/j.celrep.2017.10.087
Jackle F, Schmidt F, Wichert R, Arnold P, Prox J, Mangold M, Ohler A, Pietrzik CU, Koudelka T, Tholey A, Gutschow M, Stirnberg M, Becker-Pauly C (2015) Metalloprotease meprin beta is activated by transmembrane serine protease matriptase-2 at the cell surface thereby enhancing APP shedding. Biochem J 470(1):91–103. https://doi.org/10.1042/BJ20141417
Bien J, Jefferson T, Causevic M, Jumpertz T, Munter L, Multhaup G, Weggen S, Becker-Pauly C, Pietrzik CU (2012) The metalloprotease meprin beta generates amino terminal-truncated amyloid beta peptide species. J Biol Chem 287(40):33304–33313. https://doi.org/10.1074/jbc.M112.395608
Moller HJ, Graeber MB (1998) The case described by Alois Alzheimer in 1911. Historical and conceptual perspectives based on the clinical record and neurohistological sections. Eur Arch Psychiatry Clin Neurosci 248(3):111–122
Murphy MP, LeVine H 3rd (2010) Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis 19(1):311–323. https://doi.org/10.3233/JAD-2010-1221
Sengupta U, Nilson AN, Kayed R (2016) The role of amyloid-beta oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 6:42–49. https://doi.org/10.1016/j.ebiom.2016.03.035
Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP (1997) A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem 68(1):255–264
Abramov AY, Canevari L, Duchen MR (2004) Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta 1742(1–3):81–87. https://doi.org/10.1016/j.bbamcr.2004.09.006
McLaurin J, Chakrabartty A (1996) Membrane disruption by Alzheimer beta-amyloid peptides mediated through specific binding to either phospholipids or gangliosides. Implications for neurotoxicity. J Biol Chem 271(43):26482–26489
Han SH, Park JC, Mook-Jung I (2016) Amyloid beta-interacting partners in Alzheimer’s disease: from accomplices to possible therapeutic targets. Prog Neurobiol 137:17–38. https://doi.org/10.1016/j.pneurobio.2015.12.004
Venugopal C, Demos CM, Rao KS, Pappolla MA, Sambamurti K (2008) Beta-secretase: structure, function, and evolution. CNS Neurol Disord Drug Targets 7(3):278–294
Bohm C, Chen F, Sevalle J, Qamar S, Dodd R, Li Y, Schmitt-Ulms G, Fraser PE, St George-Hyslop PH (2015) Current and future implications of basic and translational research on amyloid-beta peptide production and removal pathways. Mol Cell Neurosci 66(Pt A):3–11. https://doi.org/10.1016/j.mcn.2015.02.016
Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB (2003) Amyloid beta -protein (Abeta) assembly: abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc Natl Acad Sci USA 100(1):330–335. https://doi.org/10.1073/pnas.222681699
Beglopoulos V, Sun X, Saura CA, Lemere CA, Kim RD, Shen J (2004) Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem 279(45):46907–46914. https://doi.org/10.1074/jbc.M409544200
Sun L, Zhou R, Yang G, Shi Y (2017) Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc Natl Acad Sci USA 114(4):E476–E485. https://doi.org/10.1073/pnas.1618657114
Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R (2001) Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci 4(3):231–232. https://doi.org/10.1038/85059
Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC (2001) BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci 4(3):233–234. https://doi.org/10.1038/85064
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M (1999) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286(5440):735–741
Vassar R (2002) Beta-secretase (BACE) as a drug target for Alzheimer’s disease. Adv Drug Deliv Rev 54(12):1589–1602
Nishitomi K, Sakaguchi G, Horikoshi Y, Gray AJ, Maeda M, Hirata-Fukae C, Becker AG, Hosono M, Sakaguchi I, Minami SS, Nakajima Y, Li HF, Takeyama C, Kihara T, Ota A, Wong PC, Aisen PS, Kato A, Kinoshita N, Matsuoka Y (2006) BACE1 inhibition reduces endogenous Abeta and alters APP processing in wild-type mice. J Neurochem 99(6):1555–1563. https://doi.org/10.1111/j.1471-4159.2006.04178.x
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 82(12):4245–4249
Wiltfang J, Esselmann H, Cupers P, Neumann M, Kretzschmar H, Beyermann M, Schleuder D, Jahn H, Ruther E, Kornhuber J, Annaert W, De Strooper B, Saftig P (2001) Elevation of beta-amyloid peptide 2-42 in sporadic and familial Alzheimer’s disease and its generation in PS1 knockout cells. J Biol Chem 276(46):42645–42657. https://doi.org/10.1074/jbc.M102790200
Kummer MP, Heneka MT (2014) Truncated and modified amyloid-beta species. Alzheimers Res Ther 6(3):28. https://doi.org/10.1186/alzrt258
Bibl M, Gallus M, Welge V, Esselmann H, Wiltfang J (2012) Aminoterminally truncated and oxidized amyloid-beta peptides in the cerebrospinal fluid of Alzheimer’s disease patients. J Alzheimers Dis 29(4):809–816. https://doi.org/10.3233/JAD-2012-111796
Schechter I, Ziv E (2011) Cathepsins S, B and L with aminopeptidases display beta-secretase activity associated with the pathogenesis of Alzheimer’s disease. Biol Chem 392(6):555–569. https://doi.org/10.1515/BC.2011.054
Hook G, Yu J, Toneff T, Kindy M, Hook V (2014) Brain pyroglutamate amyloid-beta is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer’s disease therapeutic. J Alzheimers Dis 41(1):129–149. https://doi.org/10.3233/JAD-131370
Munger JS, Haass C, Lemere CA, Shi GP, Wong WS, Teplow DB, Selkoe DJ, Chapman HA (1995) Lysosomal processing of amyloid precursor protein to A beta peptides: a distinct role for cathepsin S. Biochem J 311(Pt 1):299–305
Hamazaki H (1996) Cathepsin D is involved in the clearance of Alzheimer’s beta-amyloid protein. FEBS Lett 396(2–3):139–142
Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, Wang X, Yu G, Esposito L, Mucke L, Gan L (2006) Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron 51(6):703–714. https://doi.org/10.1016/j.neuron.2006.07.027
Letronne F, Laumet G, Ayral AM, Chapuis J, Demiautte F, Laga M, Vandenberghe ME, Malmanche N, Leroux F, Eysert F, Sottejeau Y, Chami L, Flaig A, Bauer C, Dourlen P, Lesaffre M, Delay C, Huot L, Dumont J, Werkmeister E, Lafont F, Mendes T, Hansmannel F, Dermaut B, Deprez B, Herard AS, Dhenain M, Souedet N, Pasquier F, Tulasne D, Berr C, Hauw JJ, Lemoine Y, Amouyel P, Mann D, Deprez R, Checler F, Hot D, Delzescaux T, Gevaert K, Lambert JC (2016) ADAM30 downregulates APP-Linked defects through cathepsin D activation in Alzheimer’s disease. EBioMedicine 9:278–292. https://doi.org/10.1016/j.ebiom.2016.06.002
Sevalle J, Amoyel A, Robert P, Fournie-Zaluski MC, Roques B, Checler F (2009) Aminopeptidase A contributes to the N-terminal truncation of amyloid beta-peptide. J Neurochem 109(1):248–256. https://doi.org/10.1111/j.1471-4159.2009.05950.x
Hosoda R, Saido TC, Otvos L Jr, Arai T, Mann DM, Lee VM, Trojanowski JQ, Iwatsubo T (1998) Quantification of modified amyloid beta peptides in Alzheimer disease and down syndrome brains. J Neuropathol Exp Neurol 57(11):1089–1095
Schlenzig D, Cynis H, Hartlage-Rubsamen M, Zeitschel U, Menge K, Fothe A, Ramsbeck D, Spahn C, Wermann M, Rossner S, Buchholz M, Schilling S, Demuth HU (2018) Dipeptidyl-peptidase activity of meprin beta links N-truncation of abeta with glutaminyl cyclase-catalyzed pGlu-abeta formation. J Alzheimers Dis 66(1):359–375. https://doi.org/10.3233/JAD-171183
Schonherr C, Bien J, Isbert S, Wichert R, Prox J, Altmeppen H, Kumar S, Walter J, Lichtenthaler SF, Weggen S, Glatzel M, Becker-Pauly C, Pietrzik CU (2016) Generation of aggregation prone N-terminally truncated amyloid beta peptides by meprin beta depends on the sequence specificity at the cleavage site. Mol Neurodegener 11:19. https://doi.org/10.1186/s13024-016-0084-5
Jefferson T, Causevic M, Auf dem Keller U, Schilling O, Isbert S, Geyer R, Maier W, Tschickardt S, Jumpertz T, Weggen S, Bond JS, Overall CM, Pietrzik CU, Becker-Pauly C (2011) Metalloprotease meprin beta generates nontoxic N-terminal amyloid precursor protein fragments in vivo. J Biol Chem 286(31):27741–27750. https://doi.org/10.1074/jbc.M111.252718
Muller UC, Deller T, Korte M (2017) Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci 18(5):281–298. https://doi.org/10.1038/nrn.2017.29
Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ (1992) Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 360(6405):672–674. https://doi.org/10.1038/360672a0
Elder GA, Gama Sosa MA, De Gasperi R (2010) Transgenic mouse models of Alzheimer’s disease. Mt Sinai J Med 77(1):69–81. https://doi.org/10.1002/msj.20159
Ohshima Y, Taguchi K, Mizuta I, Tanaka M, Tomiyama T, Kametani F, Yabe-Nishimura C, Mizuno T, Tokuda T (2018) Mutations in the beta-amyloid precursor protein in familial Alzheimer’s disease increase Abeta oligomer production in cellular models. Heliyon 4(1):e00511. https://doi.org/10.1016/j.heliyon.2018.e00511
Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF (2004) BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 41(1):27–33
Patel T, Brookes KJ, Turton J, Chaudhury S, Guetta-Baranes T, Guerreiro R, Bras J, Hernandez D, Singleton A, Francis PT, Hardy J, Morgan K (2017) Whole-exome sequencing of the BDR cohort: evidence to support the role of the PILRA gene in Alzheimer’s disease. Neuropathol Appl Neurobiol 44:506–521. https://doi.org/10.1111/nan.12452
Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF (2010) ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J 29(17):3020–3032. https://doi.org/10.1038/emboj.2010.167
Ohler A, Debela M, Wagner S, Magdolen V, Becker-Pauly C (2010) Analyzing the protease web in skin: meprin metalloproteases are activated specifically by KLK4, 5 and 8 vice versa leading to processing of proKLK7 thereby triggering its activation. Biol Chem 391(4):455–460. https://doi.org/10.1515/BC.2010.023
Jefferson T, Auf dem Keller U, Bellac C, Metz VV, Broder C, Hedrich J, Ohler A, Maier W, Magdolen V, Sterchi E, Bond JS, Jayakumar A, Traupe H, Chalaris A, Rose-John S, Pietrzik CU, Postina R, Overall CM, Becker-Pauly C (2013) The substrate degradome of meprin metalloproteases reveals an unexpected proteolytic link between meprin beta and ADAM10. Cell Mol Life Sci 70(2):309–333. https://doi.org/10.1007/s00018-012-1106-2
Herzog C, Haun RS, Ludwig A, Shah SV, Kaushal GP (2014) ADAM10 is the major sheddase responsible for the release of membrane-associated meprin A. J Biol Chem 289(19):13308–13322. https://doi.org/10.1074/jbc.M114.559088
Banerjee S, Bond JS (2008) Prointerleukin-18 is activated by meprin beta in vitro and in vivo in intestinal inflammation. J Biol Chem 283(46):31371–31377. https://doi.org/10.1074/jbc.M802814200
Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457(7232):981–989. https://doi.org/10.1038/nature07767
Olsen O, Kallop DY, McLaughlin T, Huntwork-Rodriguez S, Wu Z, Duggan CD, Simon DJ, Lu Y, Easley-Neal C, Takeda K, Hass PE, Jaworski A, O’Leary DD, Weimer RM, Tessier-Lavigne M (2014) Genetic analysis reveals that amyloid precursor protein and death receptor 6 function in the same pathway to control axonal pruning independent of beta-secretase. J Neurosci 34(19):6438–6447. https://doi.org/10.1523/JNEUROSCI.3522-13.2014
Peron R, Vatanabe IP, Manzine PR, Camins A, Cominetti MR (2018) Alpha-secretase ADAM10 regulation: insights into Alzheimer’s disease Treatment. Pharmaceuticals 11:1. https://doi.org/10.3390/ph11010012
Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F (1992) Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc Natl Acad Sci USA 89(22):10758–10762
Pandey P, Sliker B, Peters HL, Tuli A, Herskovitz J, Smits K, Purohit A, Singh RK, Dong J, Batra SK, Coulter DW, Solheim JC (2016) Amyloid precursor protein and amyloid precursor-like protein 2 in cancer. Oncotarget 7(15):19430–19444. https://doi.org/10.18632/oncotarget.7103
Jacobsen KT, Iverfeldt K (2009) Amyloid precursor protein and its homologues: a family of proteolysis-dependent receptors. Cell Mol Life Sci 66(14):2299–2318. https://doi.org/10.1007/s00018-009-0020-8
Sprecher CA, Grant FJ, Grimm G, O’Hara PJ, Norris F, Norris K, Foster DC (1993) Molecular cloning of the cDNA for a human amyloid precursor protein homolog: evidence for a multigene family. Biochemistry 32(17):4481–4486
Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE (1993) Isolation and characterization of APLP2 encoding a homologue of the Alzheimer’s associated amyloid beta protein precursor. Nat Genet 5(1):95–100. https://doi.org/10.1038/ng0993-95
Coulson EJ, Paliga K, Beyreuther K, Masters CL (2000) What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem Int 36(3):175–184
Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, Parker MW (1999) Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol 6(4):327–331. https://doi.org/10.1038/7562
Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, Williamson NA, White AR, Hinds MG, Norton RS, Beyreuther K, Masters CL, Parker MW, Cappai R (2003) Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J Biol Chem 278(19):17401–17407. https://doi.org/10.1074/jbc.m300629200
Kong GK, Adams JJ, Harris HH, Boas JF, Curtain CC, Galatis D, Masters CL, Barnham KJ, McKinstry WJ, Cappai R, Parker MW (2007) Structural studies of the Alzheimer’s amyloid precursor protein copper-binding domain reveal how it binds copper ions. J Mol Biol 367(1):148–161. https://doi.org/10.1016/j.jmb.2006.12.041
Dahms SO, Hoefgen S, Roeser D, Schlott B, Guhrs KH, Than ME (2010) Structure and biochemical analysis of the heparin-induced E1 dimer of the amyloid precursor protein. Proc Natl Acad Sci USA 107(12):5381–5386. https://doi.org/10.1073/pnas.0911326107
Kang J, Muller-Hill B (1990) Differential splicing of Alzheimer’s disease amyloid A4 precursor RNA in rat tissues: pre A4(695) mRNA is predominantly produced in rat and human brain. Biochem Biophys Res Commun 166(3):1192–1200
Weidemann A, Konig G, Bunke D, Fischer P, Salbaum JM, Masters CL, Beyreuther K (1989) Identification, biogenesis, and localization of precursors of Alzheimer’s disease A4 amyloid protein. Cell 57(1):115–126
Ninomiya H, Roch JM, Sundsmo MP, Otero DA, Saitoh T (1993) Amino acid sequence RERMS represents the active domain of amyloid beta/A4 protein precursor that promotes fibroblast growth. J Cell Biol 121(4):879–886
Ninomiya H, Roch JM, Jin LW, Saitoh T (1994) Secreted form of amyloid beta/A4 protein precursor (APP) binds to two distinct APP binding sites on rat B103 neuron-like cells through two different domains, but only one site is involved in neuritotropic activity. J Neurochem 63(2):495–500
Pawlik M, Otero DA, Park M, Fischer WH, Levy E, Saitoh T (2007) Proteins that bind to the RERMS region of beta amyloid precursor protein. Biochem Biophys Res Commun 355(4):907–912. https://doi.org/10.1016/j.bbrc.2007.02.047
Gralle M, Ferreira ST (2007) Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog Neurobiol 82(1):11–32. https://doi.org/10.1016/j.pneurobio.2007.02.001
Reinhard C, Hebert SS, De Strooper B (2005) The amyloid-beta precursor protein: integrating structure with biological function. EMBO J 24(23):3996–4006. https://doi.org/10.1038/sj.emboj.7600860
Turner PR, O’Connor K, Tate WP, Abraham WC (2003) Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol 70(1):1–32
Wang Y, Ha Y (2004) The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell 15(3):343–353. https://doi.org/10.1016/j.molcel.2004.06.037
Pahlsson P, Shakin-Eshleman SH, Spitalnik SL (1992) N-linked glycosylation of beta-amyloid precursor protein. Biochem Biophys Res Commun 189(3):1667–1673
Hoefgen S, Dahms SO, Oertwig K, Than ME (2015) The amyloid precursor protein shows a pH-dependent conformational switch in its E1 domain. J Mol Biol 427(2):433–442. https://doi.org/10.1016/j.jmb.2014.12.005
Hoefgen S, Coburger I, Roeser D, Schaub Y, Dahms SO, Than ME (2014) Heparin induced dimerization of APP is primarily mediated by E1 and regulated by its acidic domain. J Struct Biol 187(1):30–37. https://doi.org/10.1016/j.jsb.2014.05.006
Dahms SO, Konnig I, Roeser D, Guhrs KH, Mayer MC, Kaden D, Multhaup G, Than ME (2012) Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain. J Mol Biol 416(3):438–452. https://doi.org/10.1016/j.jmb.2011.12.057
Keil C, Huber R, Bode W, Than ME (2004) Cloning, expression, crystallization and initial crystallographic analysis of the C-terminal domain of the amyloid precursor protein APP. Acta Crystallogr D Biol Crystallogr 60(Pt 9):1614–1617. https://doi.org/10.1107/S0907444904015343
Dulubova I, Ho A, Huryeva I, Sudhof TC, Rizo J (2004) Three-dimensional structure of an independently folded extracellular domain of human amyloid-beta precursor protein. Biochemistry 43(30):9583–9588. https://doi.org/10.1021/bi049041o
Lu JX, Yau WM, Tycko R (2011) Evidence from solid-state NMR for nonhelical conformations in the transmembrane domain of the amyloid precursor protein. Biophys J 100(3):711–719. https://doi.org/10.1016/j.bpj.2010.12.3696
Botev A, Munter LM, Wenzel R, Richter L, Althoff V, Ismer J, Gerling U, Weise C, Koksch B, Hildebrand PW, Bittl R, Multhaup G (2011) The amyloid precursor protein C-terminal fragment C100 occurs in monomeric and dimeric stable conformations and binds gamma-secretase modulators. Biochemistry 50(5):828–835. https://doi.org/10.1021/bi1014002
Nadezhdin KD, Bocharova OV, Bocharov EV, Arseniev AS (2011) Structural and dynamic study of the transmembrane domain of the amyloid precursor protein. Acta Naturae 3(1):69–76
Beel AJ, Mobley CK, Kim HJ, Tian F, Hadziselimovic A, Jap B, Prestegard JH, Sanders CR (2008) Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP): does APP function as a cholesterol sensor? Biochemistry 47(36):9428–9446. https://doi.org/10.1021/bi800993c
Sato T, Tang TC, Reubins G, Fei JZ, Fujimoto T, Kienlen-Campard P, Constantinescu SN, Octave JN, Aimoto S, Smith SO (2009) A helix-to-coil transition at the epsilon-cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis. Proc Natl Acad Sci USA 106(5):1421–1426. https://doi.org/10.1073/pnas.0812261106
Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR (2012) The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336(6085):1168–1171. https://doi.org/10.1126/science.1219988
MacKenzie KR, Prestegard JH, Engelman DM (1997) A transmembrane helix dimer: structure and implications. Science 276(5309):131–133
Javadpour MM, Eilers M, Groesbeek M, Smith SO (1999) Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys J 77(3):1609–1618. https://doi.org/10.1016/S0006-3495(99)77009-8
Kim S, Jeon TJ, Oberai A, Yang D, Schmidt JJ, Bowie JU (2005) Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc Natl Acad Sci USA 102(40):14278–14283. https://doi.org/10.1073/pnas.0501234102
Anderson SM, Mueller BK, Lange EJ, Senes A (2017) Combination of Calpha-H Hydrogen Bonds and van der Waals Packing Modulates the Stability of GxxxG-Mediated Dimers in Membranes. J Am Chem Soc 139(44):15774–15783. https://doi.org/10.1021/jacs.7b07505
Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G (2007) GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J 26(6):1702–1712. https://doi.org/10.1038/sj.emboj.7601616
Decock M, Stanga S, Octave JN, Dewachter I, Smith SO, Constantinescu SN, Kienlen-Campard P (2016) Glycines from the APP GXXXG/GXXXA transmembrane motifs promote formation of pathogenic abeta oligomers in cells. Front Aging Neurosci 8:107. https://doi.org/10.3389/fnagi.2016.00107
Yano Y, Kondo K, Watanabe Y, Zhang TO, Ho JJ, Oishi S, Fujii N, Zanni MT, Matsuzaki K (2017) GXXXG-Mediated Parallel and Antiparallel Dimerization of Transmembrane Helices and Its Inhibition by Cholesterol: single-Pair FRET and 2D IR Studies. Angew Chem Int Ed Engl 56(7):1756–1759. https://doi.org/10.1002/anie.201609708
Yan Y, Xu TH, Harikumar KG, Miller LJ, Melcher K, Xu HE (2017) Dimerization of the transmembrane domain of amyloid precursor protein is determined by residues around the gamma-secretase cleavage sites. J Biol Chem 292(38):15826–15837. https://doi.org/10.1074/jbc.M117.789669
Nadezhdin KD, Bocharova OV, Bocharov EV, Arseniev AS (2012) Dimeric structure of transmembrane domain of amyloid precursor protein in micellar environment. FEBS Lett 586(12):1687–1692. https://doi.org/10.1016/j.febslet.2012.04.062
Chen W, Gamache E, Rosenman DJ, Xie J, Lopez MM, Li YM, Wang C (2014) Familial Alzheimer’s mutations within APPTM increase Abeta42 production by enhancing accessibility of epsilon-cleavage site. Nat Commun 5:3037. https://doi.org/10.1038/ncomms4037
Song Y, Hustedt EJ, Brandon S, Sanders CR (2013) Competition between homodimerization and cholesterol binding to the C99 domain of the amyloid precursor protein. Biochemistry 52(30):5051–5064. https://doi.org/10.1021/bi400735x
Dominguez L, Foster L, Straub JE, Thirumalai D (2016) Impact of membrane lipid composition on the structure and stability of the transmembrane domain of amyloid precursor protein. Proc Natl Acad Sci USA 113(36):E5281–5287. https://doi.org/10.1073/pnas.1606482113
Dominguez L, Foster L, Meredith SC, Straub JE, Thirumalai D (2014) Structural heterogeneity in transmembrane amyloid precursor protein homodimer is a consequence of environmental selection. J Am Chem Soc 136(27):9619–9626. https://doi.org/10.1021/ja503150x
Pantelopulos GA, Straub JE, Thirumalai D, Sugita Y (2018) Structure of APP-C991-99 and implications for role of extra-membrane domains in function and oligomerization. Biochim Biophys Acta Biomembr 1860:1698–1708. https://doi.org/10.1016/j.bbamem.2018.04.002
Ramelot TA, Gentile LN, Nicholson LK (2000) Transient structure of the amyloid precursor protein cytoplasmic tail indicates preordering of structure for binding to cytosolic factors. Biochemistry 39(10):2714–2725
Ando K, Iijima KI, Elliott JI, Kirino Y, Suzuki T (2001) Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. J Biol Chem 276(43):40353–40361. https://doi.org/10.1074/jbc.M104059200
Yun M, Keshvara L, Park CG, Zhang YM, Dickerson JB, Zheng J, Rock CO, Curran T, Park HW (2003) Crystal structures of the Dab homology domains of mouse disabled 1 and 2. J Biol Chem 278(38):36572–36581. https://doi.org/10.1074/jbc.M304384200
Zhang Z, Lee CH, Mandiyan V, Borg JP, Margolis B, Schlessinger J, Kuriyan J (1997) Sequence-specific recognition of the internalization motif of the Alzheimer’s amyloid precursor protein by the X11 PTB domain. EMBO J 16(20):6141–6150. https://doi.org/10.1093/emboj/16.20.6141
Radzimanowski J, Simon B, Sattler M, Beyreuther K, Sinning I, Wild K (2008) Structure of the intracellular domain of the amyloid precursor protein in complex with Fe65-PTB2. EMBO Rep 9(11):1134–1140. https://doi.org/10.1038/embor.2008.188
Arolas JL, Broder C, Jefferson T, Guevara T, Sterchi EE, Bode W, Stocker W, Becker-Pauly C, Gomis-Ruth FX (2012) Structural basis for the sheddase function of human meprin beta metalloproteinase at the plasma membrane. Proc Natl Acad Sci USA 109(40):16131–16136. https://doi.org/10.1073/pnas.1211076109
Peters F, Scharfenberg F, Colmorgen C, Armbrust F, Wichert R, Arnold P, Potempa B, Potempa J, Pietrzik CU, Hasler R, Rosenstiel P, Becker-Pauly C (2019) Tethering soluble meprin alpha in an enzyme complex to the cell surface affects IBD-associated genes. FASEB J 2019:fj201802391R. https://doi.org/10.1096/fj.201802391r
Seegar TCM, Killingsworth LB, Saha N, Meyer PA, Patra D, Zimmerman B, Janes PW, Rubinstein E, Nikolov DB, Skiniotis G, Kruse AC, Blacklow SC (2017) Structural Basis for Regulated Proteolysis by the alpha-Secretase ADAM10. Cell 171(7):1638–1648 e1637. https://doi.org/10.1016/j.cell.2017.11.014
Liu L, Ding L, Rovere M, Wolfe MS, Selkoe DJ (2019) A cellular complex of BACE1 and gamma-secretase sequentially generates Abeta from its full-length precursor. J Cell Biol 218(2):644–663. https://doi.org/10.1083/jcb.201806205
Acknowledgements
This work was supported by the Alzheimer Forschung Initiative e.V. (#18007) and the Deutsche Forschungsgemeinschaft (DFG) Project-number 125440785 SFB 877 (Proteolysis as a Regulatory Event in Pathophysiology, Projects A9 and A15) and BE 4086/2-2 (C.B.-P.).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Scharfenberg, F., Armbrust, F., Marengo, L. et al. Regulation of the alternative β-secretase meprin β by ADAM-mediated shedding. Cell. Mol. Life Sci. 76, 3193–3206 (2019). https://doi.org/10.1007/s00018-019-03179-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03179-1