
Abstract
Protein arginine methyltransferases (PRMTs) catalyze the methyl transfer to the arginine residues of protein substrates and are classified into three major types based on the final form of the methylated arginine. Recent studies have shown a strong correlation between PRMT expression level and the prognosis of cancer patients. Currently, crystal structures of eight PRMT members have been determined. Kinetic and structural studies have shown that all PRMTs share similar, but unique catalytic and substrate recognition mechanism. In this review, we discuss the structural similarities and differences of different PRMT members, focusing on their overall structure, S-adenosyl-l-methionine-binding pocket, substrate arginine recognition and catalytic mechanisms. Since PRMTs are valuable targets for drug discovery, we also rationally classify the known PRMT inhibitors into five classes and discuss their mechanisms of action at the atomic level.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Arginine methylation by protein arginine methyltransferases (PRMTs) is an abundant post-translational modification in eukaryotes [1]. PRMTs catalyze the transfer of methyl groups from the co-substrate S-adenosyl-l-methionine (AdoMet, SAM) to the guanidine nitrogen (ω-NG) of a peptidyl arginine residue, resulting in the formation of methylarginine and S-adenosyl-l-homocysteine (AdoHcy, SAH) [2]. Three major types of methlyarginines are recognized: monomethylarginine (Rme1), asymmetric dimethylarginine (Rme2a) and symmetric dimethylarginine (Rme2s). The nine PRMT members are categorized into three types according to the final form of methylarginine products: type I PRMTs (PRMT-1, 2, 3, 4, 6 and 8) catalyze formation of Rme1 and Rme2a; type II PRMTs (PRMT-5 and 9) catalyze formation of Rme1 and Rme2s; and type III PRMT (PRMT7) can only generate Rme1 [3]. In addition, a rarely occurring type IV PRMT has been described that results in monomethylation of the internal guanidine δ-nitrogen [4, 5].
PRMT1, the most predominant type I PRMT in mammalian cells, accounts for 85% of cellular PRMT activity and is involved in many biological functions [6,7,8,9]. PRMT2 acts as a coactivator of hormone receptors in a ligand-dependent manner and enhances the transcriptional activity of hormone receptors [10, 11]. The involvement of PRMT2 in breast carcinogenesis has been reported [12, 13]. PRMT3 methylates 40S ribosomal protein S2 (rpS2) and is involved in proper maturation of the 80S ribosome [14,15,16]. It is also functionally important for dendritic spine maturation in rats [17]. PRMT4, also called as CARM1 (coactivator-associated arginine methyltransferase 1), was initially identified as an enhancer of transcriptional activation by several nuclear hormone receptors [18]. As a transcriptional coactivator, it plays a major role in chromatin remodeling and gene activation [19,20,21,22]. PRMT5 methylates histones H2A and H4 as well as many other proteins [23,24,25]. Unlike other PRMTs, the activity of PRMT5 requires the presence of protein cofactors such as MEP50, RioK1 and pICln [26,27,28,29,30]. Inhibitors of PRMT5 have been developed and subsequently tested in clinical trials for solid tumors and lymphoma [31]. PRMT6, a nuclear protein that prefers substrates with glycine–arginine-rich (GAR) motifs [32], is involved in transcriptional regulation, the pathogenesis of human immunodeficiency virus, DNA excision repair and cell cycle progression [33,34,35]. The overexpression of PRMT6 in the gall bladder and lung cancers has been reported [36]. PRMT7 is associated with nuclear ribonucleoprotein particle biogenesis, DNA repair and the regulation of MLL4-mediated differentiation [37,38,39]. Full-length PRMT8 is expressed in the brain and localized to the plasma membrane via N-terminal myristoylation [40]. The correlation between PRMT8 overexpression and cancer patient survival has been described [41]. PRMT9 is identified as a non-histone methyltransferase and involved in U2 snRNP maturation [42].
Although the majority of the PRMTs share similar substrates, such as histone proteins, each PRMT member also methylates different sets of non-histone proteins. In addition, some prefer to methylate glycine–arginine-rich (GAR) motifs and the other prefers proline–glycine–methionine-rich (PGM) motifs [43, 44]. In this review, we discuss the structural similarities and differences of the eight PRMT protein family members whose structures have been determined. We focus on the overall PRMT structure, AdoMet-binding pocket, arginine recognition, catalysis mechanisms, and the mechanism of action of PMRT inhibitors.
Overall structure of PRMTs: similarities and differences
The core structure of PRMT3, a type I PRMT, was determined in 2000 and was the first PRMT core structure to be deposited in the Protein Databank (PDB) [45]. Since then, the structures of other PRMTs have been elucidated using protein crystallography (Table 1) [46]. Lately, PRMT5–MEP50 complex structure has been also determined by cryo-electron microscopy (cryoEM) approach [47]. Among the nine mammalian PRMT members, only the structure of PRMT9 has yet to be determined. A canonical PRMT core structure includes two domains: the N-terminal Rossmann fold (also referred to as the AdoMet-binding domain) and the C-terminal β-barrel domain. The α-helical dimerization arm that is responsible for PRMT dimerization protrudes from the N terminus of the β-barrel domain and contacts N terminus of Rossmann fold (Fig. 1a). The dynamic α-helices (αX, αY and αZ) at the N-terminal of Rossmann fold participates in AdoMet binding and are present in type I and type III PRMTs (Fig. 1a). The core morphology of all type I PRMTs is a doughnut-shaped homodimer arranged in a head-to-tail pattern (Fig. 1a). Both monomers bind to AdoMet and to the substrate arginine, suggesting that both protomers of the PRMT homodimer are catalytically active. However, mouse and Caenorhabditis elegans PRMT7 (MmPRMT7 and CePRMT7) are unique, as they consist of two PRMT modules in tandem and arranged as a pseudo-dimer (Fig. 1b) [48,49,50]. Structural evidence has shown that the second AdoMet-binding module of PRMT7 is occupied by a zinc ion and is unable to bind AdoHcy, so the C-terminal PRMT core module of PRMT7 is considered non-catalytically active [48, 50]. Coincidentally, PRMT7 is a type III PRMT that catalyzes only monomethylated arginine formation. PRMT7 from Trypanosoma brucei (TbPRMT7) lacks the tandem C-terminal module and in solution exists solely as a homodimer [51, 52]. Although TbPRMT7 forms a homodimer, it strictly generates Rme1, indicating that the presence of tandemly arranged PRMT modules vs. two active PRMT modules is not the determining step in Rme1 formation [48, 52]. Unlike the ring-like dimers in type I PRMT structures [45, 53,54,55,56,57,58,59], PRMT7 forms a bowl-like dimer (Fig. 1b). This anomaly in the shape of these PRMT7 dimers has been attributed to the different arrangements of the dimerization arm and β-barrel domain, such that their secondary structural elements are pushed inwards to close the central hole in the PRMT dimer. Closure of the central hole in PRMT7 is in accordance with the lack of AdoMet-binding site in the second Rossmann fold, which could possibly be one of the strategies of these enzymes for promoting monomethylation.

The structural similarities and differences of PRMTs. a The typical PRMT dimer is shown in cartoon diagram using Danio rerio PRMT2 (PDB ID: 5FUB) as an example. The N-terminal helices: αX (cyan), αY (yellow) αZ (light gray), Rossman fold (blue), β-barrel (red), dimer arm (green) are shown as cartoon diagram. The monomer B is shown in light corresponding color. The AdoHcy (black) shown as stick model, dashed line shows the dividing line for the monomer A and B. bMmPRMT7 reveals a bowel-shaped pseudo-dimer. The color scheme is same as a and the zinc-binding motif is shown as dashed circle. c The apo (top) and AdoHcy-bound (bottom) forms of MmPRMT4 show the conformation and orientation change of the N-terminal αX (color in blue) upon AdoHcy binding. The missing PH domain is labeled to shown the relative position. Monomers A and B of MmPRMT4 are colored in green and orange, respectively. dHsPRMT5 is in complex with MEP50. Catalytic domain color is same as a, whereas TIM barrel and MEP50 shown in purple and cyan color, respectively
PRMTs must maintain at least a dimer state (or pseudo-dimer for PRMT7) to be functionally active [53, 59, 60]. Higher ordered oligomerization has been also observed in PRMTs. Saccharomyces cerevisiae PRMT1 (hmt1) forms a trimer of dimers whereas vertebrate PRMT5 form a dimer of dimers [60, 61]. Despite the dimeric forms of PRMT1 and PRMT8 revealed by protein crystallography, both enzymes form higher order oligomers in solution [62,63,64]. The high-ordered oligomerization state of human PRMT8 (HsPRMT8) remains unchanged regardless of the protein concentration, but PRMT1 from Rattus norvegicus (RnPRMT1) achieves higher ordered oligomerization in a concentration-dependent manner [53, 62, 65]. In addition, the oligomerization state of RnPRMT1 decreases significantly upon AdoMet binding whereas HsPRMT8 maintains its oligomerization state upon co-substrate binding [53, 62]. Recent, crystallographic studies proposed two different oligomerization states of PRMT8, as either a helical filament or a tetramer. The different forms of HsPRMT8 oligomerization may reflect differences in the open reading frames and/or the presence of the N-terminal tag [62]. Lately, the recent cryoEM study of HsPRMT5 also revealed higher ordered oligomerization of PRMT5, in which tetrameric complexes of the enzyme associate in a side to side manner [47].
Most PRMTs deposited in the PDB contain only a structurally conserved catalytic core domain. However, the other striking difference among the PRMTs is the presence of the N-terminal non-catalytic domains. For example, PRMT2 contains an SH3 domain; PRMT3 contains a zinc finger; PRMT4 contains a pleckstrin homology (PH) domain; PRMT5 contains a TIM barrel; PRMT8 contains an N-myristoylation domain. Both of the full-length Danio rerio PRMT2 and mouse PRMT4 (MmPRMT4) crystal structures showed that the electron densities of both N-terminal non-catalytic domains were missing, which suggested the high domain flexibility of N terminus in the absence of the protein substrates [54]. Thus, the structural details of these enzymes with non-catalytic domains remain unresolved. The kinetic study showed a sevenfold reduction in the methyltransferase activity of SH3-domain-truncated PRMT2 compared to the full-length enzyme [54]. The N terminus of PRMT3 harbors a zinc finger domain. Zinc finger domains are small protein motifs and conventionally considered as a DNA-binding motif, but they are also able to bind RNA, protein and lipid substrates [66,67,68,69]. The zinc finger domain of PRMT3 is not required for methylation of glutathione S-transferase–fibrillarin amino-terminal fusion protein (GST–GAR), an artificial substrate, but is absolutely required for the recognition of RNA-associated substrates and for the binding of rpS2 [70, 71]. The PRMT4/CARM1 N-terminal domain (from residue 28–140) possesses a PH domain that is formed by two perpendicular antiparallel β-sheets followed by a C-terminal helix and is known to bind to lipids or proteins. PH domain-containing proteins are shown to be involved in a variety of biological processes, including signal transduction, cytoskeletal arrangement, nuclear transport and DNA repair [72,73,74,75,76,77]. Over 130 PRMT4 protein substrates have been identified and most of them contain proline-rich motifs [78]. Deletion of the PH domain of PRMT4 results in a global decrease in both the interaction of the PRMT4 with its substrates and methylation activities [78, 79]. Interestingly, structural studies of PRMT4 revealed striking differences in the orientation of the N terminus upon AdoHcy binding (Fig. 1c). In the absence of AdoHcy, the apo PRMT4 N terminus folds into a β-sheet with a small 310-helix rotated 180° compared to the AdoHcy-bound PRM4, which possesses only an α-helix. This difference leads to the differences in the spatial orientation of the PH domain (Fig. 1c) [55]. However, the biological and functional significance of the two alternative conformations of the PRMT4 N terminus remains unclear. In the case of PRMT8, its N terminus harbors a unique myristoylation motif that results in the association of the enzyme with the plasma membrane [40]. Both His-tagged and GST fusion species of PRMT8 lacking the initial 60 amino acid residues exhibit enhanced enzymatic activity, suggesting a role for the N terminus in regulating PRMT8 activity [80].
In PRMT5, the N-terminal extension is in the form of a TIM barrel domain that participates in dimer formation and accounts for the very large structural difference between the PRMT5 dimer and the dimers of other PRMTs [81] In addition, both human and Xenopus laevis PRMT5 (HsPRMT5 and XlPRMT5) possess a shorter dimerization arm and form a dimer with a central cavity whose diameter is approximately 30 Å [61, 82]. Four dimerization loops contributed by each protomer of PRMT5 tetramer occupy the central cavity. The N-terminal TIM barrel is also responsible for MEP50 interaction (Fig. 1d); therefore, TIM barrel domain plays dual structural roles: (1) promoting oligomerization by interacting with the catalytic domain and (2) recruiting MEP50 for substrate interaction [61, 82]. There is no obvious MEP50 ortholog seen in C. elegans and C. elegans PRMT5 (CePRMT5) only forms a dimer in solution [83].
Signature motifs of PRMTs and N-terminal helices upon the co-substrate binding
The PRMT catalytic core is remarkably similar across all type I, II and III enzymes. It is endowed with six signature motifs that are indispensable for methyltransferase function: (1) motif I (VLD/VGxGxG) forms the core of the AdoMet-binding pocket with three highly conserved glycine among all PRMTs; (2) post-motif I (V/I-X-G/A–X-D/E) forms the hydrogen bonding interactions with the ribose hydroxyl moiety of AdoMet, via a glutamic or aspartic acid residue; (3) motif II (E/K/VDII) stabilizes motif I by the formation of a β-sheet; (4) double-E motif contains SExMGxxLxxExM whose two glutamic acid residues positions substrate arginine; (5) motif III (LK/xxGxxxP) forms a parallel β-sheet with motif II and (6) the critical THW loop, located near the active site, is important for substrate binding as well as stabilization of the N-terminal α-helix (Fig. 2) [84, 85]. The typical active site of the PRMTs adopts a canonical fold with minor variations in the proximal end of the N terminus depending on the enzyme type. For example, all type I PRMTs contain dynamic α helices (αX, αY and αZ) at the N terminus whereas in PRMT5 (type II) and PRMT7 (type III) the αX helix is absent (Fig. 2a). It has been shown that αX is disordered in the absence of AdoMet/AdoHcy and stabilized upon AdoHcy binding [55]. The N-terminal motif YFxxY, which makes up part of the αX helix, is conserved in all type I PRMTs, but the phenylalanine of mouse and T. brucei PRMT6 (MmPRMT6 and TbPRMT6) substitute the conservative tyrosine residue (Fig. 2a). The YFxxY motif is missing in type II and type III PRMTs. The sequence in the corresponding region of PRMT5 homologs is PLxxN, which forms a partial loop and participates in AdoMet recognition (Fig. 2a) [61, 83]. In TbPRMT7, the corresponding sequence is VSLIE (Fig. 2a), which folds into an extension of the αY helix that protrudes outward from the AdoMet-binding domain. The latter is more inclined towards the β-barrel of the opposite molecule of the homodimer, resulting in a more solvent-exposed AdoMet-binding pocket [52].

Crystal structure-based sequence alignment of catalytic core of type I, II and III PRMTs. a The alignment is restricted to the catalytic domains of the enzymes. The AdoMet-binding domain, only important motifs secondary structures are shown as red cylinder, whereas for clarity, β-barrel domain (except its THW motif) and dimerization motif are completely removed. PRMT group I, II and III members are listed on left in green, blue and pink, respectively. Each isoform possesses six signature methyltransferase motifs: I, post-I, II, III (boxed), double-E SExMGxxLxxExM motif (indicated with bars) and the THW loop (highlighted in yellow). Highly conserved residues are highlighted in green, and conservative substitutions in cyan. The AdoMet-binding motif at the N terminus is labeled YFxxY and the glutamic acid and two methionine, which is important for methyltransferase function, shown in pink. Brown asterisk, AdoMet/AdoHcy–ribosyl H-bonding residues; red asterisk, first methionine position; blue asterisk, AdoHcy–carboxylate–arginine interactions; pink asterisk, AdoHcy–amine nitrogen H-bonding residues; green asterisk, AdoHcy–adenine nitrogen H-bonding residues. Tb, Trypanosoma brucei; Ce, Caenorhabditis elegans; Mm, Mus musculus; Rn, Rattus norvegicus; Hs, Homo sapiens and Xl, Xenopus laevis. The alignment was performed using the PROMALS3D multiple sequence and structure alignment server. b Typical type I PRMT from Danio rerio shows the six signature motifs in the active site that are important for enzyme function: I (yellow), post-I (green), II (brown), III (blue), conserved double-E SExMGxxLxxExM motif (black) and THW (pink). The FYxxY motif, which is part of the N-terminal αX helix that plays a key role in AdoMet-binding, is shown in orange. The dimerization arm is shown as a red dashed circle. Note that only the THW motif belongs to the β-barrel whereas all remaining important motifs exclusively belong to the Rossmann fold. For clarity, some of the helices of the Rossmann fold are hidden
The AdoMet-binding pocket of type-I PRMTs is highly buried and occluded. The electron density of the αX helix of RnPRMT1–AdoHcy–peptide complex is absent from the crystal structure, resulting in an exposed active site suitable as a channel for peptidyl arginine entry [53]. The superposition model of the Rattus norvegicus PRMT3 (RnPRMT3)–AdoHcy complex on the RnPRMT1–AdoHcy–peptide complex showed that the αX helix (residues 208–222) visibly occludes the peptidyl arginine entry (Fig. 3a). However, the three aromatic rings of YFxxY motif stack with the adenine ring of AdoMet and the αX helix folds onto AdoMet. YFxxY forms a hydrogen bond with conserved Glu335 of the double-E loop, which participates in correct positioning of peptidyl arginine (Fig. 3b) [45, 55, 86,87,88]. A similar interaction network has been described for the MmPRMT4–AdoHcy complex. Mutations in the second Tyr of the YFxxY motif (Tyr154 of MmPRMT4), which interacts with Glu267 (corresponding to RnPRMT3 Glu335), abolish the AdoMet-binding ability of MmPRMT4, suggesting that structural changes in the N-terminal helices (αX, αY and αZ) are necessary for the catalytic pathway [20, 55, 87].

The active site of PRMTs. aRnPRMT3 is superimposed on RnPRMT1 (blue ribbon and surface view) whose αX helix density is missing in the crystal structure. The N terminus of αX helix surface residues 208–222 (red ribbon and surface view) from RnPRMT3 occludes the P2 peptidyl arginine entry (green coil). The AdoHcy (stick form) are shown in yellow. P2 is one of the three-substrate entry channels based on the structure of the RnPRMT1–AdoHcy–peptide complex. bRnPRMT2–AdoHyc complex (PDB ID: 1F3L) shows H bonding between the αX helix (green) and the second glutamic acid (yellow) of the double-E loop. AdoHcy is shown in black. Interaction details are shown in zoomed boxes. All H bond interactions are labeled. c The electrostatic surface potential of the RnPRMT1–R3 dimer peptide complex (PDB ID: 1OR8) shows three peptide entry channels: P1 (close to the methionine moiety of AdoHcy), P2 (close to the adenine moiety of AdoHcy) and P3 (on the β-barrel). The peptide backbone is shown as green coil
Superposition of the apo-TbPRMT6 and TbPRMT6–AdoHcy structures revealed several rearrangements upon AdoHcy binding, while the isothermal titration calorimetry (ITC) data showed that the H4 peptide binds TbPRMT6 with a Kd value of 45 µM in the presence of AdoHcy [57]. By contrast, no binding was observed for the TbPRMT6 alone demonstrating that AdoHcy binding greatly enhances the affinity of the substrate peptide for TbPRMT6 [57]. It is noteworthy to pinpoint that the observation of the AdoMet-binding-induced structural changes in PRMTs is in good accordance with kinetics study of PRMT1 catalysis which shows that the formation of the catalytically competent enzyme–AdoMet–substrate ternary complex follows a kinetically ordered pathway, with AdoMet binding initially and substrate binding subsequently [65, 89]. Reciprocally, after the methyl group transfer, the product release follows a sequential order of peptide product releasing first and then AdoHcy [90, 91].
The AdoMet-binding pocket
AdoMet is a small compound that acts as a methyl group donor. After methyl transfer, its product is AdoHcy [92,93,94]. Because their chemical difference is minor and AdoHcy is chemically more stable than AdoMet, AdoHcy is often used in structural studies of PRMTs (Table 1). Although AdoMet and AdoHcy are frequently referred to in the literature as cofactors, they are actually co-substrate and co-product, respectively. Both AdoMet and AdoHcy are made up chemically of four parts: (1) an adenine ring, (2) a ribosyl ring, (3) a sulfur atom that holds the methyl group, and (4) amine and carboxylate tails. In general, the adenine ring of AdoHcy points towards the N terminus of the PRMT Rossmann fold and is stabilized by the aromatic-ring stacking interactions with the YFxxY motif from the αX helix and by hydrogen bonding with residue S/T/E/D, located opposite the αX helix (Fig. 2a and Table 2). Met155, located at the bottom of the adenine portion of the AdoMet-binding pocket provides steric constraint within the pocket, and plays a very critical role in binding of AdoMet to RnPRMT1, whereas a M155A mutant has a 27-fold higher Kd [95]. A comparison of representative of type I, II and III PRMT–AdoHcy complexes showed that a conserved glutamate residue (Glu100 in RnPRMT1) from the post-I signature motif participates in the formation of a hydrogen bond with the ribosyl moiety in all PRMTs, although human and rat PRMT3 (HsPRMT3 and RnPRMT3) have a conservative substitution in which aspartic acid replaces glutamate (Fig. 2a and Table 2). Mutagenesis followed by kinetic studies showed that this conserved glutamate–ribosyl hydrogen bonding has no effect on AdoMet binding but influences the catalytic efficiency of MmPRMT1 [95]. The second most preferred ribosyl stabilizing residue is histidine, which is also conserved among PRMTs with few exceptions, such as PRMT5, in which it is substituted by a tyrosine residue (Fig. 2a and Table 2). The amine tail of AdoMet hydrogen bonds with the backbone of a highly conserved glycine from signature motif I and the side chain of an aspartic acid residue, except in PRMT5, which lacks the aspartic acid residue in this position (Fig. 2a and Table 2). In XlPRMT5, the conserved Glu440 hydrogen bonds with the AdoMet–amine tail, which is unique in the PRMT5 family, although it may be an artifact of the crystallizations conditions [53]. The residue in the hydrogen bonding responsible for stabilizing terminal AdoMet–carboxylate interactions is R/K/H/Y/S/N/T/G among all methyltransferases [96]. However, type I/III and type II PRMTs preferably interact with the carboxylate tail via positively charged residues, such as arginine (types I/III PRMTs) and lysine (type II PRMTs), that project from the αZ helix (Fig. 2a and Table 2) [96]. Mutation of the RnPRMT1 Arg54 that interacts with AdoMet–carboxylate tail lead to 42-fold decrease in the methyltransferase catalytic efficiency, however, the Arg54 is not critical for the AdoMet binding [95]. For both PRMT type I and III, there is second hydrogen bond interaction via Ser/Thr residue present on motif I loop (Fig. 2a and Table 2). Based on sequence alignment and structural comparisons, a tyrosine residue is present within the hydrogen bond distance to AdoMet–carboxylate tail in human and C. elegans PRMT5.
Peptidyl arginine substrate recognition
The structural mechanism underlying arginine substrate recognition in PRMTs is poorly understood. Only peptide bound RnPRMT1, HsPRMT5 and TbPRMT7 structures have been deposited in the PDB and only partial peptide density is clearly revealed in these structures (Table 3) [52, 53, 61, 97]. The arginine substrate pocket of all PRMTs is endowed with two universally conserved glutamic acid residues (the “double-E loop”) such that the catalytic surface is negatively charged and thus is able to trap the positively charged guanidinium group of the target arginine residue. The widely accepted catalytic mechanism of PRMTs involves these two glutamate residues, which not only modulate the nucleophilicity of the reaction but also position the guanidine to allow methyl group transfer from AdoMet [45, 95]. Glu153 (RnPRMT1 numbering) from the double-E loop contributes to the alignment via an electrostatic interaction whereas Glu144 participates via hydrogen bonding [95]. The study also suggested that the PRMT1-catalyzed reaction is primarily driven by the alignment of the guanidine with the methyl group of AdoMet and that the deprotonation of guanidine for nucleophilic attack is not critical [95].
In the crystal structure of the RnPRMT1–AdoHcy–R3 peptide complex, only reactive arginine side chain and part of peptide backbone density are visible in the crystal structure (Table 3) [53]. Therefore, only the position of arginine substrate and part of the peptide backbone are traceable in the complex structure. Nevertheless, this information provides several clues regarding PRMT1 substrate recognition. Interactions between R3 peptide and PRMT1 occur primarily through the peptide’s backbone and the residues of the double-E loop and THW motif of PRMT1. His293 from the conserved THW motif forms a hydrogen bond with the backbone carboxyl of the R3 peptide whereas the backbone carboxyls of Glu144/Glu153 form hydrogen bonds with the guanidinium group of arginine (Fig. 4a). The catalytic efficiency of H293Q and H293A mutants for the substrate peptide and AdoMet decrease by 110-/125-fold and 256-/50-fold, respectively, compared to the wild type [95]. The involvement of residues from the THW motif and residues from the double-E loop for peptidyl arginine recognition in PRMT1 could be found for all the type I PRMTs; however, more type I PRMT–peptide complexes would be inevitably required to make further statements.

PRMT substrate recognition at the AdoHcy-binding pocket and hydrogen bond interactions. a The RnPRMT1–AdoHcy–R3 peptide complex, bHsPRMT5–OXU–H4 peptide complex and cTbPRMT7–AdoHcy–H4 peptide complex. In a–c, the active site residues, AdoHcy(or OXU) and substrate arginine are shown in stick model in green, black and cyan, respectively, and labeled. d Comparisons of the sequence of the RnPRMT1, HsPRMT5 and TbPRMT7 active-site residues and of the interactions of these residues with AdoHcy and the arginine substrate. Important motifs are underlined in black. The asterisk represents the SExMGxxLxxExM motif
Interestingly, the RnPRMT1–R3 peptide complex revealed three potential substrate entry channels (P1, P2 and P3) (Fig. 3c) [53]. The presence of multiple substrate entry channels may explain why PRMTs are able to methylate an arginine embedded within many different amino acid sequences. The surface potential of PRMT1 shows that the surface is covered by highly acidic patches favorable for binding positively charged arginine peptides (Fig. 3c) [53]. 19 acidic residues within the range of these three proposed channels have been systemically mutated for validation [98]. Single mutation of Glu46, Glu47 or Asp51 (near the P1 entry channel), Glu129, Asp164, Asp238 or Asp246 (near P2 entry) and Glu236 or Asp339 (near P3) to a positively charged residue dramatically reduces the activities of PRMT1 toward H4 peptide. Mutation of Glu47, Asp129 or Glu236 alters activity toward different protein substrates. Mutation of Asp238 or Asp246 reduces rPRMT1 binding to GST–GAR but has no effect on H4 peptide binding, whereas Asp236 mutation enhances H4 peptide binding but has no effect on GST–GAR binding. In general, these surface mutations support the idea of multi-substrate entry channels in PRMT1. Superposition of the RnPRMT1–R3 peptide complex on several type I PRMTs showed the very good fit of the substrate at the P1 entry channel at the surface of all type I PRMTs. However, this was not the case for the Mus musculus PRMT2 (MmPRMT2), Rattus norvegicus PRMT4 (RnPRMT4) and MmPRMT4 peptide complexes with the P3 substrate entry channel, as the steric clashes were observed. The fit was even worse for mPRMT6 with respect to both the P2 and P3 substrate entry channels.
The structure of the HsPRMT5–OXU–H4 peptide complex (PDB ID: 4GQB) clearly revealed the electron density of the first eight residues of the H4 peptide, including the substrate arginine at position 3. A close survey of the substrate binding area of that complex showed that the arginine guanidine of H4 forms salt bridges with Glu435 and Glu444 of the double-E loop (Fig. 4b). Glu435/444 are likely involved in deprotonating and activating the ω-N nitrogen atom for methyl transfer. Superposition of XlPRMT5–AdoHcy on HsPRMT5–OXU–H4 peptide showed the similar positions of the double-E loop residues. H4 peptide is also recognized primarily by the interactions of its peptide backbone with PRMT5 residues from the long (39 residues) flexible linker region connecting the N-terminal TIM barrel and the catalytic AdoMet methyltransferase domain. Importantly, these residues are highly conserved in PRMT5 (Fig. 2a). Phe580 in this recognition cluster is inclined towards the peptide backbone and the Leu312 backbone nitrogen forms a hydrogen bond with glycine at position 2 of the H4 peptide, thus also contributing substantially to the hydrophobic interactions at this site.
Beside interactions between PRMT5 and the peptide, the substrate recognition of vertebrate PRMT5 requires the participation of protein cofactors, such as MEP50, pICln or RioK1 [27, 30, 99]. Human and X. laveis PRMT5 form a complex with MEP50 consisting of a tetramer of heterodimers. HsPRMT5/ME50 complex shows positive cooperatively with the H4 substrate [100]. The presence of MEP50 is required for PRMT5 to methylate histones [101]. Disrupting the association with MEP50 by the phosphorylation of Tyr297, 304 and 306 of PRMT5 reduces the PRMT5 activities [102]. These kinetic data suggest MEP50 is involved in substrate interactions, which is further supported by low-resolution PRMT5–MEP50–nucleoplasmin complex by electron microscopy [82].
In the TbPRMT7–H4 peptide complex, only the first four residues of the H4 peptide have been revealed (Fig. 4c). Glu172 and Glu181 of the TbPRMT7 double-E loop show stereospecificities and interactions with guanidine similar to those of the HsPRMT5–H4 peptide complex (Fig. 4b, c). However, Gln329, which substitutes for histidine in the THW loop for the type I PRMT, forms a unique hydrogen bond with the substrate arginine. In addition, Thr176, which is part of the double-E loop, hydrogen bonds with the H4 peptide backbone [52]. Superposition of the TbPRMT7–AdoHcy complex with the TbPRMT7–AdoHcy–H4 peptide complex showed that movement of the αY helix and Asp70 move close to the peptide substrate and hydrogen bond with the Ser2 and Gly3 residues of the H4 peptide backbone [52]. This model of the downward movement of αY upon H4 peptide binding is supported by the finding that deletion of residues 33–55 of TbPRMT7 completely inactivates the enzyme [52]. Studies have shown that human and mouse PRMT7 specifically recognize RXRXR (or RXR) motif for arginine methylation [103, 104]. However, the TbPRMT7–H4 peptide structure overlaid with apo MmPRMT7 could not reveal the mechanism of RXRXR motif preference in mammalian PRMT7. One possible reason is that PRMT7 may also require more than one substrate entry channels. The active site of TbPRMT7 is surrounded by a cluster of hydrophobic and acidic residues. Contributing to these patches are residues from αX and αY helices, residues of the SExMGxxLxxExM motif (double-E loop), β7 strand of the β-barrel and conserved Trp330 from the THW motif (Fig. 2a). Residues of the signature motif SExMGxxLxxExM and the THW motif as well as residues of the β7 motif are hydrophobic and form the floor of the substrate-binding surface, whereas the αY helix residues pack the substrate from the top.
In summary, RnPRMT1–AdoHcy–R3, HsPRMT5–OXU–H4 and TbPRMT7–AdoHcy–H4 peptide complex structure consistently showed the importance of the double-E loop and THW motif of these PRMTs for substrate binding and recognition (Fig. 4d). However, the long linker in PRMT5 and the αY helix downward movement in PRMT7 clearly show the differences in substrate recognition among the type I, II and III PRMTs. In addition, all PRMT–peptide complex structures shown the presence of high population of glycine residues surrounding the arginine substrate [52, 53, 61, 97]. The presence of these glycine residues are thought to provide the conformational flexibility and less steric hindrance around peptidyl arginine around the peptidyl arginine that may be necessary for peptidyl substrate binding, because PRMTs have been shown to favor unstructured peptidyl regions, such as GAR and PGM motifs, loops or terminal parts of proteins [105].
PRMT methylation specificities and product selectivity
As noted in the introduction, PRMT type I, II and III also differ in their methylation reactions, which yield Rme2a, Rme2s and Rme1, respectively. Comparisons of their structures show that the peptidyl arginine is surrounded by two methionine residues in PRMT1 (type I), by phenylalanine/serine residues in PRMT5 (type II) and by methionine/alanine residues in PRMT7 (type III), which contribute to the product specificities PRMTs (Fig. 4) [51, 52, 83, 106, 107].
The conserved Met155 of type I PRMTs is an important determinant of Rme2a formation, as it provides steric bulk at the active site and prevents free rotation of the Rme1 intermediate (Fig. 4a) [45, 108]. Two methionine residues of PRMT1 (Met48 and Met155) were mutated to determine the specificity of the type I enzyme for Rme2a vs. Rme2s. Both mutations led to a higher ratio of Rme1 as the final product but neither produced Rme2s; hence M48 and M155 alone do not dictate Rme2a over Rme2s [106]. However, M48A activity was only 1.4% of that of the wild type (WT) but substitution with leucine, which has a bulkier side chain, increased activity to 40%. This result clearly indicated that conserved Met48 play a vital role in enzymatic activity and that smaller or larger side chains inhibit type I activity [106]. Met155, which is located very close to the substrate arginine, is highly conserved in all type I PRMTs but substituted by residues with a small side chain, such as alanine or serine, in type II and III PRMTs, respectively. This strongly suggests that the steric bulk provided by methionine blocks the binding of Rme1 in a conformation that would allow symmetric dimethylation (Fig. 4a) [45, 108].
In structural investigations of the several non-catalytically conserved residues in the vicinity of the PRMT active site, CePRMT5 was mutated with a view to understand if the surrounding residues near the catalytic site have any influence in controlling the product formation of symmetric demethylation [83]. Mutant F379 M (corresponding to F327 in HsPRMT5) exhibited increased enzyme activity whereas mutant F379Y was completely inactive, probably because the larger tyrosine interfered with AdoMet binding. Conversely, substitution of Phe379 with smaller side chain residues, such as alanine or glycine, severely reduced enzyme activity. Thus, residues larger or smaller than phenylalanine at position 379 make the catalytic site unfit for type II PRMT activity (Fig. 4b) [83]. In addition, mutation of the conserved Ser503 residue or a V188T/S669H double mutation greatly diminished enzymatic activity. Ser503, situated on the tip of the double-E loop, is involved in hydrogen bonding with the amide group of Phe671 from the THW motif. Both Val188 and Ser669 are located away from the active site, suggesting that inter-domain contacts are required to precisely position key residues for catalysis [83]. The same research group further showed that only the F379 M mutant produces both Rme2s and Rme2a as final products. The corresponding F327 M mutant in HsPRMT5 also produces Rme2s and Rme2a. Phe379 from αY is located near the arginine substrate but does not directly contact the guanidinium group. The ability of the F379 M mutant to conduct both symmetric and asymmetric dimethylation implies that (1) symmetric and asymmetric dimethylation share a common catalytic mechanism; (2) Phe379 occupies a critical position for Rme2s production specificity of PRMT5 [83].
In case of TbPRMT7, ITC assays were carried out to determine the affinities of the H4 peptide and the monomethylated H4R3me peptide. The 30-fold decrease in the Kd of the H4R3me peptide suggested that the PRMT7–guanidine-binding pocket is relatively small and cannot support the addition of a second methyl group as this would result in steric hindrance (Fig. 4c) [52]. Binding of the H4R3Rme1 peptide by the TbPRMT7 E181D mutant was sixfold higher than by the wild type. A double mutant in E181D (from the double-E loop) and Q329A (from the THW motif) converted TbPRMT7 to a PRMT that produced Rme2s. This mutation, therefore, resulted in the accommodation of a bulkier methylated peptide, due to increased space inside the binding pocket, which enabled a second methylation resulting in dimethylation reaction [51, 109].
The possibility that a single residue can alter product specificity originates from a Phe/Tyr switch model of lysine methyltransferases [108, 110, 111]. In general, the two conserved methionine residues of type I PRMTs discussed above (Met48 and Met155) sandwich the incoming guanidine in the active site to provide steric bulk. Although Met48 or Met155 mutation shifts product specificity to Rme1, neither mutant produces Rme2s. In type II PRMTs, the conserved Phe379 from αY does not contact the guanidinium group directly, but methionine substitution allows PRMT5 to produce both Rme2a and Rme2s. As for type III PRMTs, the Rme1 product specificity is due to a smaller active site that cannot accommodate a second methyl group.
PRMT inhibitor classification and mechanism of inhibition
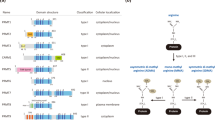
Clinical and preclinical studies showing a strong correlation between PRMT expression levels and the survival of cancer patients support the targeting of PRMTs in cancer treatment [112, 113]. Several PRMT chemical inhibitors have been developed in recent years from both academic laboratories and pharmaceutical companies worldwide (see recent reviews [91, 112, 114,115,116]). To date, 40 PRMT–inhibitor entries are deposited in the PDB databank. To simplify the discussion of these inhibitors, we have roughly divided them into five major classes (I–V). Most inhibitors belong to class I, II, or III, in which the AdoMet or arginine substrate-binding pocket or both pockets, respectively, are occupied (Fig. 5a–c) and Table 4). Class IV inhibitors inhibit PRMT5 selectively by blocking the αY/THW motif and partially the arginine substrate pocket (Fig. 5d). Class V inhibitors are different because they bind to the PRMT dimerization arm and influence enzyme activity allosterically (Fig. 5e). The single class V inhibitor (PDB ID: 4QPP) occupies the substrate entry position P3, but there is as yet no published investigations of 4QPP. In the following, we present examples of class I–V inhibitors to illustrate the general concept of inhibition.

Representative examples of PRMT class I–V inhibition. The surface maps are depicted with the inhibitor. The overlaid P1 entry peptide from RnPRMT1–peptide complex is shown as green coil to indicate position of substrate arginine inhibitor and PRMT surface is gray in color. Interaction details are shown in zoomed boxes. The inhibitor (black) and the interacting residues (cyan) shown in stick model. The water molecule is drawn as a red dot. The key interacting residues are labeled. a PRMT5–inhibitor complex (PDB ID: 6CKC) as an example for class I inhibition: AdoMet pocket inhibition. The TIM barrel of the PRMT5 surface has been hidden for clarity. b PRMT6–inhibitor complex (PDB ID: 4Y2H) as an example for class II inhibition: substrate pocket inhibition. c PRMT4–inhibitor complex (PDB ID: 5TBJ) as an example for class III inhibition: AdoMet and substrate arginine simultaneous inhibition. d PRMT5–inhibitor complex (PDB ID: 4X60) as an example of class IV inhibition: partial substrate pocket inhibition. e PRMT3–inhibitor complex (PDB ID: 4RLY) as an example of class V inhibition: dimerization arm inhibition
Class I The AdoMet analog LLY-283 is a selective PRMT5 inhibitor that occupies the AdoMet-binding pocket. Interactions of the adenine and ribose moieties of the inhibitor are similar to those described for PRMT5:MEP50 structures with AdoMet analogs [117]. Asp419 of PRMT5 hydrogen bonds with the adenine ring of LLY-283, and Glu392 and Tyr324 with its ribosyl moiety (Fig. 5a). LLY-283 was shown to potently inhibit PRMT5:MEP50 activity, as evidenced by an IC50 of 22 nM whereas its stereoisomer was 50-fold less inhibitory [117].
Class II EPZ0220411 is a class II inhibitor of PRMT6 that specifically occupies the arginine substrate binding region of the enzyme (Fig. 5b) [118]. The diamine side chain occupies the putative site of the substrate arginine side chain, and the terminal nitrogen is 3.4 Å away from the sulfur atom of AdoHcy. The terminal NH2 of EPZ0220411 directly hydrogen bonds with Glu155 whereas hydrogen bonding with Glu164 and Trp156 is water mediated. The pyrazole ring of this inhibitor forms hydrogen bonds with Glu59 and Tyr159 of the enzyme, and the tertiary amine of its diamine side chain with His317. The aryl ring of the inhibitor exhibits π–π interactions with Tyr159 (Fig. 5b). In tests of EPZ0220411 and a series of analogs against PRMT1 and PRMT8, IC50 values < 1 µM were reported [118]. Treatment with EPZ0220411 resulted in a dose-dependent decrease in H3R2 methylation (IC50 = 0.67 µM) [118].
Class III A series of 5-methylcytosine–adenosine complexes able to mimic the DNA methyltransferase transition state analog act as PRMT4 inhibitors in the micromolar range [119]. An analysis of co-crystals of PRMT4 with the class III inhibitor compound 4 (PDB ID: 5TBJ) revealed that the adenosine moiety of the inhibitor occupies the AdoMet pocket of the enzyme and the methylcytosine moiety accommodates the arginine substrate binding site (Fig. 5c) [119]. Compound 4 is clustered by a network of hydrogen bonds in which Glu244 and Ser272 of PRMT4 hydrogen bond with its adenine nitrogen while Glu215 and Gln160 connect with its ribosyl moiety (Fig. 5c). In the arginine pocket, Glu258 from the double-E loop of PRMT4 forms a hydrogen bond with the cytosine nitrogen of compound 4 whereas Tyr154 connects to its junction nitrogen. In addition, π–π interactions with the inhibitor are mediated by Trp416 and Tyr262 of PRMT4 (Fig. 5c). Compound 4 shows the strongest potency against PRMT4 in the inhibition assays and IC50 calculated 1.5 µM and 81% inhibition at 10 µM concentration. However, at this concentration, the inhibition of PRMT1, PRMT6 and PRMT7 was 13, 20 and 33%, respectively [119]. We have identified a set of diamidine compounds for selective inhibition of PRMT1 [120, 121]. Although no X-ray co-crystal structures have yet been obtained, molecular docking suggests that these inhibitors likely target the active site across both AdoMet- and arginine-binding regions.
Class IV These inhibitors especially act on PRMT5, positioning themselves in the vicinity of the clustered hydrophobic residues Phe327 (αY motif) and Trp579/Phe580 from the THW motif (Fig. 5d). Partial occupation of the substrate pocket and π–π interactions seem to be the dominant features of class IV inhibitor binding (Fig. 5d). However, Glu444 from the double-E loop of the enzyme is also involved in water-mediated hydrogen bonding with the inhibitor’s nitrogen [122]. Interestingly, the class IV inhibitor EPZ015666 does not bind to the apo form of PRMT5 whereas the presence of the aminonucleoside induces EPZ015666 binding, thus evidencing the AdoMet-uncompetitive mode of inhibition [122]. EPZ015666 has only modest affinity for AdoHcy-bound PRMT5 (Kd = 171 nm) whereas the affinity for AdoMet- or sinefungin-bound PRMT5 is much higher (Kd < 1 nM), indicating that the cation–π interaction of EPZ015666 and AdoMet contributes > 3 kcal mol−1 of binding energy [122]. EPZ015666 showed dose-dependent inhibition with nearly 95% tumor growth inhibition after 21 days in mantle cell lymphoma models [122].
Class V The mode of inhibition of the class V PRMT3 inhibitor 1-(benzo[d] [1,2,3] thiadiazol-6-yl)-3-(2-cyclohexenylethyl) urea differs from that of inhibitors of other classes. The inhibitor occupies the dimerization arm, where it exerts allosteric effects (Fig. 5e) [123], probably by preventing arginine peptide positioning in the catalytically active conformation. The PRMT3 residues Arg396, Glu422 and Thr466 are the major hydrogen bonding partners of the inhibitor and essential for inhibitor binding. Val420 also plays a vital role by providing hydrophobic interactions (Fig. 5e) [123]. Based on these findings, several PRMT3 inhibitors have been designed by optimizing allosteric inhibition [124,125,126]. They include SGC707, which has an IC50 of 31 nM and exhibits outstanding selectivity against 31 other protein-, DNA-, and RNA-methyltransferases [125].
Summary and perspectives
Although protein arginine methylation was first reported around 1970, it has only gained substantial interest in the past 10–20 years [127]. Recent efforts in structural and biochemical studies reveal key information of how PRMTs catalyze the methyl transfer reaction and how PRMTs achieve their substrate/product specificities. In addition, PRMT inhibitors have been designed for cancer treatment at preclinical and clinical trial stages. Due to technical limitations, the PRMT studies so far have largely relied on the PRMT core domains and peptide-based substrates. Since the N-terminal domain of PRMTs is critical for protein substrate recognition, it would be rational to hypothesize that the as-yet uncharacterized interactions between the N-terminal domain and respective protein substrates will steer which areas of a protein substrate to approach the active site pocket, resulting in the methylation of specific arginine residues in the protein substrate. Furthermore, PRMT protein substrates have different primary and secondary sequences, molecular shapes, and surface potentials. It is envisaged that the PRMT core domain may interact with varied substrates in different modes, thus providing additional means to alter PRMT substrate specificity. To date, the regulatory mechanisms of PRMT-catalyzed methylation of arginine residues on histone and various non-histone proteins remain elusive due to the lack of structural elucidation of full-length PRMT and protein substrate complexes. Moreover, the dynamic αX motif whose structural changes are critically needed for PRMT activity connects the N-terminal domain to the catalytic domain. Likely, interactive proteins that bind to the N-terminal domain may alter the αX conformation and thus allosterically regulates the enzymatic activity of PRMT. Recent development in methylarginine antibody and cryo-electron microscopy may provide enabling technology to approve or disapprove these putative mechanisms using full-length PRMTs and protein-based substrates.
References
Boffa LC, Karn J, Vidali G, Allfrey VG (1977) Distribution of NG, NG,-dimethylarginine in nuclear protein fractions. Biochem Biophys Res Commun 74:969–976
Lee HW, Kim S, Paik WK (1977) S-adenosylmethionine: methyltransferase. Purification and mechanism of the enzyme. Biochemistry 16:78–85
Zurita-Lopez CI, Sandberg T, Kelly R, Clarke SG (2012) Human Protein Arginine Methyltransferase 7 (PRMT7) Is a Type III Enzyme Forming omega-N-G-Monomethylated Arginine Residues. J Biol Chem 287:7859–7870
Dhayalan A, Kudithipudi S, Rathert P, Jeltsch A (2011) Specificity analysis-based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chem Biol 18:111–120
Horowitz S et al (2013) Conservation and functional importance of carbon-oxygen hydrogen bonding in AdoMet-dependent methyltransferases. J Am Chem Soc 135:15536–15548
Tsai YJ et al (2011) The predominant protein arginine methyltransferase PRMT1 is critical for zebrafish convergence and extension during gastrulation. FEBS J 278:905–917
Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, Clarke S, Herschman HR (2000) PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem 275:7723–7730
Pawlak MR, Scherer CA, Chen J, Roshon MJ, Ruley HE (2000) Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol Cell Biol 20:4859–4869
Cimato TR, Tang J, Xu Y, Guarnaccia C, Herschman HR, Pongor S, Aletta JM (2002) Nerve growth factor-mediated increases in protein methylation occur predominantly at type I arginine methylation sites and involve protein arginine methyltransferase 1. J Neurosci Res 67:435–442
Qi C, Chang J, Zhu YW, Yeldandi AV, Rao SM, Zhu YJ (2002) Identification of protein arginine methyltransferase 2 as a coactivator for estrogen receptor alpha. J Biol Chem 277:28624–28630
Meyer R, Wolf SS, Obendorf M (2007) PRMT2, a member of the protein arginine methyltransferase family, is a coactivator of the androgen receptor. J Steroid Biochem Mol Biol 107:1–14
Zhong J et al (2014) Nuclear loss of protein arginine N-methyltransferase 2 in breast carcinoma is associated with tumor grade and overexpression of cyclin D1 protein. Oncogene 33:5546–5558
Oh TG et al (2014) PRMT2 and ROR gamma expression are associated with breast cancer survival outcomes. Mol Endocrinol 28:1166–1185
Bachand F, Silver PA (2004) PRMT3 is a ribosomal protein methyltransferase that affects the cellular levels of ribosomal subunits. EMBO J 23:2641–2650
Swiercz R, Person MD, Bedford MT (2005) Ribosomal protein S2 is a substrate for mammalian PRMT3 (protein arginine methyltransferase 3). Biochem J 386:85–91
Di Lorenzo A, Bedford MT (2011) Histone arginine methylation. Febs Lett 585:2024–2031
Miyata S, Mori Y, Tohyama M (2010) PRMT3 is essential for dendritic spine maturation in rat hippocampal neurons. Brain Res 1352:11–20
Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR (1999) Regulation of transcription by a protein methyltransferase. Science 284:2174–2177
Feng Q, Yi P, Wong JM, O’Malley BW (2006) Signaling within a coactivator complex: methylation of SRC-3/AIB1 is a molecular switch for complex disassembly. Mol Cell Biol 26:7846–7857
Lee YH, Koh SS, Zhang X, Cheng XD, Stallcup MR (2002) Synergy among nuclear receptor coactivators: selective requirement for protein methyltransferase and acetyltransferase activities. Mol Cell Biol 22:3621–3632
Naeem H, Cheng DH, Zhao QS, Underhill C, Tini M, Bedford MT, Torchia J (2007) The activity and stability of the transcriptional coactivator p/CIP/SRC-3 are regulated by CARM1-dependent methylation. Mol Cell Biol 27:120–134
Xu W, Cho H, Kadam S, Banayo EM, Anderson S, Yates JR, Emerson BM, Evans RM (2004) A methylation-mediator complex in hormone signaling. Genes Dev 18:144–156
Wilczek C, Chitta R, Woo E, Shabanowitz J, Chait BT, Hunt DF, Shechter D (2011) Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2A and H4 and the histone chaperone nucleoplasmin in Xenopus laevis eggs. J Biol Chem 286:42221–42231
Migliori V et al (2012) Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol 19:136–144
Karkhanis V, Hu YJ, Baiocchi RA, Imbalzano AN, Sif S (2011) Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem Sci 36:633–641
Ancelin K, Lange UC, Hajkova P, Schneider R, Bannister AJ, Kouzarides T, Surani MA (2006) Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat Cell Biol 8:623–630
Guderian G, Peter C, Wiesner J, Sickmann A, Schulze-Osthoff K, Fischer U, Grimmler M (2011) RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J Biol Chem 286:1976–1986
Friesen WJ et al (2001) The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol Cell Biol 21:8289–8300
Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WAM, Cohen A, Lasonder E, Stunnenberg HG (2006) MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol 26:843–851
Friesen WJ, Wyce A, Paushkin S, Abel L, Rappsilber J, Mann M, Dreyfuss G (2002) A novel WD repeat protein component of the methylosome binds Sm proteins. J Biol Chem 277:8243–8247
Wang YX, Hu WH, Yuan YQ (2018) Protein arginine methyltransferase 5 (PRMT5) as an anticancer target and its inhibitor discovery. J Med Chem 61:9429–9441
Frankel A, Yadav N, Lee JH, Branscombe TL, Clarke S, Bedford MT (2002) The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J Biol Chem 277:3537–3543
Kleinschmidt MA, de Graaf P, van Teeffelen HAAM, Timmers HTM (2012) Cell cycle regulation by the PRMT6 arginine methyltransferase through repression of cyclin-dependent kinase inhibitors. Plos One 7:41446–41452
Phalke S et al (2012) p53-independent regulation of p21Waf1/Cip1 expression and senescence by PRMT6. Nucleic Acids Res 40:9534–9542
Stein C, Riedl S, Ruthnick D, Notzold RR, Bauer UM (2012) The arginine methyltransferase PRMT6 regulates cell proliferation and senescence through transcriptional repression of tumor suppressor genes. Nucleic Acids Res 40:9522–9533
Yoshimatsu M et al (2011) Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer 128:562–573
Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X, Reinberg D, Lee MG (2012) Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev 26:2749–2762
Gonsalvez GB, Tian L, Ospina JK, Boisvert FM, Lamond AI, Matera AG (2007) Two distinct arginine methyltransferases are required for biogenesis of Sm-class ribonucleoproteins. J Cell Biol 178:733–740
Karkhanis V, Wang L, Tae S, Hu YJ, Imbalzano AN, Sif S (2012) Protein arginine methyltransferase 7 regulates cellular response to DNA damage by methylating promoter histones H2A and H4 of the polymerase delta catalytic subunit gene, POLD1. J Biol Chem 287:29801–29814
Lee J, Sayegh J, Daniel J, Clarke S, Bedford MT (2005) PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J Biol Chem 280:32890–32896
Hernandez SJ, Dolivo DM, Dominko T (2017) PRMT8 demonstrates variant-specific expression in cancer cells and correlates with patient survival in breast, ovarian and gastric cancer. Oncol Lett 13:1983–1989
Yang YZ et al. (2015) PRMT9 is a type II methyltransferase that methylates the splicing factor SAP145. Nat Commun 6:6428–6439
Najbauer J, Johnson BA, Young AL, Aswad DW (1993) Peptides with sequences similar to glycine, arginine-rich motifs in proteins interacting with RNA are efficiently recognized by methyltransferase(s) modifying arginine in numerous proteins. J Biol Chem 268:10501–10509
Bedford MT, Clarke SG (2009) Protein arginine methylation in mammals: who, what, and why. Mol Cell 33:1–13
Zhang X, Zhou L, Cheng X (2000) Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J 19:3509–3519
Schapira M, Ferreira de Freitas R (2014) Structural biology and chemistry of protein arginine methyltransferases. Medchemcomm 5:1779–1788
Timm DE, Bowman V, Madsen R, Rauch C (2018) Cryo-electron microscopy structure of a human PRMT5:MEP50 complex. Plos One 13(3):01932205–0193218
Cura V, Troffer-Charlier N, Wurtz JM, Bonnefond L, Cavarelli J (2014) Structural insight into arginine methylation by the mouse protein arginine methyltransferase 7: a zinc finger freezes the mimic of the dimeric state into a single active site. Acta Crystallogr Sect D-Biol Crystallogr 70:2401–2412
Chen YY, Wang BQ, Lan XX, Zhou ZY, Wu XQ (2018) Increased protein arginine methyl transferase 7 expression is correlated with the occurrence and development of endometrial carcinoma. Int J Clin Exp Med 11:4883–4890
Hasegawa M, Toma-Fukai S, Kim JD, Fukamizu A, Shimizu T (2014) Protein arginine methyltransferase 7 has a novel homodimer-like structure formed by tandem repeats. FEBS Lett 588:1942–1948
Debler EW, Jain K, Warmack RA, Feng Y, Clarke SG, Blobel G, Stavropoulos P (2016) A glutamate/aspartate switch controls product specificity in a protein arginine methyltransferase. Proc Natl Acad Sci USA 113:2068–2073
Wang CY et al (2014) structural determinants for the strict monomethylation activity by trypanosoma brucei protein arginine methyltransferase 7. Structure 22:756–768
Zhang X, Cheng X (2003) Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 11:509–520
Cura V et al (2017) Structural studies of protein arginine methyltransferase 2 reveal its interactions with potential substrates and inhibitors. FEBS J 284:77–96
Troffer-Charlier N, Cura V, Hassenboehler P, Moras D, Cavarelli J (2007) Functional insights from structures of coactivator-associated arginine methyltransferase 1 domains. EMBO J 26:4391–4401
Bonnefond L, Stojko J, Mailliot J, Troffer-Charlier N, Cura V, Wurtz JM, Cianferani S, Cavarelli J (2015) Functional insights from high resolution structures of mouse protein arginine methyltransferase 6. J Struct Biol 191:175–183
Wang C et al (2014) Crystal structure of arginine methyltransferase 6 from Trypanosoma brucei. PLoS One 9:e87267
Wu H et al (2016) Structural basis of arginine asymmetrical dimethylation by PRMT6. Biochem J 473:3049–3063
Cheng Y, Frazier M, Lu FL, Cao XF, Redinbo MR (2011) Crystal structure of the plant epigenetic protein arginine methyltransferase 10. J Mol Biol 414:106–122
Weiss VH, McBride AE, Soriano MA, Filman DJ, Silver PA, Hogle JM (2000) The structure and oligomerization of the yeast arginine methyltransferase, Hmt1. Nat Struct Biol 7:1165–1171
Antonysamy S et al (2012) Crystal structure of the human PRMT5:MEP50 complex. Proc Natl Acad Sci USA 109:17960–17965
Toma-Fukai S et al (2016) Novel helical assembly in arginine methyltransferase 8. J Mol Biol 428:1197–1208
Lin WJ, Gary JD, Yang MC, Clarke S, Herschman HR (1996) The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J Biol Chem 271:15034–15044
Lee WC et al (2015) Protein arginine methyltransferase 8: tetrameric structure and protein substrate specificity. Biochemistry 54:7514–7523
Feng Y, Xie N, Jin M, Stahley MR, Stivers JT, Zheng YG (2011) A transient kinetic analysis of PRMT1 catalysis. Biochemistry 50:7033–7044
Klug A (1999) Zinc finger peptides for the regulation of gene expression. J Mol Biol 293:215–218
Hall TM (2005) Multiple modes of RNA recognition by zinc finger proteins. Curr Opin Struct Biol 15:367–373
Brown RS (2005) Zinc finger proteins: getting a grip on RNA. Curr Opin Struct Biol 15:94–98
Matthews JM, Sunde M (2002) Zinc fingers–folds for many occasions. IUBMB Life 54:351–355
Swiercz R, Person MD, Bedford MT (2005) Ribosomal protein S2 is a substrate for mammalian PRMT3 (protein arginine methyltransferase 3). Biochem J 386:85–91
Frankel A, Clarke S (2000) PRMT3 is a distinct member of the protein arginine N-methyltransferase family. conferral of substrate specificity by a zinc-finger domain. J Biol Chem 275:32974–32982
Blomberg N, Baraldi E, Nilges M, Saraste M (1999) The PH superfold: a structural scaffold for multiple functions. Trends Biochem Sci 24:441–445
Ball LJ, Jarchau T, Oschkinat H, Walter U (2002) EVH1 domains: structure, function and interactions. FEBS Lett 513:45–52
Lemmon MA, Ferguson KM, Abrams CS (2002) Pleckstrin homology domains and the cytoskeleton. FEBS Lett 513:71–76
Gervais V et al (2004) TFIIH contains a PH domain involved in DNA nucleotide excision repair. Nat Struct Mol Biol 11:616–622
She M, Decker CJ, Sundramurthy K, Liu Y, Chen N, Parker R, Song H (2004) Crystal structure of Dcp1p and its functional implications in mRNA decapping. Nat Struct Mol Biol 11:249–256
Lemmon MA (2007) Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp 74:81–93
Shishkova E, Zeng H, Liu F, Kwiecien NW, Hebert AS, Coon JJ, Xu W (2017) Global mapping of CARM1 substrates defines enzyme specificity and substrate recognition. Nat Commun 8:15571
Berberich H et al (2017) Identification and in silico structural analysis of Gallus gallus protein arginine methyltransferase 4 (PRMT4). FEBS Open Biol 7:1909–1923
Sayegh J, Webb K, Cheng D, Bedford MT, Clarke SG (2007) Regulation of protein arginine methyltransferase 8 (PRMT8) activity by its N-terminal domain. J Biol Chem 282:36444–36453
Antonysamy S et al (2012) Crystal structure of the human PRMT5:MEP50 complex. Proc Natl Acad Sci USA 109:17960–17965
Ho MC et al. (2013) Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. Plos One 8(2):57008–57023
Sun LT, Wang MZ, Lv ZY, Yang N, Liu YF, Bao SL, Gong WM, Xu RM (2011) Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc Natl Acad Sci USA 108:20538–20543
Yang YZ, Bedford MT (2013) Protein arginine methyltransferases and cancer. Nat Rev Cancer 13:37–50
Fuhrmann J, Clancy KW, Thompson PR (2015) Chemical biology of protein arginine modifications in epigenetic regulation. Chem Rev 115:5413–5461
Morales Y, Caceres T, May K, Hevel JM (2016) Biochemistry and regulation of the protein arginine methyltransferases (PRMTs). Arch Biochem Biophys 590:138–152
Yue WW, Hassler M, Roe SM, Thompson-Vale V, Pearl LH (2007) Insights into histone code syntax from structural and biochemical studies of CARM1 methyltransferase. EMBO J 26:4402–4412
Feng Q, He B, Jung SY, Song YC, Qin J, Tsai SY, Tsai MJ, O’Malley BW (2009) Biochemical control of CARM1 enzymatic activity by phosphorylation. J Biol Chem 284:36167–36174
Obianyo O, Osborne TC, Thompson PR (2008) Kinetic mechanism of protein arginine methyltransferase 1. Biochemistry 47:10420–10427
Hu H, Luo C, Zheng YG (2016) Transient kinetics define a complete kinetic model for protein arginine methyltransferase 1. J Biol Chem 291:26722–26738
Fulton MD, Brown T, Zheng YG (2018) Mechanisms and inhibitors of histone arginine methylation. Chem Rec 18:1792–1807
Bottiglieri T (1997) Ademetionine (S-adenosylmethionine) neuropharmacology: implications for drug therapies in psychiatric and neurological disorders. Expert Opin Investig Drugs 6:417–426
Schubert HL, Blumenthal RM, Cheng X (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci 28:329–335
Fontecave M, Atta M, Mulliez E (2004) S-adenosylmethionine: nothing goes to waste. Trends Biochem Sci 29:243–249
Rust HL, Zurita-Lopez CI, Clarke S, Thompson PR (2011) Mechanistic studies on transcriptional coactivator protein arginine methyltransferase 1. Biochemistry 50:3332–3345
Gana R, Rao S, Huang H, Wu C, Vasudevan S (2013) Structural and functional studies of S-adenosyl-l-methionine binding proteins: a ligand-centric approach. BMC Struct Biol 13:6
Mavrakis KJ et al (2016) Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351:1208–1213
Lee DY, Ianculescu I, Purcell D, Zhang X, Cheng X, Stallcup MR (2007) Surface-scanning mutational analysis of protein arginine methyltransferase 1: roles of specific amino acids in methyltransferase substrate specificity, oligomerization, and coactivator function. Mol Endocrinol 21:1381–1393
Pesiridis GS, Diamond E, Van Duyne GD (2009) Role of pICLn in methylation of Sm proteins by PRMT5. J Biol Chem 284:21347–21359
Jain K, Jin CY, Clarke SG (2017) Epigenetic control via allosteric regulation of mammalian protein arginine methyltransferases. Proc Natl Acad Sci USA 114:10101–10106
Burgos ES, Wilczek C, Onikubo T, Bonanno JB, Jansong J, Reimer U, Shechter D (2015) Histone H2A and H4 N-terminal tails are positioned by the MEP50 WD repeat protein for efficient methylation by the PRMT5 arginine methyltransferase. J Biol Chem 290:9674–9689
Liu F et al (2011) JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 19:283–294
Feng Y et al (2013) Mammalian protein arginine methyltransferase 7 (PRMT7) specifically targets RXR sites in lysine- and arginine-rich regions. J Biol Chem 288:37010–37025
Feng Y, Hadjikyriacou A, Clarke SG (2014) Substrate specificity of human protein arginine methyltransferase 7 (PRMT7): the importance of acidic residues in the double E loop. J Biol Chem 289:32604–32616
Kolbel K et al (2012) Peptide backbone conformation affects the substrate preference of protein arginine methyltransferase I. Biochemistry 51:5463–5475
Gui S, Wooderchak WL, Daly MP, Porter PJ, Johnson SJ, Hevel JM (2011) Investigation of the molecular origins of protein-arginine methyltransferase I (PRMT1) product specificity reveals a role for two conserved methionine residues. J Biol Chem 286:29118–29126
Wu H et al (2016) Structural basis of arginine asymmetrical dimethylation by PRMT6. Biochem J 473:3049–3063
Cheng X, Collins RE, Zhang X (2005) Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct 34:267–294
Jain K, Warmack RA, Debler EW, Hadjikyriacou A, Stavropoulos P, Clarke SG (2016) Protein arginine methyltransferase product specificity is mediated by distinct active-site architectures. J Biol Chem 291:18299–18308
Collins RE et al (2005) In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. J Biol Chem 280:5563–5570
Takahashi YH, Lee JS, Swanson SK, Saraf A, Florens L, Washburn MP, Trievel RC, Shilatifard A (2009) Regulation of H3K4 trimethylation via Cps40 (Spp1) of COMPASS is monoubiquitination independent: implication for a Phe/Tyr switch by the catalytic domain of Set1. Mol Cell Biol 29:3478–3486
Hu H, Qian K, Ho MC, Zheng YG (2016) Small molecule inhibitors of protein arginine methyltransferases. Expert Opin Investig Drugs 25:335–358
Poulard C, Corbo L, Le Romancer M (2016) Protein arginine methylation/demethylation and cancer. Oncotarget 7:67532–67550
Luo M (2015) Inhibitors of protein methyltransferases as chemical tools. Epigenomics 7:1327–1338
Kaniskan HU, Konze KD, Jin J (2015) Selective inhibitors of protein methyltransferases. J Med Chem 58:1596–1629
Qian K, Zheng YG (2016) Discovery and development of small molecule epigenetic drugs. Chap 8, Current development of protein arginine methyltransferase inhibitors. Epi-informatics: discovery and development of small molecule epigenetic drugs and probes, pp 231–256
Bonday ZQ et al (2018) LLY-283, a potent and selective inhibitor of arginine methyltransferase 5, PRMT5, with antitumor activity. ACS Med Chem Lett 9:612–617
Mitchell LH et al (2015) Aryl pyrazoles as potent inhibitors of arginine methyltransferases: identification of the first PRMT6 tool compound. Acs Med Chem Lett 6:655–659
Halby L et al. (2018) Hijacking DNA methyltransferase transition state analogues to produce chemical scaffolds for PRMT inhibitors. Philos Trans R Soc Lond B Biol Sci 373:1748–1762
Yan L et al (2014) Diamidine compounds for selective inhibition of protein arginine methyltransferase 1. J Med Chem 57:2611–2622
Zhang J, Qian K, Yan C, He M, Jassim BA, Ivanov I, Zheng YG (2017) Discovery of decamidine as a new and potent PRMT1 inhibitor. Medchemcomm 8:440–444
Chan-Penebre E et al (2015) A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 11:432
Siarheyeva A et al (2012) An allosteric inhibitor of protein arginine methyltransferase 3. Structure 20:1425–1435
Kaniskan HU et al (2018) Discovery of potent and selective allosteric inhibitors of protein arginine methyltransferase 3 (PRMT3). J Med Chem 61:1204–1217
Kaniskan HU et al (2015) A potent, selective and cell-active allosteric inhibitor of protein arginine methyltransferase 3 (PRMT3). Angewandte Chemie-Int Edit 54:5166–5170
Liu F et al (2013) Exploiting an allosteric binding site of PRMT3 yields potent and selective inhibitors. J Med Chem 56:2110–2124
Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439:811–816
Acknowledgements
S.K.T. was supported by Taiwan Protein Project (Grant no. AS-KPQ-105-TPP). Y.G.Z. was supported by NIH Grant R01GM126154. M.C.H. was supported by the Academia Sinica and Ministry of Science and Technology (MOST 107-2311-B-001-002).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tewary, S.K., Zheng, Y.G. & Ho, MC. Protein arginine methyltransferases: insights into the enzyme structure and mechanism at the atomic level. Cell. Mol. Life Sci. 76, 2917–2932 (2019). https://doi.org/10.1007/s00018-019-03145-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03145-x