
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that affects normal functions of the brain. Currently, AD is one of the leading causes of death in developed countries and the only one of the top ten diseases without a means to prevent, cure, or significantly slow down its progression. Therefore, newer therapeutic concepts are urgently needed to improve survival and the quality of life of AD patients. Microtubule affinity-regulating kinases (MARKs) regulate tau-microtubule binding and play a crucial role in neurons. However, their role in hyperphosphorylation of tau makes them potential druggable target for AD therapy. Despite the relevance of MARKs in AD pathogenesis, only a few small molecules are known to have anti-MARK activity and not much has been done to progress these compounds into therapeutic candidates. But given the diverse role of MARKs, the specificity of novel inhibitors is imperative for their successful translation from bench to bedside. In this regard, a recent co-crystal structure of MARK4 in association with a pyrazolopyrimidine-based inhibitor offers a potential scaffold for the development of more specific MARK inhibitors. In this manuscript, we review the biological role of MARKs in health and disease, and draw attention to the largely unexplored area of MARK inhibitors for AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The last few decades have seen a tremendous life-span improvement due to better healthcare; however, escalating population and unhealthy lifestyles have brought in new challenges to age-related disorders in the elderly population. Alzheimer’s disease (AD) is one of the most prevalent forms of age-related disorders, clinically characterized by dementia and a progressive loss of mental, behavioral, and functional decline of the brain. It has become the leading cause of death in elderly over the age of 60 compared to other diseases like cancers, heart diseases, stroke, and others [1].
Historically, the primary hallmarks of AD are the formation of Hirano’s bodies, neurofibrillary tangles (NFTs), senile plaques, and cortical Lewy bodies [2,3,4,5,6]. Extracellular senile plaques of amyloid-beta (Aβ) mainly accumulate in the neocortical areas followed by deposition in allocortical regions, entorhinal cortex, and limbic structures [4, 5]. In the advanced stages, Aβ deposits are also observed in the subpial surface of the cerebral cortex [4]. However, unlike Aβ, the intracytoplasmic NFT deposits of hyperphosphorylated microtubule-associated protein (MAP) tau show a conventional pattern of accumulation [4]. NFTs first start to accumulate in the perirhinal and entorhinal regions of the cortex followed by CA1 region of the hippocampus, limbic system, and isocortical areas (reviewed extensively in Ref. [4]).
Tau is primarily found in abundance in axons of neurons and plays a key role in regulating microtubule (MT) dynamics, axonal growth, and a number of other MT-dependent neuronal functions [7, 8]. Under normal conditions, the physiological functions of tau are tightly regulated by alternating cycles of phosphorylation and dephosphorylation [9]. However, a dysfunction in this cycle is believed to be responsible, in part, for the pathological alterations in AD [10, 11]. While tau hyperphosphorylation is the most widely studied post-translational modification, tau can also undergo a number of other modifications that alter its MT-binding affinity [12]. Protein kinases that play a major role in tau hyperphosphorylation include glycogen synthase kinase 3 (GSK-3), cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAPKs), and MT affinity-regulating kinases (MARKs) [11, 13, 14]. Among these tau kinases, MARKs have recently gained significant interest due to their role in Aβ- and tau-mediated toxicity. Although MARKs have also been associated with cancers and metabolic disorder, to limit the scope of this review, we mainly focus on their association to tau pathology in AD. In addition, we present an update on small molecule inhibitors of MARKs that have the potential to develop into promising lead candidates.
Biological functions of MARKs
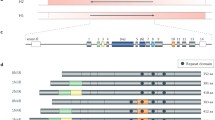
Microtubule affinity-regulating kinases belong to the calcium/calmodulin-dependent protein kinase superfamily and phosphorylate tau at AD-specific Ser262 site on KXGS motif located in the MT-binding domain (Fig. 1a) [15]. The human MARK family consists of four members (MARK1–4; Fig. 1b) that play a major role in a number of physiological processes [16,17,18,19,20,21,22]. MARK4 is an unusual member of the MARK family and lacks a hydrophobic pocket adjacent to the ATP-binding domain [23]. Alternate splicing of MARK4 gene results in the expression of two additional isoforms (MARK4L and MARK4S) that differ in carboxyl termini [19]. MARK1 and MARK2 expression levels are higher in fetal than adult tissues presumably due to the active proliferation of cells in developing tissues that require highly dynamics MTs [24]. MARK4L is highly expressed in testis, brain, kidney, liver, and lungs [25]. Although the expression of MARK4L remains relatively unchanged in postmitotic cells, upregulation of MARK4L in hepatocarcinomas and gliomas implicates MARK4 in neoplastic transformation [19, 26]. In contrast to MARK4L, MARK4S is predominantly expressed in the central nervous system (CNS) [25], in addition to other non-CNS tissues, such as testis and heart [27].

Full length tau and MARK isoforms expression in AD. a Bar diagram showing the structure of full length human tau40 (hTau40) with KXGS motifs (Ser262, Ser293, Ser324, and Ser356) phosphorylated by MARKs. The domain structure of MARK2 phosphorylated by LKB1/MARKK at Thr208 residue is shown. The bar diagrams were generated using IBS 1.0. b A summary of expression of different MARK isoforms in AD brain and normal tissue. NDE non-demented elderly
MARKs regulate key cellular functions
Overexpression of MARK2 and MARK4 in rat hippocampal neurons results in tau hyperphosphorylation, and loss of dendritic spines and synaptic markers [28, 29]. Similarly, MARK1 and MARK2 when overexpressed in Chinese hamster ovary CHO phosphorylate endogenous MAPs, causing their detachment from MTs [30]. Heterologous overexpression of MARK4S in hepatocytes reduces their viability [26]. In contrast, blocking MARK2 activity inhibits neurite outgrowth and differentiation in mouse neuroblastoma N2a cells, indicating the role of MARK in regulating neuronal plasticity [31]. This is also evident following knockdown of MARK2 that induces axonal growth in rat hippocampal neurons [32]. Co-transfection of MARK with Par-3/Par-6/atypical protein kinase C (aPKC) reverses the toxic effect of MARK on axonal growth, presumably due to the inactivation of MARK effect on tau phosphorylation [32]. These data indicate that optimal level of MARK protein and activity is crucial for normal cellular functions.
Trinczek et al. show that in contrast to MARK kinase (MARKK) that shows cytoplasmic distribution, MARK4 co-localizes with MTs [15]. Proteomic analysis of protein complexes from cells suggests a potential interaction between MARK4 and γ-tubulin [33]. Furthermore, co-localization of MARK4 with clathrin-coated vesicles suggests that MARK also regulate MT-dependent cellular transport [34]. Overexpression of MARK4 (either S or L isoform) affects MT density, and silencing MARK4S alters centrosomes orientation in G1-arrested cells [35]. Additionally, MARK4L has been also reported to co-localize with vimentin [35]. Par-1b/MARK2 has also been reported to regulate the actin cytoskeleton [21], and a recent study shows the importance of MARK4 in regulating ciliogenesis [22]. Additionally, FEZ1, a kinesin-1 adapter, involved in neuronal transport is regulated by MARK/Par-1 through phosphorylation of a Ser58 regulatory site [36]. These studies clearly indicate to a broader role of MARK in regulating cell cytoskeleton.
Association of MARKs to tauopathy in AD
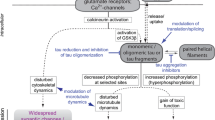
Although toxic effects of tau are frequently attributed to their aggregation into NFTs, this aggregation is preceded by a number of ‘pre-phosphorylation’ events involving MARKs and other tau kinases [37, 38]. Figure 2 shows a schematic of the role of MARKs in the events of tau hyperphosphorylation. MARKs are activated following their phosphorylation at Thr208 residue by upstream effector kinases, such as liver serine/threonine kinase B1 (LKB1) and MARKK [33, 39,40,41]. A recent study further shows that MARK/Par-1 is activated downstream of NMDA receptors in hippocampal neurons through protein kinase A-dependent phosphorylation of LKB1 Ser431 [42]. Activated MARKs phosphorylate tau at Ser262 and make MT-unbound/bound tau susceptible to hyperphosphorylation by other MAP kinases, such as GSK-3, CDKs, and MAPKs, including MARKs [43, 44]. These events are clearly evident in a study by Ando et al. [45] that shows that MARK/Par-1 initiates the phosphorylation of MT-unbound tau at Ser262/356 in a Drosophila model of AD. This ‘pre-phosphorylation’ stabilizes the less phosphorylated tau, making them more prone to Aβ-mediated processing into toxic tau species. Interestingly, co-expression of amyloid precursor protein (APP) and MARK/Par-1 results in the phosphorylation of Par-1 Thr408 (Drosophila Par-1 Thr408 is the same conserved site as Thr208 in MARK2) and tau [46]. This study also provides the first evidence to show how activated MARK/Par-1 is regulated in vivo [46].

Aβ-MARK-Tau axis of neurodegeneration. A schematic showing the role played by MARKs in the process of hyperphosphorylation of tau
Members of MARK3 family (EMK and C-TAK1) have also been reported to phosphorylate Cdc25c phosphatase, PTPH1 tyrosine phosphatase, MAPK-scaffolding KSR1 protein, plakophilin 2, and class IIa histone deacetylases [47,48,49,50]. C-TAK1-induced phosphorylation of histone deacetylases alters their subcellular localization, which is associated with a number of disorders [51, 52]. Additionally, C-TAK1 partially alters the activity of protein phosphatase 2 (PP-2), resulting in the inhibition of GSK-3β-mediated phosphorylation of PP-2 [53]. Defects in PP-2 activity and function are well-reported to cause tau hyperphosphorylation in AD [54]. Intriguingly, MARKs have also been reported to phosphorylate doublecortin, an MAP implicated in X-linked lissencephaly [55].
Given the key role of MARKs, it is not surprising that AD and non-demented elderly (NDE) brains show significant differences in the levels of individual MARK isoforms [16]. Gu et al. report an increased co-localization of phosphorylated tau with MARK2 in AD brains compared to NDE controls [56]. Although at mRNA level, all isoforms of MARK show a uniform neuronal distribution. At the protein level, only MARK1 and MARK2 are evenly distributed in the cytoplasm and neuropils of NDE and AD brains [37]. MARK3 shows only a weak neuronal cytoplasmic staining in AD and NDE brains, in contrast to MARK4, which is largely absent in NDE neuronal cytoplasm [37]. Elevated levels of MARK4 protein and MARK4-tau interaction in AD brains correlate with Braak stages of the disease, directly implicating MARK4 to AD progression [16]. This is also indicated by a progressive accumulation of phosphorylated MARK4 and Ser262-phosphorylated tau in granulovacuolar degeneration bodies (GVDs) of AD brains [16, 37]. In contrast to MARK4, only a subset of GVD-containing neurons show the presence of MARK3 [37].
MARKs in cancers and metabolic disorder
In addition to AD, overexpression of MARKs in different cancer types is associated with metastasis and chemoresistance [26, 27, 57,58,59]. MARK2 has been associated with cisplatin resistance in lung cancer cell lines [60], and a recent study by Hubaux et al. shows that MARK2 is altered at both DNA and RNA levels in non-small cell lung cancer patient cohorts [58]. Additional studies in lung cancer cell lines suggest that the oncogenic role of MARK2 is not kinase dependent, implicating MARK2 overexpression to cisplatin resistance [58]. The data from cell lines studies also correlate with clinical outcome of cisplatin therapy in lung cancer patients showing high expression of MARK2 [58, 60]. Pardo et al. [61] show that miR-515-5p directly regulates MARK4 activity at post-transcriptional level and results in the inhibition of migration of breast cancer cells. Interestingly, high miR-515-5p and low MARK4 expression levels correlate with increased survival of breast and lung cancer patients [61]. A recent study also shows that knockdown of MARK4 decreases tumorigenic properties of breast cancer MDA-MB-231 cells due to attenuation of MARK4-mediated inhibition of Hippo signaling [62].
In addition to cancers, other studies indicate a potential role of MARKs in metabolic disorders [20, 63, 64]. MARK4 knockout mice show increased metabolic activity and resistance to high fat diet-induced obesity [20, 65]. MARK4 induces oxidative stress and aggravates adipose inflammatory responses by activating the IKKα/NF-κB pathway [65]. However, transcriptional suppression of MARK4 by PPARγ significantly reduces the oxidative stress induced by MARK4.
Rationale to target MARKs for AD
The evidence from various studies clearly present the multifaceted role of MARKs under physiological and diseased conditions [32, 36, 42]. The data from the dual-gene Drosophila model indicate that MARKs act as initiators of tau hyperphosphorylation, suggesting their role in the early events of tauopathy [45]. This is also evident from the presence of MARK3 and MARK4 in NFTs of AD brains [16, 66, 67]. Therefore, altering the activity/overexpression of MARKs in AD may prevent MARK-mediated tau toxicity as evident in the study by Chen et al. [32]. Additionally, APP-mediated LKB1-induced activation of MARK/Par-1 suggests that MARKs might be the link that connect Aβ-tau pathologies in AD [46]. Knockdown of endogenous LKB1 prevents APP-mediated phosphorylation of MARK/Par-1 and tau toxicity [46], further suggesting the potential of targeting aberrant MARK activity for preventing tau-related toxicities. Besides, the involvement of MARK4 in tumorigenesis, metastasis, and drug resistance highlights the therapeutic potential of anti-MARK molecules for lung and breast cancer therapy [23, 61, 62]. In the section below, we present an update on currently known inhibitors of MARKs. Although a number of compounds are known to have anti-MARK activity (Fig. 3), their translation into potential anti-MARK drug candidate still lags behind due to under-exploration of these inhibitors.

Small molecule inhibitors of MARKs
Staurosporine
Staurosporine (STS) is a pan-kinase inhibitor that induces apoptosis and G1 cell cycle arrest in various cell types [68]. In addition to its effect on protein kinases C, A and G, STS alters the expression of cyclin-dependent kinase inhibitors, and leucine-rich repeat kinase 2 associated with Parkinson’s disease at nanomolar range [69]. STS treatment results in neurite outgrowth in dopaminergic neuron [70] and elevates choline acetyltransferase activity following basal forebrain lesion in cholinergic neurons [71]. In addition to its neurotropic effects in a number of cell types [70], STS has been reported to attenuate impaired learning in rats [71]. Recently, STS was shown to inhibit MARK-tau interaction, reducing tau phosphorylation at Ser262 in mouse embryonic 3T3 fibroblast cells [16, 56]. Moreover, there was no effect of STS on MARK2 and tau protein levels [56]. However, dose-limiting toxicities and poor pharmacokinetics (PK) [72,73,74,75] make STS a less likely choice for development as an anti-MARK therapeutic agent.
Interestingly, STS analog 7-hydroxystaurosporine (UCN-01) shows better anti-tumor effects in animal models of epidermoid carcinoma, fibrosarcoma, and acute myeloid leukemia over STS [74]. UCN-01 is currently under clinical trials for cancer therapy, in addition to another analog, N-benzoyl-staurosporine (PKC412) [76]. See Monnerat et al. [76] and Sausville et al. [77] for detailed PK properties of PKC412 and UCN-01, respectively. In addition to analogs, liposomal encapsulation has been reported to enhance the anti-tumor activity of STS in mice without resulting in adverse effects [78]. Moreover, liposomal encapsulation reduces the circulation half-life of STS in comparison to free STS [75, 78]. These properties make STS analogs and liposomal formulation promising compounds for further exploration in relation to MARKs. Although currently there are no data on anti-MARK effects of liposomal STS, a single study in a cell-free system reports the anti-MARK3 activity of UCN-01 [79].
Methylene blue
The therapeutic effects of methylene blue (MB), a phenothiazine dye, against a number of diseases including AD are well-reported in the literature [80,81,82]. MB permeates the blood–brain barrier (BBB) following either intravenous, intraperitoneal, or intraduodenal dosing; however, the level of MB uptake is higher in the CNS when administrated intravenously [83]. Although MB is known to target multiple AD-specific molecular targets [84], a recent study reported the first anti-MARK4 effects of MB in a fly model of MARK4/Par-1 overexpression [85]. MB decreased Par-1 protein levels at neuromuscular junctions and rescued synaptic loss without affecting the expression of human leucine-rich repeat serine/threonine-protein kinase 2, suggesting MB specifically targets MARK4/Par-1. Similarly, MB also downregulated MARK4-induced tau Ser262 phosphorylation in a cell-free assay and 293T cells expressing human 4R2N tau in a dose-dependent manner. The study also shows that MB inhibits tau phosphorylation at Ser396 and Ser214 in 293T cells, presumably due to MB effect on GSK-3β and CDK5. MB decreased MARK4 protein level plausibly by proteasomal degradation of ubiquitinated MARK4, suggesting that the ubiquitin–proteasome pathway is also a potential target of MB [85]. Since MB has been reported to induce autophagy and decrease tau protein levels [86], it is also likely that the observed effect of MB on MARK4 may have been partly due to the induction of autophagy [85].
Proline-directed hymenialdisine
The marine natural product hymenialdisine (HD01) was reported to competitively inhibit proline-directed kinases, such as GSK-3β- and CDK, and tau hyperphosphorylation at AD-specific sites [87, 88]. Additionally, HD01 inhibits non-proline-directed MARK2 at nanomolar concentration by binding at the catalytic pocket of MARK [31]. However, since the discovery of MARK inhibitory effects of HD01, there has been no progress in the development of HD01 as an anti-MARK agent. Although a number of HD01 analogs were synthesized [89], none of these compounds were reported to have any anti-MARK activity. Two other proline-directed potent GSK-3β inhibitors, SB-216763 and SD-415286, are known to interact with MARK3 and its activating kinase, MARKK [79, 88]. Although there are no additional data on anti-MARK activity, both SB-216763 and SD-415286 are known to have neuroprotective effects [90].
9-OXO-9H-acridin-10-yl compounds
The Mendelkow group at the Max-Planck-Unit for Structural Molecular Biology in Hamburg, Germany, reported the identification of four low-molecular weight anti-MARK2 compounds (30019, 30195, 30197, 30199) sharing a common 9-OXO-9H-acridin-10-yl functional group (Fig. 3) [88]. The compound 30019 inhibited all MARK isoforms in a dose-dependent manner. Further, the compounds were not effective against GSK-3β and showed little to no cross-reactivity against close relatives of MARK, such as SAD kinase B and AMP-activated protein kinase (AMPK). Abolition of MT disruption in CHO cells overexpressing MARK2 following treatment with the compounds suggests that this class of compounds possibly inhibit MARK2-mediated hyperphosphorylation of tau [88]. Interestingly, a recent study also highlights the specificity of 9-OXO-9H-acridin-10-yl derivatives for MARK4 and their presumable development into potent anti-prostate cancer candidates [23]. This provides a strong rationale for further exploration of 9-OXO-9H-acridin-10-yl derivatives given the role of MARKs in AD and cancers.
CagA peptide
A toxin of Helicobacter pylori, CagA, was shown to inhibit the activity of MARK/Par-1 and disrupt cell polarity in canine kidney MDCK cells [91]. CagA is also reported to affect a number of other kinases involved in different signaling pathways, including protein kinase C-related kinase 2 (PRK2) [91, 92]. CagA lacking the Par-1/MARK-binding domain continues to inhibit PRK2, suggesting that CagA interacts with PRK2 and Par-1/MARK via two different domains [92]. This is evident from the study of Neišić et al. that shows that a sub-domain of CagA containing a short chain of 14 amino acid occupies the substrate binding site of MARK2 [93]. A mutation in the amino acid sequence abolishes CagA interaction with MARK2. This peptide of CagA—termed as MARK2 kinase inhibitor (MKI)—inhibits basal kinase and MARKK-activated activity of MARK2 at micromolar range [93]. MKI dramatically inhibits MARK4 activity in rat hippocampal neurons, reducing tau hyperphosphorylation at Ser262 residue [28]. Additionally, MKI-mediated inhibition of MARK4 activity abrogates Aβ-mediated loss of dendritic spines and synapses in neurons [32]. The lack of effect of MKI on acetyl-CoA carboxylase phosphorylation by AMPK suggests that MKI specifically targets MARK/Par-1 [32]. Although the above studies provide encouraging evidence to develop MKI as an anti-MARK agent [28, 93], whether MKI will have any therapeutic benefits in MARK-directed AD therapy requires extensive studies.
Other known protein kinase inhibitors with anti-MARK effects
C16 is a known neuroprotective protein kinase R (PKR) inhibitor that prevents PKR-induced neuronal death and inflammatory cytokines release in animal models of brain injury [94, 95]. Additionally, C16 prevents nuclear translocation of Fas-associated protein with a death domain and Aβ-induced apoptosis in human neuroblastoma SH-SY5Y cells [96]. Intraperitoneal administration of C16 results in dose-dependent decrease in PKR phosphorylation in rat brain, indicating the ability of C16 to penetrate the BBB [95]. A recent study reports the promising effects of C16 as a lead anti-MARK4 candidate [92]. This inhibitory effect results from the interaction of C16 with the hydrophobic cavity of MARK4 [97].
Three other kinase inhibitors (BX-912, BX-795, and OTSSP167) already known to have anti-cancer effects were also reported to have anti-MARK activity (Fig. 3). The inhibitors have a similar binding pattern to MARK4 kinase domain [98]; however, OTSSP167 forms a more stable complex with MARK4 [98]. Not surprisingly, OTSSP167 results in better inhibition of MARK4 compared to BX-912, BX-795, and C16 [99]. A similar inhibitory effect of BX-795 was previously reported against MARK3 [79]. BX-795 and BX-919 are known inhibitors of 3-phosphoinositide-dependent kinase-1/AKT signaling [100], whereas, BX-795 also affects kinases that are involved in immune responses [79, 100, 101]. OTSSP167 inhibits maternal embryonic leucine zipper kinase overexpressed in a number of aggressive tumors presumably explaining the potent anti-cancer effects in different tumor cell types and xenograft models [102]. Although these data provide a new insight into anti-MARK4 activity of BX-912, BX-795, OTSSP167, and C16 [98], additional studies are necessary to advance these compounds as anti-AD candidates. Since these agents are already known to have anti-cancer effects, their potential use for reversing MARK-induced drug resistance in cancers will benefit cancer therapy.
Pyrazolopyrimidines are protein tyrosine kinase inhibitors with potent anti-tumor effects [103, 104]. Recently, Sloman et al. reported the preferential anti-MARK activity of a pyrazolopyrimidine inhibitor [105]. Furthermore, the derivative was reported to have better PK and cell anti-proliferative properties than the parental compound (see Fig. 3 for biochemical and cellular properties of this compound). A recent co-crystal structure study revealed that pyrazolopyrimidine interacts with MARK4 at the ATP site and in close proximity to a position that potentially alters the catalytic loop of MARK4 [106]. This is likely the reason for the potent MARK inhibitory effects of pyrazolopyrimidines [105, 106]. Given the improved PK properties and CNS penetration of the pyrazolopyrimidine derivative [105], additional studies in cell and animal models of AD are required to show the potential therapeutic benefits of pyrazolopyrimidines.
Targeting MARKs via indirect pathways: a perspective
The activity of MARK is regulated by activating kinases and interaction with scaffolding and other cellular proteins, making them potential indirect targets for altering the activity of MARKs [107, 108]. LKB1 regulates MARK activity by phosphorylating Thr residue at activation loop [40, 46]. Knockdown of LKB1 in mouse embryonic fibroblasts markedly reduces the catalytic activity of MARK1–4 [40]. In contrast, ectopic expression of LKB1 increases MARK4 phosphorylation [33]. Likewise, synthetic LKB1 peptides also alleviate the activity of all MARK types. Aβ-mediated increase in MARK activity occurs in a LKB1-dependent manner and contributes to tauopathy in AD [46]. These data indicate that LKB1 is a master regulator of MARK and interventions that can modulate LKB1 activity/expression may alter MARK-induced tau hyperphosphorylation [109]. Although LKB1 signaling is being investigated as druggable target, and LKB1 knockout has been reported to reduce MARK activity [40], a number of studies also show the deleterious effects of targeting LKB1 [110, 111]. This is not surprising given the role of LKB1 in several vital physiological functions.
Heterodimerization of MARK2 with protein kinase PAK5 results in the inhibition of MARK activity, indicating dimerization as another potential indirect mechanism for targeting MARK activity. This is in fact evident from the study of Sun et al. that suggests that MB-induced inhibition of MARK4 activity presumably results from the stabilization of covalently dimerized MARK4 [85]. The other mechanisms that are known to inhibit MARK activity are polyubiquitination and binding with scaffold protein 14-3-3 [112,113,114]. Both MARK2 and MARK3 are negatively regulated by aPKC [63], and MARK4 that was previously known to interact with aPKC [33], also undergoes aPKC-mediated phosphorylation at serine and threonine sites [115]. In addition to aPKC, Watkins et al. show the regulation of Par-1b/MARK2 activity by novel protein kinase C (PKC) and protein kinase D [116]. This is further confirmed in cells following treatment with PKC-activating agent phorbol-12-myristate-13-acetate which results in phosphorylation of Par-1b/MARK2 at Ser400 [116]. These studies clearly show that members of PKC family regulate MARKs, and modulating their functions may indirectly alter MARK activity.
In addition to tau, GSK-3β also phosphorylates MARK at Ser212 and results in the inhibition of its kinase activity under in vitro condition [117]. Interestingly, there is a marked change in the pattern of tau phosphorylation in AD following a concomitant activation of GSK-3β and inactivation of MARK [117]. In contrast to Timm et al. [117], Kosuga et al. show that phosphorylation of MARK2/Par-1 at Ser212 activates MARK2, resulting in tau phosphorylation at Ser262 [118]. However, co-expression of cells with wild-type and constitutively active mutants of GSK-3β and MARK2 reduces tau phosphorylation, confirming that GSK-3β rather inhibits the activation of MARK2 [117]. The aPKC/GSK-3β and MARK signaling cascades present another potential window for exploring methods that can indirectly alter MARK activity under pathological conditions.
Conclusion
Although AD is a multifactorial disorder, the precise mechanism that contributes to the development and progression of the disease remains ambiguous [119]. Studies now clearly indicate that the members of MARK family are clearly associated with cancers and AD in humans [20, 27, 37]. Moreover, it is also evident that MARKs have a larger role in the pathological events of AD [19, 45]. Therefore, development of specific inhibitors of MARKs will not only serve as promising therapeutic candidates, but also broaden our understanding of the role of each isoform in disease and health. In this context, the recently reported ligand-bound crystallographic structure of MARK4 could potentially serve as a scaffold for the rational design of MARK inhibitors [106]. Besides, pathways that are known to regulate MARK activity may serve as potential indirect strategies for targeting MARKs. However, given the myriad role of MARKs and MARK-regulating kinases, such as LKB1, further studies are essential to validate the implications of these strategies in relation to physiological functions. This is particularly evident from the study of Yu et al. [28]. Although MKI restored neurotransmission in diseased neurons, the decrease in excitatory potential in healthy neurons suggests that maintaining MARK activity at a physiological level is essential for normal neurotransmission.
References
Tayeb HO, Yang HD, Price BH, Tarazi FI (2012) Pharmacotherapies for Alzheimer’s disease: beyond cholinesterase inhibitors. Pharmacol Ther 134:8–25. doi:10.1016/j.pharmthera.2011.12.002
Braak H, Del Tredici K (2016) Potential pathways of abnormal tau and α-synuclein dissemination in sporadic Alzheimer’s and Parkinson’s diseases. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a023630
Minati L, Edginton T, Grazia Bruzzone M, Giaccone G (2009) Reviews: Current concepts in Alzheimer’s disease: a multidisciplinary review. Am J Alzheimers Dis Other Dement 24:95–121. doi:10.1177/1533317508328602
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1:a006189
Stone J, Casadesus G, Gustaw-Rothenberg K et al (2011) Frontiers in Alzheimer’s disease therapeutics. Ther Adv Chronic Dis 2:9–23
Wang W-Y, Tan M-S, Yu J-T, Tan L (2015) Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 3:136. doi:10.3978/j.issn.2305-5839.2015.03.49
Kahn OI, Baas PW (2016) Microtubules and growth cones: motors drive the turn. Trends Neurosci 39:433–440. doi:10.1016/j.tins.2016.04.009
Sarma T, Koutsouris A, Yu JZ et al (2015) Activation of microtubule dynamics increases neuronal growth via the nerve growth factor (NGF)- and Gαs-mediated signaling pathways. J Biol Chem 290:10045–10056. doi:10.1074/jbc.M114.630632
Billingsley ML, Kincaid RL (1997) Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J 323:577–591
Di J, Cohen LS, Corbo CP et al (2016) Abnormal tau induces cognitive impairment through two different mechanisms: synaptic dysfunction and neuronal loss. Sci Rep 6:20833. doi:10.1038/srep20833
Mietelska-Porowska A, Wasik U, Goras M et al (2014) Tau protein modifications and interactions: their role in function and dysfunction. Int J Mol Sci 15:4671–4713. doi:10.3390/ijms15034671
Thomas S, Funk K, Wan Y et al (2012) Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol 123:105–117
Martin L, Latypova X, Terro F (2011) Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem Int 58:458–471. doi:10.1016/j.neuint.2010.12.023
Roder HM, Hutton ML (2007) Microtubule-associated protein tau as a therapeutic target in neurodegenerative disease. Expert Opin Ther Targets 11:435–442. doi:10.1517/14728222.11.4.435
Trinczek B, Brajenovic M, Ebneth A, Drewes G (2004) MARK4 is a novel microtubule-associated proteins/microtubule affinity-regulating kinase that binds to the cellular microtubule network and to centrosomes. J Biol Chem 279:5915–5923. doi:10.1074/jbc.M304528200
Gu GJ, Lund H, Wu D et al (2013) Role of individual MARK isoforms in phosphorylation of tau at Ser262 in Alzheimer’s disease. Neuromol Med 15:458–469. doi:10.1007/s12017-013-8232-3
Mandelkow E-M, Thies E, Trinczek B et al (2004) MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol 167:99. doi:10.1083/jcb.200401085
Matenia D, Mandelkow E-M (2009) The tau of MARK: a polarized view of the cytoskeleton. Trends Biochem Sci 34:332–342. doi:10.1016/j.tibs.2009.03.008
Naz F, Anjum F, Islam A et al (2013) Microtubule affinity-regulating kinase 4: structure, function, and regulation. Cell Biochem Biophys 67:485–499. doi:10.1007/s12013-013-9550-7
Sun C, Tian L, Nie J et al (2012) Inactivation of MARK4, an AMP-activated protein kinase (AMPK)-related kinase, leads to insulin hypersensitivity and resistance to diet-induced obesity. J Biol Chem 287:38305–38315. doi:10.1074/jbc.M112.388934
Yamahashi Y, Saito Y, Murata-Kamiya N, Hatakeyama M (2011) Polarity-regulating kinase partitioning-defective 1b (PAR1b) phosphorylates guanine nucleotide exchange factor H1 (GEF-H1) to regulate RhoA-dependent actin cytoskeletal reorganization. J Biol Chem 286:44576–44584. doi:10.1074/jbc.M111.267021
Kuhns S, Schmidt KN, Reymann J et al (2013) The microtubule affinity regulating kinase MARK4 promotes axoneme extension during early ciliogenesis. J Cell Biol 200:505. doi:10.1083/jcb.201206013
Jenardhanan P, Mannu J, Mathur PP (2014) The structural analysis of MARK4 and the exploration of specific inhibitors for the MARK family: a computational approach to obstruct the role of MARK4 in prostate cancer progression. Mol Biosyst 10:1845–1868. doi:10.1039/C3MB70591A
Drewes G, Ebneth A, Preuss U et al (1997) MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. doi:10.1016/S0092-8674(00)80208-1
Moroni RF, De Biasi S, Colapietro P et al (2006) Distinct expression pattern of microtubule-associated protein/microtubule affinity-regulating kinase 4 in differentiated neurons. Neuroscience 143:83–94. doi:10.1016/j.neuroscience.2006.07.052
Magnani I, Novielli C, Fontana L et al (2011) Differential signature of the centrosomal MARK4 isoforms in glioma. Anal. Cell, Pathology, p 34
Kato T, Satoh S, Okabe H et al (2001) Isolation of a novel human gene, MARKL1, homologous to MARK3 and its involvement in hepatocellular carcinogenesis. Neoplasia N Y 3:4–9. doi:10.1038/sj.neo.7900132
Yu W, Polepalli J, Wagh D et al (2012) A critical role for the PAR-1/MARK-tau axis in mediating the toxic effects of Aβ on synapses and dendritic spines. Hum Mol Genet 21:1384–1390. doi:10.1093/hmg/ddr576
Chen YM, Wang QJ, Hu HS et al (2006) Microtubule affinity-regulating kinase 2 functions downstream of the PAR-3/PAR-6/atypical PKC complex in regulating hippocampal neuronal polarity. Proc Natl Acad Sci USA 103:8534–8539. doi:10.1073/pnas.0509955103
Ebneth A, Drewes G, Mandelkow E-M, Mandelkow E (1999) Phosphorylation of MAP2c and MAP4 by MARK kinases leads to the destabilization of microtubules in cells. Cell Motil Cytoskelet 44:209–224. doi:10.1002/(SICI)1097-0169(199911)44:3<209:AID-CM6>3.0.CO;2-4
Biernat J, Wu Y-Z, Timm T et al (2002) Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol Biol Cell 13:4013–4028. doi:10.1091/mbc.02-03-0046
Chen YM, Wang QJ, Hu HS et al (2006) Microtubule affinity-regulating kinase 2 functions downstream of the PAR-3/PAR-6/atypical PKC complex in regulating hippocampal neuronal polarity. Proc Natl Acad Sci 103:8534–8539. doi:10.1073/pnas.0509955103
Brajenovic M, Joberty G, Küster B et al (2004) Comprehensive proteomic analysis of human Par protein complexes reveals an interconnected protein network. J Biol Chem 279:12804–12811. doi:10.1074/jbc.M312171200
Schmitt-Ulms G, Matenia D, Drewes G, Mandelkow E-M (2009) Interactions of MAP/microtubule affinity regulating kinases with the adaptor complex AP-2 of clathrin-coated vesicles. Cell Motil Cytoskelet 66:661–672. doi:10.1002/cm.20394
Rovina D, Fontana L, Monti L et al (2014) Microtubule-associated protein/microtubule affinity-regulating kinase 4 (MARK4) plays a role in cell cycle progression and cytoskeletal dynamics. Eur J Cell Biol 93:355–365. doi:10.1016/j.ejcb.2014.07.004
Butkevich E, Härtig W, Nikolov M et al (2016) Phosphorylation of FEZ1 by microtubule affinity regulating kinases regulates its function in presynaptic protein trafficking. Sci Rep 6:26965. doi:10.1038/srep26965
Lund H, Gustafsson E, Svensson A et al (2014) MARK4 and MARK3 associate with early tau phosphorylation in Alzheimer’s disease granulovacuolar degeneration bodies. Acta Neuropathol Commun 2:22. doi:10.1186/2051-5960-2-22
Dolan PJ, Johnson GV (2010) The role of tau kinases in Alzheimer’s disease. Curr Opin Drug Discov Dev 13:595–603
Timm T, Li X-Y, Biernat J et al (2003) MARKK, a Ste20-like kinase, activates the polarity-inducing kinase MARK/PAR-1. EMBO J 22:5090–5101. doi:10.1093/emboj/cdg447
Lizcano JM, Göransson O, Toth R et al (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 23:833–843. doi:10.1038/sj.emboj.7600110
Lee S, Wang J-W, Yu W, Lu B (2012) Phospho-dependent ubiquitination and degradation of PAR-1 regulates synaptic morphology and tau-mediated Aβ toxicity in Drosophila. Nat Commun 3:1312
Bernard LP, Zhang H (2015) MARK/Par1 kinase is activated downstream of NMDA receptors through a PKA-dependent mechanism. PLoS One 10:e0124816. doi:10.1371/journal.pone.0124816
Mazanetz MP, Fischer PM (2007) Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov 6:464–479. doi:10.1038/nrd2111
Fischer D, Mukrasch MD, Biernat J et al (2009) Conformational changes specific for pseudophosphorylation at Serine 262 selectively impair binding of tau to microtubules. Biochemistry (Mosc) 48:10047–10055. doi:10.1021/bi901090m
Ando K, Maruko-Otake A, Ohtake Y et al (2016) Stabilization of microtubule-unbound tau via tau phosphorylation at Ser262/356 by Par-1/MARK contributes to augmentation of AD-related phosphorylation and Aβ42-induced tau toxicity. PLoS Genet 12:e1005917. doi:10.1371/journal.pgen.1005917
Wang J-W, Imai Y, Lu B (2007) Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J Neurosci 27:574. doi:10.1523/JNEUROSCI.5094-06.2007
Dequiedt F, Martin M, Von Blume J et al (2006) New role for hPar-1 kinases EMK and C-TAK1 in regulating localization and activity of class IIa histone deacetylases. Mol Cell Biol 26:7086–7102. doi:10.1128/MCB.00231-06
Müller J, Ritt DA, Copeland TD, Morrison DK (2003) Functional analysis of C-TAK1 substrate binding and identification of PKP2 as a new C-TAK1 substrate. EMBO J 22:4431. doi:10.1093/emboj/cdg426
Müller J, Ory S, Copeland T et al (2001) C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell 8:983–993. doi:10.1016/S1097-2765(01)00383-5
Zhang S-H, Kobayashi R, Graves PR et al (1997) Serine phosphorylation-dependent association of the band 4.1-related protein-tyrosine phosphatase PTPH1 with 14-3-3β protein. J Biol Chem 272:27281–27287. doi:10.1074/jbc.272.43.27281
Mathias RA, Guise AJ, Cristea IM (2015) Post-translational modifications regulate class IIa histone deacetylase (HDAC) function in health and disease. Mol Cell Proteom MCP 14:456–470. doi:10.1074/mcp.O114.046565
Platholi J, Federman A, Detert JA et al (2014) Regulation of protein phosphatase 1I by Cdc25C-associated kinase 1 (C-TAK1) and PFTAIRE protein kinase. J Biol Chem 289:23893–23900. doi:10.1074/jbc.M114.557744
Gong C-X, Singh TJ, Grundke-Iqbal I, Iqbal K (1993) Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem 61:921–927. doi:10.1111/j.1471-4159.1993.tb03603.x
Sontag J-M, Sontag E (2014) Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front Mol Neurosci 7:16. doi:10.3389/fnmol.2014.00016
Schaar BT, Kinoshita K, McConnell SK (2004) Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron 41:203–213. doi:10.1016/S0896-6273(03)00843-2
Gu GJ, Wu D, Lund H et al (2013) Elevated MARK2-dependent phosphorylation of Tau in Alzheimer’s disease. J Alzheimers Dis JAD 33:699–713. doi:10.3233/jad-2012-121357
Beghini A, Magnani I, Roversi G et al (2003) The neural progenitor-restricted isoform of the MARK4 gene in 19q13.2 is upregulated in human gliomas and overexpressed in a subset of glioblastoma cell lines. Oncogene 22:2581–2591
Hubaux R, Thu KL, Vucic EA et al (2015) Microtubule affinity-regulating kinase 2 is associated with DNA damage response and cisplatin resistance in non-small cell lung cancer. Int J Cancer 137:2072–2082. doi:10.1002/ijc.29577
Marshall EA, Ng KW, Anderson C et al (2015) Gene expression analysis of microtubule affinity-regulating kinase 2 in non-small cell lung cancer. Genom Data 6:145–148. doi:10.1016/j.gdata.2015.08.011
Wu Z-Z, Lu H-P, Chao CC-K (2010) Identification and functional analysis of genes which confer resistance to cisplatin in tumor cells. Biochem Pharmacol 80:262–276. doi:10.1016/j.bcp.2010.03.029
Pardo OE, Castellano L, Munro CE et al (2016) miR-515-5p controls cancer cell migration through MARK4 regulation. EMBO Rep 17:570. doi:10.15252/embr.201540970
Arash EH, Shiban A, Song S, Attisano L (2017) MARK4 inhibits Hippo signaling to promote proliferation and migration of breast cancer cells. EMBO Rep. doi:10.15252/embr.201642455
Hurov JB, Watkins JL, Piwnica-Worms H (2004) Atypical PKC phosphorylates PAR-1 kinases to regulate localization and activity. Curr Biol 14:736–741. doi:10.1016/j.cub.2004.04.007
Lennerz JK, Hurov JB, White LS et al (2010) Loss of Par-1a/MARK3/C-TAK1 kinase leads to reduced adiposity, resistance to hepatic steatosis, and defective gluconeogenesis. Mol Cell Biol 30:5043–5056. doi:10.1128/MCB.01472-09
Liu Z, Gan L, Chen Y et al (2016) Mark4 promotes oxidative stress and inflammation via binding to PPARγ and activating NF-κB pathway in mice adipocytes. Sci Rep 6:21382. doi:10.1038/srep21382
Chin JY, Knowles RB, Schneider A et al (2000) Microtubule-affinity regulating kinase (MARK) is tightly associated with neurofibrillary tangles in Alzheimer brain: a fluorescence resonance energy transfer study. J Neuropathol Amp Exp Neurol 59:966. doi:10.1093/jnen/59.11.966
Mocanu M-M, Nissen A, Eckermann K et al (2008) The potential for β-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous tau in inducible mouse models of tauopathy. J Neurosci 28:737. doi:10.1523/JNEUROSCI.2824-07.2008
Murray MM, Bui T, Smith M et al (2013) Staurosporine is chemoprotective by inducing G(1) arrest in a Chk1- and pRb-dependent manner. Carcinogenesis 34:2244–2252. doi:10.1093/carcin/bgt186
Lee BD, Shin J-H, VanKampen J et al (2010) Inhibitors of leucine rich repeat kinase 2 (LRRK2) protect against LRRK2-models of Parkinson’s disease. Nat Med 16:998–1000. doi:10.1038/nm.2199
Wakita S, Izumi Y, Nakai T et al (2014) Staurosporine induces dopaminergic neurite outgrowth through AMP-activated protein kinase/mammalian target of rapamycin signaling pathway. Neuropharmacology 77:39–48. doi:10.1016/j.neuropharm.2013.09.012
Nabeshima T, Ogawa S, Nishimura H et al (1991) Staurosporine facilitates recovery from the basal forebrain-lesion-induced impairment of learning and deficit of cholinergic neuron in rats. J Pharmacol Exp Ther 257:562
Mainardes R, Gremiao M (2009) Reversed phase HPLC determination of zidovudine in rat plasma and its pharmacokinetics after a single intranasal dose administration. Biol Res 42:357–364
Fuse E, Tanii H, Kurata N et al (1998) Unpredicted clinical pharmacology of UCN-01 caused by specific binding to human α1-acid glycoprotein. Cancer Res 58:3248
Akinaga S, Gomi K, Morimoto M et al (1991) Antitumor activity of UCN-01, a selective inhibitor of protein kinase C, in murine and human tumor models. Cancer Res 51:4888
Gurley L, Umbarger K, Kim J et al (1995) Development of a high-performance liquid chromatographic method for the analysis of staurosporine. J Chromatogr B Biomed Sci Appl 670:125–138
Monnerat C, Henriksson R, Le Chevalier T et al (2004) Phase I study of PKC412 (N-benzoyl-staurosporine), a novel oral protein kinase C inhibitor, combined with gemcitabine and cisplatin in patients with non-small-cell lung cancer. Ann Oncol 15:316–323. doi:10.1093/annonc/mdh052
Sausville EA, Arbuck SG, Messmann R et al (2001) Phase I trial of 72-hour continuous infusion UCN-01 in patients with refractory neoplasms. J Clin Oncol 19:2319–2333. doi:10.1200/JCO.2001.19.8.2319
Mukthavaram R, Jiang P, Saklecha R et al (2013) High-efficiency liposomal encapsulation of a tyrosine kinase inhibitor leads to improved in vivo toxicity and tumor response profile. Int J Nanomed 8:3991–4006. doi:10.2147/IJN.S51949
Bain J, Plater L, Elliott M et al (2007) The selectivity of protein kinase inhibitors: a further update. Biochem J 408:297–315. doi:10.1042/BJ20070797
Pakavathkumar P, Sharma G, Kaushal V et al (2015) Methylene blue inhibits caspases by oxidation of the catalytic cysteine. Sci Rep 5:13730
Wainwright M, Crossley KB (2002) Methylene blue—a therapeutic dye for all seasons? J Chemother 14:431–443. doi:10.1179/joc.2002.14.5.431
Rodriguez P, Zhou W, Barrett DW et al (2016) Multimodal randomized functional MR imaging of the effects of methylene blue in the human brain. Radiology 281:516–526. doi:10.1148/radiol.2016152893
Walter-Sack I, Rengelshausen J, Oberwittler H et al (2009) High absolute bioavailability of methylene blue given as an aqueous oral formulation. Eur J Clin Pharmacol 65:179–189. doi:10.1007/s00228-008-0563-x
Schirmer RH, Adler H, Pickhardt M, Mandelkow E (2011) “Lest we forget you—methylene blue…”. Neurobiol Aging 32:2325.e7–2325.e16. doi:10.1016/j.neurobiolaging.2010.12.012
Sun W, Lee S, Huang X et al (2016) Attenuation of synaptic toxicity and MARK4/PAR1-mediated Tau phosphorylation by methylene blue for Alzheimer’s disease treatment. Sci Rep 6:34784
Congdon EE, Wu JW, Myeku N et al (2012) Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 8:609–622. doi:10.4161/auto.19048
Meijer L, Thunnissen A-M, White A et al (2000) Inhibition of cyclin-dependent kinases, GSK-3β and CK1 by hymenialdisine, a marine sponge constituent. Chem Biol 7:51–63. doi:10.1016/S1074-5521(00)00063-6
Timm T, von Kries JP, Li X et al (2011) Microtubule affinity regulating kinase activity in living neurons was examined by a genetically encoded fluorescence resonance energy transfer/fluorescence lifetime imaging-based biosensor: inhibitors with therapeutic potential. J Biol Chem 286:41711–41722. doi:10.1074/jbc.M111.257865
Wan Y, Hur W, Cho CY et al (2004) Synthesis and target identification of hymenialdisine analogs. Chem Biol 11:247–259. doi:10.1016/j.chembiol.2004.01.015
Eldar-Finkelman H, Martinez A (2011) GSK-3 inhibitors: preclinical and clinical focus on CNS. Front Mol Neurosci 4:32. doi:10.3389/fnmol.2011.00032
Saadat I, Higashi H, Obuse C et al (2007) Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 447:330–333. doi:10.1038/nature05765
Mishra JP, Cohen D, Zamperone A et al (2015) CagA of Helicobacter pylori interacts with and inhibits the serine-threonine kinase PRK2. Cell Microbiol 17:1670–1682. doi:10.1111/cmi.12464
Neišić D, Miller MC, Quinkert ZT et al (2010) Helicobacter pylori CagA inhibits PAR1/MARK family kinases by mimicking host substrates. Nat Struct Mol Biol 17:130–132. doi:10.1038/nsmb.1705
Tronel C, Page G, Bodard S et al (2014) The specific PKR inhibitor C16 prevents apoptosis and IL-1β production in an acute excitotoxic rat model with a neuroinflammatory component. Neurochem Int 64:73–83. doi:10.1016/j.neuint.2013.10.012
Ingrand S, Barrier L, Lafay-Chebassier C et al (2007) The oxindole/imidazole derivative C16 reduces in vivo brain PKR activation. FEBS Lett 581:4473–4478. doi:10.1016/j.febslet.2007.08.022
Couturier J, Morel M, Pontcharraud R et al (2010) Interaction of double-stranded RNA-dependent protein kinase (PKR) with the death receptor signaling pathway in amyloid β (Aβ)-treated cells and in APP(SL)PS1 knock-in mice. J Biol Chem 285:1272–1282. doi:10.1074/jbc.M109.041954
Naz F, Shahbaaz M, Khan S et al (2015) PKR-inhibitor binds efficiently with human microtubule affinity-regulating kinase 4. J Mol Graph Model 62:245–252. doi:10.1016/j.jmgm.2015.10.009
Naz F, Shahbaaz M, Bisetty K et al (2015) Designing new kinase inhibitor derivatives as therapeutics against common complex diseases: structural basis of microtubule affinity-regulating kinase 4 (MARK4) inhibition. OMICS J Integr Biol 19:700–711. doi:10.1089/omi.2015.0111
Naz F, Sami N, Naqvi AT et al (2016) Evaluation of human microtubule affinity-regulating kinase 4 inhibitors: fluorescence binding studies, enzyme, and cell assays. J Biomol Struct Dyn. doi:10.1080/07391102.2016.1249958
Feldman RI, Wu JM, Polokoff MA et al (2005) Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J Biol Chem 280:19867–19874. doi:10.1074/jbc.M501367200
Clark K, Plater L, Peggie M, Cohen P (2009) Use of the pharmacological inhibitor bx795 to study the regulation and physiological roles of TBK1 and IκB kinase ϵ: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem 284:14136–14146. doi:10.1074/jbc.M109.000414
Chung S, Suzuki H, Miyamoto T et al (2012) Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget 3:1629–1640
Fraser C, Dawson JC, Dowling R et al (2016) Rapid discovery and structure-activity relationships of pyrazolopyrimidines that potently suppress breast cancer cell growth via SRC kinase inhibition with exceptional selectivity over ABL kinase. J Med Chem 59:4697–4710. doi:10.1021/acs.jmedchem.6b00065
Tandon M, Johnson J, Li Z et al (2013) New pyrazolopyrimidine inhibitors of protein kinase D as potent anticancer agents for prostate cancer cells. PLoS One 8:e75601. doi:10.1371/journal.pone.0075601
Sloman DL, Noucti N, Altman MD et al (2016) Optimization of microtubule affinity regulating kinase (MARK) inhibitors with improved physical properties. Bioorg Med Chem Lett 26:4362–4366. doi:10.1016/j.bmcl.2016.02.003
Sack JS, Gao M, Kiefer SE et al (2016) Crystal structure of microtubule affinity-regulating kinase 4 catalytic domain in complex with a pyrazolopyrimidine inhibitor. Acta Crystallogr Sect F 72:129–134
Gan R-Y, Li H-B (2014) Recent progress on liver kinase B1 (LKB1): expression, regulation, downstream signaling and cancer suppressive function. Int J Mol Sci 15:16698–16718. doi:10.3390/ijms150916698
Timm T, Marx A, Panneerselvam S et al (2008) Structure and regulation of MARK, a kinase involved in abnormal phosphorylation of Tau protein. BMC Neurosci 9:S9. doi:10.1186/1471-2202-9-S2-S9
Kodamullil AT, Younesi E, Naz M et al (2015) Computable cause-and-effect models of healthy and Alzheimer’s disease states and their mechanistic differential analysis. Alzheimers Dement J Alzheimers Assoc 11:1329–1339. doi:10.1016/j.jalz.2015.02.006
Ozcan C, Battaglia E, Young R, Suzuki G (2015) LKB1 knockout mouse develops spontaneous atrial fibrillation and provides mechanistic insights into human disease process. J Am Heart Assoc. doi:10.1161/JAHA.114.001733
Shan T, Xiong Y, Kuang S (2016) Deletion of Lkb1 in adult mice results in body weight reduction and lethality. Sci Rep 6:36561
Marx A, Nugoor C, Panneerselvam S, Mandelkow E (2010) Structure and function of polarity-inducing kinase family MARK/Par-1 within the branch of AMPK/Snf1-related kinases. FASEB J 24:1637–1648. doi:10.1096/fj.09-148064
Matenia D, Griesshaber B, Li X et al (2005) PAK5 kinase is an inhibitor of MARK/Par-1, which leads to stable microtubules and dynamic actin. Mol Biol Cell 16:4410–4422. doi:10.1091/mbc.E05-01-0081
Benton R, Palacios IM, Johnston DS (2002) Drosophila 14-3-3/PAR-5 is an essential mediator of PAR-1 function in axis formation. Dev Cell 3:659–671. doi:10.1016/S1534-5807(02)00320-9
Naz F, Islam A, Ahmad F, Hassan MI (2015) Atypical PKC phosphorylates microtubule affinity-regulating kinase 4 in vitro. Mol Cell Biochem 410:223–228. doi:10.1007/s11010-015-2555-3
Watkins JL, Lewandowski KT, Meek SEM et al (2008) Phosphorylation of the Par-1 polarity kinase by protein kinase D regulates 14-3-3 binding and membrane association. Proc Natl Acad Sci 105:18378–18383. doi:10.1073/pnas.0809661105
Timm T, Balusamy K, Li X et al (2008) Glycogen synthase kinase (GSK) 3β directly phosphorylates serine 212 in the regulatory loop and inhibits microtubule affinity-regulating kinase (MARK) 2. J Biol Chem 283:18873–18882. doi:10.1074/jbc.M706596200
Kosuga S, Tashiro E, Kajioka T et al (2005) GSK-3beta directly phosphorylates and activates MARK2/PAR-1. J Biol Chem. doi:10.1074/jbc.M507941200
Kumar A, Singh A, Ekavali (2015) A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep 67:195–203. doi:10.1016/j.pharep.2014.09.004
Acknowledgements
This work was supported by Grants from the Czech Ministry of Education, Youth and Sports (Grant Numbers: LO1304, LM2011024) and Ministry of Health of the Czech Republic (Grant Number: NV15-31984A).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interests. All authors read and approved the final manuscript.
Rights and permissions
About this article

Cite this article
Annadurai, N., Agrawal, K., Džubák, P. et al. Microtubule affinity-regulating kinases are potential druggable targets for Alzheimer’s disease. Cell. Mol. Life Sci. 74, 4159–4169 (2017). https://doi.org/10.1007/s00018-017-2574-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2574-1