
Abstract
The gut microbiota is essential to health and has recently become a target for live bacterial cell biotherapies for various chronic diseases including metabolic syndrome, diabetes, obesity and neurodegenerative disease. Probiotic biotherapies are known to create a healthy gut environment by balancing bacterial populations and promoting their favorable metabolic action. The microbiota and its respective metabolites communicate to the host through a series of biochemical and functional links thereby affecting host homeostasis and health. In particular, the gastrointestinal tract communicates with the central nervous system through the gut–brain axis to support neuronal development and maintenance while gut dysbiosis manifests in neurological disease. There are three basic mechanisms that mediate the communication between the gut and the brain: direct neuronal communication, endocrine signaling mediators and the immune system. Together, these systems create a highly integrated molecular communication network that link systemic imbalances with the development of neurodegeneration including insulin regulation, fat metabolism, oxidative markers and immune signaling. Age is a common factor in the development of neurodegenerative disease and probiotics prevent many harmful effects of aging such as decreased neurotransmitter levels, chronic inflammation, oxidative stress and apoptosis—all factors that are proven aggravators of neurodegenerative disease. Indeed patients with Parkinson’s and Alzheimer’s diseases have a high rate of gastrointestinal comorbidities and it has be proposed by some the management of the gut microbiota may prevent or alleviate the symptoms of these chronic diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The gut microbiota and the gut–brain axis
The gut microbiota is composed of a vast plethora of bacterial species residing within the gastrointestinal tract (GIT). The importance of the gut microbiota to human health has recently been recognized due to the bacterial community’s bilateral connectivity to the rest of the body, and notably, the brain. The GIT and the central nervous system (CNS) are intricately connected through a network of signaling pathways collectively known as the gut–brain axis. The gut–brain axis is a dynamic bidirectional neuroendocrine system consisting of direct neurological connections, endocrine signals and immunological factors [1,2,3]. The gut microbiota conveys information contained in the ingested components passing through the GIT (i.e., vitamins, minerals, carbohydrates, fats, etc.) with the CNS via the aforementioned routes to elicit a systemic response reflecting nutritional and energy states. When the relative microbial populations fall out of balance (dysbiosis), the messages sent to the brain propagate unhealthy signals manifesting in low-grade inflammation, increased oxidative stress, unbalanced energy homeostasis and a general increase in cellular degeneration [4]. Many recent studies suggest that microbial dysbiosis contributes to the pathology of multiple neurological diseases including depression, anxiety and neurodegeneration [5, 6]. This article describes several possible mechanisms of gut–brain axis communication and how manipulation of the gut microbiota with probiotics can influence neurodegenerative diseases by improving inflammatory markers, modulating neurological signaling and reducing the levels of oxidative stress: the main common features of idiopathic neurodegeneration.
The gut microbiota
The gut microbiota consists of a diverse community of bacterial species in the GIT existing symbiotically with the human host. The majority of the microbiota belongs to the phyla Firmicutes (~51%) including the Clostridium coccoides and Clostridium leptum groups and the well-known Lactobacillus genera and the phyla Bacteroidetes (~48%) including the well-known genera Bacteroides and Prevotella [7]. The remaining 1% is constituted by other less populous phyla, including Proteobacteria, Actinobacteria (including the Bifidobacteria genera), Fusobacteria, Spirochaetes, Verrucomicrobia and Lentisphaerae [8]. Modern sequencing technology has identified at least 1000 species and more than 7000 strains of bacteria composing the 1013–1014 microorganisms of the microbiota [9]. There are high inter-individual variations in the gut microbial populations; however, the overall functionality is conserved, suggesting that a core gut microbiota is required to maintain a basic set of physiological functions [10]. The gut microbiota may be considered an organ onto itself, being responsible for a variety of physiological activities including host metabolism, neurological development, energy homeostasis, immune regulation, vitamin synthesis and digestion [11].
The gut–brain axis
The gut–brain axis is a dynamic bidirectional neuroendocrine system describing the connections between the GIT and the nervous system. There are many common regulatory factors between the enteric nervous system (ENS) and the CNS [12]. Many of the hormones and metabolites secreted by the microbiota and intestinal enterochromaffin (EC) cells intersect with biochemical pathways that influence CNS processes creating a means of direct communication between the external environment in contact with the gut microbiota and the brain, which is isolated from the environment by the blood–brain barrier (BBB).
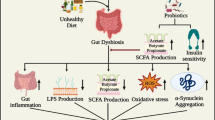
The gut–brain axis consists of the entirety of the intestinal microbiota, ENS, parasympathetic and sympathetic nervous systems, CNS, neuroendocrine connections, humoral pathways, cytokines, neuropeptides and signaling molecules [13]. There are three main modes of communication between the gut and the brain, namely (1) neuronal messages carried by vagal afferents, (2) endocrine messages carried by gut hormones and (3) immune messages carried by cytokines [1, 14] (Fig. 1). The impact of the gut microbiota on the brain is profound and has been recognized to affect behavior (anxiety, depression, learning and memory, sociability), microglial activity, BBB integrity, neurogenesis, and neurotransmitter production (reviewed in Ref. [15]). Recently, it has been realized that brain injury and different psychological states can also affect the composition of the gut microbiota and possibly precipitate disease. For example, brain injury in the form of stroke was shown to alter the composition of the caecal microbiota in mice with specific changes to Peptococcaceae and Prevotellaceae, correlating to the extent of injury [16].
Gut dysbiosis is linked to disease
Evidence suggests that the gut microbiota, especially when in a state of dysbiosis, can influence neurological disease progress and even initiate disease onset [17]. There is also a growing realization that the reduced diversity in the aging gut microbiota may be a major factor in the development of neurodegeneration [18, 19]. One of the major mechanisms linking the microbiota to age-related diseases is neuroinflammation [20]. The gut microbiota plays a key role in the activation of microglia [21] and it has been suggested that manipulation of the gut microbiome, especially with short-chain fatty acid (SCFA)-producing bacteria, could modulate neuroimmune activation [18, 22]. This relationship could explain the high percentage of gastrointestinal disturbances comorbid with neurodegenerative disease including microbial dysbiosis, constipation, diarrhea, vitamin deficiencies, obesity and diabetes [23,24,25]. The prevalence of these comorbidities is indisputable, indicating strong functional consequences of the gut–brain axis in neurodegeneration [26].
Neurodegenerative diseases commonly have a sporadic pathology meaning that the disease is triggered by an accumulation of harmful and random interactions with the environment. For example, Parkinson’s disease (PD) and Alzheimer’s disease (AD) are both linked to the exposure of environmental toxins, such as herbicides, fungicides and pesticides [27], in addition to lifestyle habits such as diet and stress [28]. Notably, in response to environmental stressors such as oxidative stress, the relative balance of microbiota population, and consequently its metabolic and genomic expression is altered implementing broad physiological changes in metabolism, endocrine signaling and innervation in the human host [29]. Further, many of the early symptoms of neurodegenerative disease reside in the GIT, suggesting that dysbiosis may even trigger neurodegenerative disease [30].
Neurological disorders and their connection to gut microbiota
AD, PD, multiple sclerosis (MS) and amyloid lateral sclerosis (ALS) are categorized as neurodegenerative diseases. Although each of these diseases has distinct physiological manifestations, they do have common underlying etiologies linking their pathology, most of which are associated with normal aging. Interestingly, the gut microbiota and its downstream effectors broadly intersect many of these pathways, indicating that management of the microbiota could have therapeutic potential for the prevention and treatment of neurodegenerative diseases.

Gut microbiota influences neurodegenerative diseases through various pathways. Research linking changes in the gut microbiota to neurodegenerative diseases is still emerging. There are many established studies linking the pathways influenced by the gut microbiota (neurological, endocrine and immune) to the pathogenesis of neurodegeneration; however, further studies confirming these microbiota-related linkages are required. In addition, there are many studies that confirm a relationship between the endocrine, neurological and immune signaling pathways contributing to the complexity of the neurodegenerative pathology. In the figure, solid lines represent pathways that are confirmed and the dotted lines have yet to be fully established
Common pathways in neurodegeneration
Oxidative damage and inflammation are two major systemic conditions that aggravate neurodegeneration and both states are fueled by the normal physiological decline that occurs with age. Generation of reactive oxidative species (ROS) primarily occurs in the mitochondria where 0.4–4% of electrons traveling through the electron transport chain (ETC) escape and react with an oxygen molecule to create a superoxide radical [31]. Normally, these escaped radicals are converted to harmless species by the cells’ anti-oxidant defense systems; however, with age, the progressive loss of cellular defenses leads to an accumulation of cellular, genetic and membrane damage and eventually cell death [32]. The brain is particularly sensitive to oxidative damage as neurons have high energy demands and are almost exclusively post-mitotic cells making their polyunsaturated fatty acid rich membranes more sensitive to ROS-induced peroxidative damage [33]. Indeed, oxidative damage in PD and AD is a major factor in their progression, especially considering that the areas affected by the degeneration are selectively sensitive to oxidative stress, particularly in AD [34]. The slow accumulation of ROS in neurons stimulates cytokine release and consequently microglial activation and neuroinflammation. The pathology of oxidative damage and inflammation creates a vicious cycle cumulatively known as ‘inflamm-aging’, which is defined as a chronic low-grade systemic proinflammatory state characterized by elevated cytokines and inflammatory mediators with no precipitated cause [35]. Inflamm-aging describes a common basis for a broad spectrum of age-related pathologies, including neurodegeneration [36].
Recently, disrupted energy metabolism, such as that present in diabetes and obesity, has been linked to the development and prognosis of neurodegenerative disease. Several comprehensive reviews have been written on this topic [5, 6, 18, 19, 37], so further elaboration will not be done here.
Age-related changes in the gut microbiota observed in neurodegenerative diseases
Aging is associated with clear shifts in the composition of the gut microbiota. In general, there is a loss of gut microbial diversity in the aging gut [38, 39]. The phyla Bacteriodetes and Firmicutes remain dominant, although their relative proportions may change. There may also be an increase in pathogenic bacteria (pathobionts) at the expense of beneficial bacteria (symbionts), an increase in Proteobacteria spp. with a decrease in Bifidobacteria spp., a reduction in butyrate-producing species (Ruminococcus spp., Faecalibacterium spp., etc.) and an increase in microbiota known to stimulate an inflammatory response (Escherichia spp., Enterobacteriaceae spp., Bacteroides spp., Clostridium difficile, etc.) [36, 38, 39]. Interestingly, centenarians typically do not experience these harmful changes and have marked differences in their microbiota populations compared to other elderly populations, indicating that a healthy microbiota may be one of the keys to longevity [36, 39].
Striking variations in the composition of the gut microbiota of aging patients suffering from neurodegenerative diseases has also been observed. One study found that the bacterial metabolite indican, a marker of intestinal dysbiosis, was significantly elevated in PD patients indicating a broad microbial dysbiosis [40]. In a large study cohort including 72 PD and 72 healthy subjects, high-resolution 16S sequencing revealed a 77.6% decrease in Prevotellaceae in the PD patients. This is significant as Prevotellaceae is one of the main producers of mucin, a highly glycosylated protein that produces a barrier along the epithelial wall against invading pathogens. This group also found a significant increase in Enterobacteriaceae, which was positively correlated with postural instability [41]. In a similar study, intestinal biopsies of PD patients indicated marked differences in the sigmoid mucosa, significant reductions in anti-inflammatory butyrate-producing bacteria (i.e., Roseburia and Faecalibacterium spp.) and a clear increase in proinflammatory Proteobacteria species of the genus Ralstonia compared to healthy age-matched controls [42]. This group has also shown that the accumulation of α-synuclein (α-Syn) neurons tend to first occur in the sigmoid mucosa of patients, 2–5 years before developing neurological symptoms of PD [43]. Based on these findings, this group hypothesized that PD pathology is subsequently manifested to the brain via α-Syn translocation in a prion-like fashion or through the induction of inflammation and oxidative stress. Variations in the PD gut microbiota have also been associated with reduced levels of fecal SCFAs, which were postulated to induce alterations in the ENS contributing to the reduced gastrointestinal motility observed in PD patients [44]. Reductions in butyrate is notable in PD patients as sodium butyrate is a histone deacetylase (HDAC) inhibitor that protects dopaminergic neurons from degeneration by upregulating neurotrophic factors including brain-derived growth factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) [45, 46]. Interestingly, metagenomic studies also indicated lower counts of metabolic genes indicating metabolic dysregulation in PD patients [42]. In PD patients, it has been found that fecal transplants from healthy donors improve both the motor and non-motor symptoms of PD outlining a novel therapeutic option by modifying of the gut microbiota [47]. To test this effect directly, it was found that germ-free α-Syn overexpressing mice retained higher physical coordination than their wild-type counterparts, indicating that the microbiota is responsible for the manifestation of the physical symptoms of PD. α-Syn mice with a complex microbiota developed the same physical impairments and physiological effects as the germ-free PD mice; however, the effects were significantly delayed by 12 weeks. In addition, it was found that microbes in the disease model promote α-Syn-dependent activation of microglia within the brain regions affected in PD, which exacerbates the disease phenotype by promoting inflammation [22]. Interestingly, transplant of fecal samples from PD patients into GF mice promoted significant α-Syn-mediated motor dysfunctions in the humanized PD mice, but not in mice inoculated with fecal matter from healthy individuals [22].
In AD, there is also evidence for gut dysbiosis, however, less direct. Several bacterial species have been found to produce or aggravate the production of amyloid β (Aβ) plaques including B. subtilis, E. coli, Klebsiella pneumonia, Mycobacterium spp., Salmonella spp., Staphylococcus aureus and Streptococcus spp. [6]. There have also been reports of increased proportions of Gram-negative bacteria in AD patients coupled with mucosal disruption in response to this dysbiosis. Nonetheless, there are clear connections between a disrupted gut microbiota and the pathology of AD that could be targeted with probiotic and prebiotic therapy to alleviate its underlying symptoms.
The influence of gut microbiota, gut–brain axis and probiotics in neurodegenerative disease
There are many interlocking hormonal and biochemical pathways relating the health of the GIT to the brain creating a strong therapeutic potential for the use of probiotics against neurodegeneration. One common theme linking specific microbiota to the prevention of neurodegeneration is a broad and potent anti-inflammatory action. Embedded in the subepithelial lamina propria tissue of the GIT are antigen-presenting innate immune cells including dendritic cells and macrophages. This positioning puts the immune cells in close proximity to the gut microbiota, invading pathogens and antigens that breach the protective epithelial barrier allowing efficient immunological communication between the external environment and the systemic immune system [48]. Toll-like receptors (TLRs) and NOD-like receptors (NLRs) on these cells recognize the microbe-associated molecular patterns (MAMPs) from bacteria and other microbes, which trigger signaling cascades leading to pro- or anti-inflammatory cytokine expression. This is highly significant as neuroinflammation is highly correlated with neurodegeneration, behavioral and other neurological deficits [49, 50]. In addition, through the production of secondary metabolites, the microbiota can orchestrate several levels of communication with host physiology including insulin control, lipogenesis, apoptosis, neuronal and hormonal signaling. The action of several microbial species on each of these aspects is outlined in Table 1.
Biomolecules in neurological disease that can be targeted by manipulating gut microbiota
Commensal microbiota produces a variety of neuroactive molecules that directly or indirectly impact signaling in the CNS (rev in Ref. [51]). In addition, there are extensive endocrine and molecular signaling cascades interlacing the gut and brain that co-regulate key processes. Below is an overview of how biomolecules derived from the gut microbiota impact various hormonal and molecular signaling pathways in the CNS and can be important towards the development of neurodegenerative disease.
Gut-derived ferulic acid impacts neurological health
Ferulic acid (FA), or trans-4-hydroxy-3-methoxycinnamic acid, is a phenolic compound abundantly found in seed plants (rice, wheat and oats), vegetables (tomatoes and carrots) and fruits (pineapple and orange). Traditionally, plants and herbs containing high levels of FA have been used in Chinese medicine for its potent inhibition of ROS generation and anti-inflammatory properties [52, 53]. In modern medicine, FA is recognized as a potent ROS scavenger with therapeutic potential in various chronic diseases including neurodegeneration, cancer, accelerated cell aging, obesity and diabetes [54]. Considering neurodegeneration, FA directly impacts neurons and can stimulate proliferation of neural stem cells both in vitro and in vivo [55]. For the latter, FA increased the number of neurons in the dentate gyrus of corticosterone-treated mice, indicating the potent ability of FA to stimulate neurogenesis in vivo. Therapeutically, FA was shown to reverse the morphological damage sustained through a chronic mild stress depression paradigm in rats by inducing neurogenesis via upregulation of nerve growth factor (NGF) and BDNF [56]. More recently, FA has become a key target for mediating communication between the commensal microbiota and the brain. Apart from dietary sources, FA is rapidly and abundantly synthesized by some gut microbiota via a ferulic acid esterase gene, the most potent being L. fermentum NCIMB 5221 [57] and B. animalis [58]. There are several other species that contain feruloyl esterase, an enzyme that hydrolyzes and releases FA from its bound state including L. plantarum NCIMB 8826 [57, 59], indicating the necessity of having a complete community of healthy microbiota to support the action of ferulic acid.
There has been extensive research linking the pathology of AD with the therapeutic potential of FA. FA was shown to prevent Aβ-related toxicity both in vitro and in vivo AD models by directly inhibiting Aβ aggregation and β-secretase activity [60, 61]. Indeed, oral FA treatment administered for six months reduced several typical AD behavioral phenotypes in mice while simultaneously reducing Aβ fibril formation, the cleavage of β-carboxy-terminal amyloid precursor protein (APP), neuroinflammation and oxidative stress [61]. In a similar long-term administration model, FA was shown to destabilize Aβ1–42-induced learning and memory deficits and amyloid deposition [62].
Accumulation of ROS underlines a key pathological feature of most neurodegenerative diseases and FA has been shown to be protective against oxidative neurological damage in several disease models. For example, FA is neuroprotective against cerebral ischemia in rats, whose major pathological feature is oxidative stress [63], via its anti-inflammatory and anti-oxidant effects [64]. To delineate these effects, FA administered to rats 2 h prior, concurrently or 2 h following induction of cerebral ischemia was found to be neuroprotective and downregulated glial fibrillary acidic protein (GFAP) and several apoptotic markers including mitochondrial Bax, cytochrome c and cleaved caspase c [65]. One mechanism explaining FA’s protective action against ROS can be explained by the regulation of peroxiredoxins (PRX) and thioredoxin (Trx) [66]. PRX and Trx are ubiquitous anti-oxidant proteins that regulate cell proliferation and apoptosis while providing neuroprotection (rev. in Ref. [67]). Notably, the expression of PRX proteins is inversely correlated with aging, which could correlate with the simultaneous rise of ROS [67]. PRX-2 is important to maintain cellular redox homeostasis in neurons and it was shown that transgenic expression of PRX-2 in a mouse model of cerebral ischemia protected neurons from stressful ischemic insults by preventing apoptosis and improving neurological recovery [68]. PRX-2 normally keeps certain peroxides, such as Trx, in a reduced state, thereby signaling a pro-survival state and resistance to oxidative stress. When there is an over-consumption of PRX-2 due to increased oxidative stress, Trx is converted to its oxidized state leading to the activation of pro-death cascades including apoptosis signaling kinase 1 (ASK1) and the downstream MKK/JNK pro-death signaling pathway [68, 69]. Interestingly, PRX and Trx protein expression are consistently dysregulated in neurodegenerative diseases and are related to elevated microglial activation and reduced anti-oxidant activity (rev in Ref. [67]). Based on these mechanisms and relation to neurodegeneration, it is significant to note that FA prevents both the ischemia-mediated attenuation of PRX and the corresponding oxidation of Trx and the dissociation of Trx from ASK1, therefore preventing apoptosis and providing neuroprotection [66].
FA also inhibits caspase-3 expression following ischemic damage, which is a major source of apoptotic cell death in neurodegenerative diseases [70]. For example, neuronal ischemic stress is associated with reduced levels of phosphorylated Akt [71], and correspondingly, elevated levels of glycogen synthase kinase 3 beta (GSK3β), which is the main activator of collapsing response mediator protein 2 (CRMP-2), an initiator of apoptosis [72] and mediator of neurite retraction [73]. Indeed, phosphorylated CRMP-2 is associated with high neurofibrillary tangle formation in AD [74] and prevention of neurite outgrowth in damaged neurons [75], and may even precede physiological symptoms of AD [76]. FA can inhibit apoptosis by preventing CRMP-2 expression by upregulating signaling through Akt consequently inhibiting the GSK3β pathway [71]. Not only does this have direct implications for stress-induced apoptosis, but the Akt pathway is highly integrated in the pathophysiology of PD outlining a possible broad mechanism of action in neurodegeneration.
Short-chain fatty acids manipulate neurodegenerative disease via the gut microbiota
The microbiota is responsible for the production of several metabolites formed by the fermentation of soluble fibers such as galacto-oligosaccharides and fructo-oligosaccharides. These metabolites include the SCFAs acetate, propionate and butyrate and are produced by fermentation mediated by Bacteroides, Bifidobacterium, Propionibacterium, Eubacterium, Lactobacillus, Clostridium, Roseburia and Prevotella species [77]. The type of SCFAs produced depends both on the type of fiber consumed and the relative population of microbiota in the gut. For instance, microbes in the Firmicutes phyla, particularly the genera Roseburia, Eubacterium and Lachnospiraceae of the Clostridia class actively produce butyrate while Bifidobacteria spp. produce lactate and acetate [78]. In contrast, the actions of SCFAs also influence the functional profile of gut microbiota, especially with regards to endocrine signaling.
SCFAs have a range of regulatory activities beneficial to the host, notably the regulation of systemic energy homeostasis and colonocyte metabolism [79]. In the GIT, SCFAs implement signaling through the G-protein coupled free fatty acid receptors (FFAR)2 and FFAR3 on the gut epithelium, but can also be passively or actively transported into central circulation where they have broad physiological effects [80] which play a role in lipid, glucose and cholesterol metabolism [81,82,83]. SCFAs are also well-known to have potent anti-inflammatory effects, which have been described in detail in several reviews and will not be further elaborated here [84,85,86].
In particular, propionate initiates intestinal gluconeogenesis via FFAR3 signaling [87]. The released glucose from the gluconeogenesis processes enters directly into the portal vein where glucose sensors transduce the glucose satiation signals to the brain [88]. It is through these afferent circuits that the SCFA’s action on the glucose regulation in the gut lumen influences central signaling processes. One of the direct targets of propionate-induced intestinal gluconeogenesis is the dorsal motor nucleus of the vagus (DMV), which receives inputs from the ventral vagus nerve [89]. This is a notable interaction as the activity in the DMV is an early indicator of PD and AD [90], indicating that the production and regulation of propionate intestinal gluconeogenesis could be a major factor in the early stages of neurodegeneration. Butyric acid also has a direct effect on vagal afferents [91], a stimulation that has been shown in clinical trials to be beneficial for cognition in AD patients [92, 93].
Apart from energy homeostasis and direct vagal stimulation, SCFAs can act as endocrine signaling molecules and influence a number of biochemical pathways systemically and in the brain. SCFAs can easily enter circulation from the gut and be transported across the BBB by monocarboxylate transporters [94], directly influencing brain biochemistry [87, 95]. For example, butyrate inhibits HDAC activity resulting in hyper acetylation and loosening of the chromatin. Consequently, there is an alteration of epigenetic signatures facilitating access of DNA repair enzymes [96] to transcribe various regulatory genes. Through this mechanism, butyrate, upregulates the regulatory regions of the Forkhead box (Foxo) gene locus, which are particularly sensitive to regulation by acetylation [97]. FOXO is a central factor to longevity as it induces expression of several life-promoting processes including anti-oxidant genes, autophagic factors and stress-response genes [98]; however, in disease states, such as patients afflicted with PD, FOXO, which also transcriptionally regulates apoptotic genes, can induce cell death [99], indicating that the tight regulation of FOXO is critical for the balance between cell death and cell survival. Particularly in neurodegeneration, the induction of autophagic genes by FOXO has proven to be a key neuroprotective function [99, 100].
There have been several studies indicating the direct neuroprotective potential of butyrate through its HDAC inhibitory action and effects on FOXO expression in both in vitro and in vivo models of neurodegenerative disease. In mouse primary cultured neurons, the ketone body β-hydroxybutyrate, also an HDAC inhibitor, was shown to be neuroprotective by inhibiting apoptosis pathways in an NMDA-induced excitotoxicity model [101]. Further, in an aged C. elegans model engineered to express a temperature-sensitive human transgene of Aβ, β-hydroxybutyrate delayed AD’s Aβ toxicity and decreased Parkinson’s α-Syn aggregation in a DAF-16/FOXO-dependent manner [102]. A similar result was seen with butyrate itself through the histone acetyltransferase action of CREB-binding protein in the hypothalamus [103]. Similarly in C. elegans, inhibition of the insulin signaling pathway (daf-2 RNAi) reduced Aβ1–42 toxicity due to aging in a DAF-16/FOXO-dependent manner indicating that FOXO is essential for regulating the age-induced toxicity of Aβ aggregation [104]. In PD, it was shown in a Drosophila melanogaster early onset PD model that FOXO was protective against PD by managing mitochondrial dynamics [105]. Altogether, manipulation of FOXO, possible through the HDAC inhibitory activity of butyrate, can be a potential therapeutic and preventative target for neurodegenerative diseases.
SCFAs modulate neurotransmitter synthesis and expression of several neurotransmitter receptors including nicotinic and GABA receptors [106]. The ability of SCFAs to have such a broad effect on neurogenesis genes is attributed to their HDAC inhibitory activity and the corresponding increase in acetylation of neurotrophic genes including BDNF and NGF [107]. Interestingly, only propionic acid and not butyric acid was able to modulate serotonergic signaling by inducing the expression of tryptophan 6-hydroxylase 1 in PC12 rat pheochromocytoma cells [106]. Similarly, in a conventional and GF mouse humanized with a healthy human microbiota, an increase in tryptophan hydroxylase 1 was noted, an effect deemed to be dependent on the action of SCFAs on EC cells [108]. Further, in an ex vivo rat colonic model, SCFAs were shown to increase serotonin secretion into the lumen possibly from the stimulation of serotonin receptors on the vagal sensory fibers [109]. These findings are significant as the majority (>90%) of the body’s serotonin is produced in the intestinal EC cells making SCFA regulation imperative for serotonin regulation in the brain [110]. SCFAs, especially butyrate and propionate, can also control the production of catecholamines by regulating tyrosine hydroxylase gene expression [111] in addition to several dopamine biosynthesis, degradation and transport genes [106]. This is very significant in the pathology of PD as tyrosine hydroxylase is the rate-limiting step to dopamine synthesis, which is often downregulated in the substantia nigra region of affected patients. A further study found in PC12 cells that both butyric and propionic acid significantly downregulated amyloid beta A4 protein precursor expression by approximately six- and threefold, respectively [106], indicating the potential neuroprotective roles against AD. Indeed, butyrate through its HDAC inhibitory activity has been shown to improve memory function in a late-stage AD mouse model and increase expression of genes implicated in associative learning [112].
The role of microbiota-produced gut histamine in neurodegenerative disease
Histamine was recently identified as a possible therapeutic agent against neurodegenerative diseases, notably MS and AD [113]. Histamine is a biogenic monoamine that is produced by EC cells in the GIT and is directly released from certain Lactobacillus spp. It plays a role in a wide variety of physiological functions including cell proliferation, allergic reactions, wound healing and regulation of immune cells as well as acting as a neurotransmitter in the brain [114]. There is a high density of histamine receptors on neurons of the striatum, thalamus, hippocampus, substantia nigra, amygdala and other areas, indicating the broad effects of histamine throughout the CNS.
Depending on the receptor that it acts upon, histamine can have either pro- or anti-inflammatory properties [113, 115]. In the brain, histamine induces an allergic inflammatory response by increasing the production of proinflammatory cytokines such as IL-1α, IL-1β, IL-6 and various chemokines. However, the action of histamine through a different receptor, namely H4R, induces an anti-inflammatory response which is particularly critical in the CNS [113]. H4R leads to the activation of several signaling factors including the JAK-STAT, MAPK/ERK and PI3 K, ultimately leading to the regulation of cytokine release, dendritic cell function and recruitment of T regulatory cells to sites of acute inflammation [116, 117]. Interestingly, histamine was recently found to be a product of gut microbial metabolism. Lactobacillus, Lactococcus, Streptococcus, Pediococcus and Enterococcus spp. all have the histidine decarboxylase gene and can produce histamine [118]. L. reuteri, which converts the dietary l-histidine into histamine, was previously identified as an immunomodulatory probiotic that can suppress TLR signaling and ultimately reduce the expression of TNFα through the modulation of PKA and ERK signaling [119].
In the context of neurodegeneration, elevated levels of histamine have been found associated with AD and are thought to elevate nitric oxide levels, thereby stimulating neuroinflammation [120]. This parallels the hypothesis that low-grade inflammation contributes to the development of neurodegenerative diseases. However, there have also been reports of deficit histaminergic signaling in the rats afflicted with vascular dementia [121]. Clearly, the concentration and localization of particular histamine receptors both centrally and systemically have a wide range of effects on the development of neurodegeneration making its regulation through the gut microbiota a possible route for therapy [114].
Microbiota-modulated ghrelin impacts neurological function
Ghrelin is a peptide produced in the GIT that acts both as a hormone to convey satiety signals and as a neuropeptide in the CNS. Ghrelin is secreted when the stomach is empty to facilitate the feeling of hunger. Apart from this, ghrelin is a key regulatory factor for many metabolic processes including energy homeostasis, inflammation and neuromodulation [122, 123]. Ghrelin receptors are diffused throughout the brain but have particularly high concentrations in the hippocampus, substantia nigra [124], raphe nuclei and ventral tegmental area [125]. Gut hormones such as PYY and cholecystokinin produced by the EC cells under the influence of the microbiota, interact with ghrelin signaling to induce feelings of satiety and direct other regulatory events [126]. Notably, the administration of prebiotics that alter microbiota populations such as Bifidobacterium spp., have been shown to reduce ghrelin secretion in humans [127]. Further, there is a clear correlation between ghrelin and the composition of the gut microbiota in rats under various nutritional statuses and levels of physical activity indicating the influence of gut microbiota dynamics on ghrelin secretion [128]. Ghrelin secretion is significant in the context of neurodegeneration as ghrelin has been shown to elicit neuroprotective effects in both AD and PD [129].
The neuroprotective abilities of ghrelin were first shown in ischemic damage models in rats where exogenous ghrelin treatment reduced ROS accumulation, protected mitochondrial integrity, and therefore promoted an anti-apoptotic environment [130]. Since then, ghrelin has been implicated in promoting synaptic plasticity and rescuing memory deficits in AD models [123]. In addition, in an Aβ mouse model of AD, ghrelin was shown to reduce the toxic accumulation of Aβ while inhibiting the excessive inflammatory response [131]. Correspondingly, AD patients have a reduced ghrelin representation in the brain, indicating that ghrelin does play a key role in its pathology [123].
In PD, activation of ghrelin receptors on substantia nigra neurons stimulates tyrosine hydroxylase expression, the rate-limiting step in dopamine synthesis. Further, ghrelin provided protection against a toxic model of PD by reducing dopaminergic cell loss and protecting mitochondrial integrity [132]. This and other studies were followed up by more mechanistic-based approaches to understand the role of ghrelin in PD, and it was found that acylated ghrelin protected 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced loss of tyrosine hydroxylase in mice while protecting GFAP expression [133]. It is clear that ghrelin has both broad-acting and local responses in the brain leading to its neuroprotective effects and the ghrelin-producing abilities of gut bacteria make it a promising therapeutic target for neurodegeneration.
Kynurenine pathway signaling in the gut–brain axis
Tryptophan is an essential amino acid critical for the synthesis of serotonin. Over 90% of serotonin is found in the GIT where it is absorbed from the diet or synthesized by EC cells from tryptophan [110]. Serotonin plays a key role in secretion, peristalsis, vasodilation, perception of pain and nausea through the 5-HT receptors in the GIT. Notably, tryptophan from the GIT can enter circulation, cross the BBB and initiate serotonin synthesis in the brain making tryptophan metabolism in the GIT critical for central serotonergic signaling.
The kynurenine pathway (KP) is the major route of tryptophan catabolism. This pathway is of interest not only because it regulates the amount of bioavailable serotonin, but also because it produces several neuroactive intermediates that have implications in neurodegenerative disorders [134]. Dysregulation of serotonergic and kynurenine routes of tryptophan metabolism influences CNS pathological conditions including dementia, Huntington’s disease and AD [135]. Of interest, probiotic treatment alters kynurenine levels [136].
Two of the key intermediate metabolites of the KP are quinolinic acid (QA) and kynurenic acid (KA). QA stimulates the overactivation of NMDA receptors and consequently stimulates excitotoxicity and neuronal cell death [137]. KA, on the other hand, is an endogenous NMDA receptor antagonist that can modulate the neurotoxic effects of QA and provide neuroprotection [138]. Notably, kynurenine produced in the gut can effectively cross the BBB and contribute to the production of these metabolites directly in the brain. Another metabolite of the KP, 3-hydroxyanthranilic acid (3-HAA), induces oxidative stress and promotes ROS production contributing to the pathogenesis of neurodegenerative diseases [139]. The balance of QA and KA levels determines the level 3-HAA toxicity in the CNS and their relative abundance in controlled by the expression of the rate-limiting enzymes in the KP.
The rate-limiting enzymes responsible for the initiation of the KP are the hepatic tryptophan 2,3-dioxygenase (TDO) and the extra hepatic indoleamine 2,3-dioxygenase (IDO). Both enzymes catalyze the conversion of tryptophan to l-kynurenine. TDO is the more processive and dominant enzyme which is inducible by glucocorticoids, whereas IDO is ubiquitously expressed and induced by inflammatory mediators such as IFN-γ [135]. The activity of IDO and TDO leads to the production of the neuroactive metabolites KA, QA and 3-HAA, via separate downstream pathways. A marker of IDO/TDO activity is the kynurenine per tryptophan quotient (KYN/TRP ratio) and it has been shown that the increase in this quotient is proportional to the level of cognitive impairment [140].
The composition of the gut microbiota has a profound effect on the metabolism of tryptophan and regulation of the KP. GF mice have increased levels of tryptophan, which can be normalized following the colonization of mice immediately post-weaning [141, 142]. Interestingly, administration of B. infantis in rats reduced the levels of 5-HIAA, the main metabolite of serotonin and reliable marker of its abundance, in the frontal cortex and also increased plasma tryptophan and KA [136].
There is also evidence that the gut microbiota can directly regulate/impact the activity of the key enzymes in KP. GF mice have reduced IDO activity, which is normalized following the induction of gut microbiota immediately post-weaning [141]. Administration of L. johnsonii leads to the reduction of serum kynurenine levels and was also shown to reduce IDO activity in vitro. The possible mechanism could be linked to the increased secretion of hydrogen peroxide, which activates the peroxidase function of IDO inhibiting its enzymatic activity [143]. This is intriguing as hydrogen peroxide is commonly released in many lactic acid bacteria adding another level of regulation [144].
Apart from excitotoxicity, increased levels of QA promote tau phosphorylation and tangle formation, therefore directly linking this pathway to AD pathogenesis [145]. IDO activity is also upregulated in the AD hippocampus, enriched in the senile plaques of AD [146] and the level of IDO activity is correlated with the level of cognitive impairment [140].
IDO inhibitors are currently being tested for their ability to protect neurons against oxidative damage, and thereby alleviate symptoms of cognitive impairment. Inhibitors of IDO can counter balance their inflammatory-induced induction and consequently reduce QA induction while increasing KA production [147]. There are also other inhibitors that can be exploited in altering the balance of KA and QA such as Ro61-8048 which inhibits kynurenine hydroxylase and has been shown to be protective against HD [148]. Considering the influence of the gut microbiota on regulation of the KP and tryptophan availability, it is possible that proper probiotic therapy could be beneficial in regulating KP dynamics either prophylactically or therapeutically in patients with neurodegenerative disorders.
Microbial-produced neurometabolites
There are many neurometabolites secreted directly from the microbiota and produced by the stimulatory action of the microbiota on secretory epithelial cells. These neurometabolites include neurotransmitters that act directly on CNS signaling cascades and through other biochemical effectors that have direct or indirect implications on CNS health [149, 150]. For example, Lactobacillus and Bifidobacterium strains can produce large quantities of GABA in the presence of a suitable substrate [151]. Gut-derived neurometabolites communicate to the CNS by local stimulation of vagal afferents and through their distal endocrine action after being absorbed into the blood stream. These variations in neurotransmitter levels manifest in behavioral changes, such as increased spontaneous motor activity from the elevated levels of dopamine, noradrenaline and serotonin in the striatum [150]. This is critical in the management of neurodegenerative disease as there is often a dysregulation of neurotransmitter production in these conditions that ultimately fuel disease progression. In Table 2, a list of neurotransmitters directly secreted by various probiotics is listed.
Conclusion
The gut–brain axis encompasses several biochemical pathways that functionally link the health of the gut microbiota and the CNS (Fig. 2). Any imbalance in the commensal gut microbiota leads to aberrant endocrine, immunological and neuronal signals that ultimately harm neuronal development and aggravate the age-related symptoms of neurodegenerative disease. The major common feature of neurodegeneration is the gradual failure of physiological systems with age, and this includes the shifting populations of the gut microbiota that propagate an inflammatory response, stimulate oxidative stress, unfavorably alter production of neuroactive molecules and modulate metabolic signals that disrupt energy metabolism in the brain. Biotherapy using probiotics shows immense potential as therapeutic or prophylactic agents against neurodegenerative disease as they reinstate balance to the microbiota and the corresponding pathways that link microbial and host metabolism. The further development and characterization of the biochemical effects of probiotic consumption on people suffering from neurodegenerative disease needs to be investigated to fully elucidate the scope of probiotics for these debilitating diseases.

Investigating the mechanisms of probiotic treatment in neurological signaling. The gut microbiota impacts neurological disease via three main modalities: (1) neuronal factors, (2) endocrine pathways and (3) immunological signals. The microbiota present in the gut lumen plays a specific role in influencing these three pathways. (1) Individual bacteria can both produce certain neurotransmitters such as DA and ACh while the same and others stimulate neurotransmitter production via the secretory ECs such as 5HT and GABA. The ECs cells can also produce several neuroactive factors including PYY, Typ and His. The neurotransmitters and neuroactive molecules enter blood circulation and cross the BBB influencing CNS signaling. Some neuroactive components also go further to stimulate the production of gut hormones in the CNS such as ghrelin and IPA that have dual roles in the CNS including neuroprotection. Further, individual microbiota species can directly stimulate electrical signals in the ENS, thereby propagating signals through the vagus nerve to stimulate the DMV. Finally, the microbiota through the production of SCFAs and FFAR signaling, releases glucose which also propagates signals through the ENS. (2) The gut microbiota directly and indirectly produces a battery of endocrine signaling molecules. SCFAs, including propionate, butyrate and acetate, are major signaling molecules produced by the microbiota that have many roles including stimulation of neurotransmitter synthesis in the periphery and centrally, inhibiting ROS production by upregulating FoxP + transcription and inhibiting apoptosis through caspase cascades. A major mechanism instigated by the gut microbiota is HPA axis stimulation and the consequent release of cortisol. Cortisol suppresses the inflammatory response and influences a number of neurological processes. Ferulic acid is another key molecule produced directly by the microbiota that has a variety of functions including suppression of ROS both directly and by indirectly by PRX/Trx signaling, suppression of apoptosis by inhibiting CRMP2 and caspase 3 expression and either directly or indirectly, suppressing the inflammatory response. (3) A healthy gut microbiota suppresses inflammation, both chronic and pathological. The MAMPs such as LTA and SlpA on the surface of microbiota directly stimulate receptors (TLR and ICAM, respectively) on immunological cells such as DCs. This interaction propagates an anti-inflammatory response with an upregulation of anti-inflammatory factors (IL-10 and IL-4) while suppressing proinflammatory cytokines (TNFα, IL-1β and IL-6). In addition, some microbiota also directly suppresses proinflammatory cytokines such as the action of B. animalis on IL-6. Finally, the gut microbiota influences the production of mucin, an inhibitory chemical gel that blocks the penetrance of pathogens through the gut. 5HT serotonin, Ach acetylcholine, BBB blood–brain barrier, CRMP2 collapsin response mediator protein family, DA dopamine, DHA docosahexaenoic acid, DMV dorsal motor nucleus of the vagus, EC enterochromaffin cell, ENS enteric nervous system, EPA eicosapentaenoic acid, FFAR free fatty acid receptor, GABA gamma-aminobutyric acid, His histamine, HPA hypothalamic–pituitary–adrenal axis, IFNγ interferon gamma, IL-10 interleukin 10, IL-12 interleukin 12, IPA indole-3-propionic acid, NA noradrenaline, PRX peroxiredoxin, PYY peptide YY, ROS reactive oxygen species, SCFAs short-chain fatty acids, TNFα tumor necrosis factor alpha, Trp tryptophan, Trx thioredoxin
References
Burokas A, Moloney RD, Dinan TG, Cryan JF (2015) Microbiota regulation of the mammalian gut–brain axis. Adv Appl Microbiol 91:1–62. doi:10.1016/bs.aambs.2015.02.001
Forsythe P, Bienenstock J, Kunze WA (2014) Vagal pathways for microbiome–brain–gut axis communication. Microbial endocrinology: the microbiota–gut–brain axis in health and disease. Springer, New York, pp 115–133
Bauer KC, Huus KE, Finlay BB (2016) Microbes and the mind: emerging hallmarks of the gut microbiota–brain axis. Cell Microbiol 18:632–644. doi:10.1111/cmi.12585
Noble EE, Hsu TM, Kanoski SE (2017) Gut to brain dysbiosis: mechanisms linking western diet consumption, the microbiome, and cognitive impairment. Front Behav Neurosci 11:9. doi:10.3389/fnbeh.2017.00009
Mulak A, Bonaz B (2015) Brain–gut–microbiota axis in Parkinson’s disease. WJG 21:10609–10620. doi:10.3748/wjg.v21.i37.10609
Friedland RP (2015) Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J Alzheimers Dis 45:349–362. doi:10.3233/JAD-142841
Qin J, Li R, Raes J et al (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65. doi:10.1038/nature08821
Rajilic-Stojanovic M, Smidt H, de Vos WM (2007) Diversity of the human gastrointestinal tract microbiota revisited. Environ Microbiol 9:2125–2136. doi:10.1111/j.1462-2920.2007.01369.x
The Human Microbiome Project Consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. doi:10.1038/nature11234
Mandal RS, Saha S, Das S (2015) Metagenomic surveys of gut microbiota. Genom Proteom Bioinform 13:148–158. doi:10.1016/j.gpb.2015.02.005
Everard A, Cani PD (2014) Gut microbiota and GLP-1. Rev Endocr Metab Disord 15:189–196. doi:10.1007/s11154-014-9288-6
Burns AJ (2005) Migration of neural crest-derived enteric nervous system precursor cells to and within the gastrointestinal tract. Int J Dev Biol 49:143–150. doi:10.1387/ijdb.041935ab
O’Mahony SM, Clarke G, Borre YE et al (2014) Serotonin, tryptophan metabolism and the brain–gut–microbiome axis. Behav Brain Res. doi:10.1016/j.bbr.2014.07.027
Ochoa-Reparaz J, Mielcarz DW, Begum-Haque S, Kasper LH (2011) Gut, bugs, and brain: role of commensal bacteria in the control of central nervous system disease. Ann Neurol 69:240–247. doi:10.1002/ana.22344
Luczynski P, McVey Neufeld K-A, Oriach CS et al (2016) Growing up in a bubble: using germ-free animals to assess the influence of the gut microbiota on brain and behavior. Int J Neuropsychopharmacol 19(8):234–248. doi:10.1093/ijnp/pyw020
Houlden A, Goldrick M, Brough D et al (2016) Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun 57:10–20. doi:10.1016/j.bbi.2016.04.003
Catanzaro R, Anzalone MG, Calabrese F et al (2014) The gut microbiota and its correlations with the central nervous system disorders. Panminerva Med 57(3):127–143
Dinan TG, Cryan JF (2017) Gut instincts: microbiota as a key regulator of brain development, ageing and neurodegeneration. J Physiol 595:489–503. doi:10.1113/JP273106
Kohler CA, Maes M, Slyepchenko A et al (2016) The gut–brain axis, including the microbiome, leaky gut and bacterial translocation: mechanisms and pathophysiological role in Alzheimer’s disease. Curr Pharm Des 22:6152–6166
Jyothi HJ, Vidyadhara DJ, Mahadevan A et al (2015) Aging causes morphological alterations in astrocytes and microglia in human substantia nigra pars compacta. Neurobiol Aging 36:3321–3333. doi:10.1016/j.neurobiolaging.2015.08.024
Erny D, Hrabe de Angelis AL, Jaitin D et al (2015) Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci 18:965–977. doi:10.1038/nn.4030
Sampson TR, Debelius JW, Thron T et al (2016) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167(1469–1480):e12. doi:10.1016/j.cell.2016.11.018
Alam MZ, Alam Q, Kamal MA et al (2014) A possible link of gut microbiota alteration in type 2 diabetes and Alzheimer’s disease pathogenicity: an update. CNS Neurol Disord Drug Targets 13:383–390
Naseer MI, Bibi F, Alqahtani MH et al (2014) Role of gut microbiota in obesity, type 2 diabetes and Alzheimer’s disease. CNS Neurol Disord Drug Targets 13:305–311
Bekkering P, Jafri I, van Overveld FJ, Rijkers GT (2013) The intricate association between gut microbiota and development of type 1, type 2 and type 3 diabetes. Expert Rev Clin Immunol 9:1031–1041. doi:10.1586/1744666X.2013.848793
Rao M, Gershon MD (2016) The bowel and beyond: the enteric nervous system in neurological disorders. Nat Rev Gastroenterol Hepatol 13:517–528. doi:10.1038/nrgastro.2016.107
Baltazar MT, Dinis-Oliveira RJ, de Lourdes Bastos M et al (2014) Pesticides exposure as etiological factors of Parkinson’s disease and other neurodegenerative diseases—a mechanistic approach. Toxicol Lett 230:85–103. doi:10.1016/j.toxlet.2014.01.039
Virmani A, Pinto L, Binienda Z, Ali S (2013) Food, nutrigenomics, and neurodegeneration–neuroprotection by what you eat! Mol Neurobiol 48:353–362. doi:10.1007/s12035-013-8498-3
Ley RE, Hamady M, Lozupone C et al (2008) Evolution of mammals and their gut microbes. Science 320:1647–1651. doi:10.1126/science.1155725
Goldman JG, Postuma R (2014) Premotor and nonmotor features of Parkinson’s disease. Curr Opin Neurol 27:434–441. doi:10.1097/WCO.0000000000000112
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13. doi:10.1042/BJ20081386
Mitsuma T, Odajima H, Momiyama Z et al (2008) Enhancement of gene expression by a peptide p(CHWPR) produced by Bifidobacterium lactis BB-12. Microbiol Immunol 52:144–155. doi:10.1111/j.1348-0421.2008.00022.x
Wang X, Michaelis EK (2010) Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 2:12. doi:10.3389/fnagi.2010.00012
Zhu X, Raina AK, Lee H-G et al (2004) Oxidative stress signalling in Alzheimer’s disease. Brain Res 1000:32–39
Franceschi C, Campisi J (2014) Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69(Suppl 1):S4–S9. doi:10.1093/gerona/glu057
Biagi E, Nylund L, Candela M et al (2010) Through ageing, and beyond: gut microbiota and inflammatory status in seniors and centenarians. PLoS One 5:e10667. doi:10.1371/journal.pone.0010667
Steen E, Terry BM, Rivera EJ et al (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J Alzheimers Dis 7:63–80
Cheng J, Palva AM, de Vos WM, Satokari R (2013) Contribution of the intestinal microbiota to human health: from birth to 100 years of age. In: Dobrindt U, Hacker JH, Svanborg C (eds) Between pathogenicity and commensalism. Springer, Berlin, pp 323–346
Claesson MJ, Cusack S, O’Sullivan O et al (2011) Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA 108(Suppl 1):4586–4591. doi:10.1073/pnas.1000097107
Cassani E, Barichella M, Cancello R et al (2015) Increased urinary indoxyl sulfate (indican): new insights into gut dysbiosis in Parkinson’s disease. Parkinsonism Relat Disord 21:389–393. doi:10.1016/j.parkreldis.2015.02.004
Scheperjans F, Aho V, Pereira PAB et al (2014) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30(3):350–358. doi:10.1002/mds.26069
Keshavarzian A, Green SJ, Engen PA et al (2015) Colonic bacterial composition in Parkinson’s disease. Mov Disord 30:1351–1360. doi:10.1002/mds.26307
Shannon KM, Keshavarzian A, Dodiya HB et al (2012) Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord 27:716–719. doi:10.1002/mds.25020
Unger MM, Spiegel J, Dillmann K-U et al (2016) Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 32:66–72. doi:10.1016/j.parkreldis.2016.08.019
Kidd SK, Schneider JS (2010) Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain Res 1354:172–178. doi:10.1016/j.brainres.2010.07.041
Wu X, Chen PS, Dallas S et al (2008) Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol 11:1123–1134. doi:10.1017/S1461145708009024
Guseo A (2012) The Parkinson puzzle. Orv Hetil 153:2060–2069. doi:10.1556/OH.2012.29461
Smith PD, Smythies LE, Shen R et al (2011) Intestinal macrophages and response to microbial encroachment. Mucosal Immunol 4:31–42. doi:10.1038/mi.2010.66
Faden AI, Loane DJ (2015) Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics 12:143–150. doi:10.1007/s13311-014-0319-5
Vivekanantham S, Shah S, Dewji R et al (2015) Neuroinflammation in Parkinson’s disease: role in neurodegeneration and tissue repair. Int J Neurosci 125:717–725. doi:10.3109/00207454.2014.982795
Clarke G, Stilling RM, Kennedy PJ et al (2014) Minireview: gut microbiota: the neglected endocrine organ. Mol Endocrinol 28:1221–1238. doi:10.1210/me.2014-1108
Mancuso C, Santangelo R (2014) Ferulic acid: pharmacological and toxicological aspects. Food Chem Toxicol 65:185–195. doi:10.1016/j.fct.2013.12.024
Trombino S, Cassano R, Ferrarelli T et al (2013) Trans-ferulic acid-based solid lipid nanoparticles and their antioxidant effect in rat brain microsomes. Colloids Surf B Biointerfaces 109:273–279. doi:10.1016/j.colsurfb.2013.04.005
Hu C-T, Wu J-R, Cheng C-C et al (2011) Reactive oxygen species-mediated PKC and integrin signaling promotes tumor progression of human hepatoma HepG2. Clin Exp Metastasis 28:851–863. doi:10.1007/s10585-011-9416-6
Yabe T, Hirahara H, Harada N et al (2010) Ferulic acid induces neural progenitor cell proliferation in vitro and in vivo. Neuroscience 165:515–524. doi:10.1016/j.neuroscience.2009.10.023
Yu L, Zhang Y, Liao M et al (2011) Neurogenesis-enhancing effect of sodium ferulate and its role in repair following stress-induced neuronal damage. WJNS 1(2):9–18. doi:10.4236/wjns.2011.12002
Tomaro-Duchesneau C, Saha S, Malhotra M et al (2012) Probiotic ferulic acid esterase active Lactobacillus fermentum NCIMB 5221 APA microcapsules for oral delivery: preparation and in vitro characterization. Pharmaceuticals (Basel) 5:236–248. doi:10.3390/ph5020236
Szwajgier D, Dmowska K (2010) Novel ferulic acid esterases from Bifidobacterium sp. produced on selected synthetic and natural carbon sources. Acta Sci Pol Technol Aliment 9:305–318
Bhathena J, Martoni C, Kulamarva A et al (2009) Orally delivered microencapsulated live probiotic formulation lowers serum lipids in hypercholesterolemic hamsters. J Med Food 12:310–319. doi:10.1089/jmf.2008.0166
Durairajan SSK, Yuan Q, Xie L et al (2008) Salvianolic acid B inhibits Abeta fibril formation and disaggregates preformed fibrils and protects against Abeta-induced cytotoxicty. Neurochem Int 52:741–750. doi:10.1016/j.neuint.2007.09.006
Mori T, Koyama N, Guillot-Sestier M-V et al (2013) Ferulic acid is a nutraceutical β-secretase modulator that improves behavioral impairment and Alzheimer-like pathology in transgenic mice. PLoS One 8:e55774. doi:10.1371/journal.pone.0055774
Yan J-J, Jung J-S, Kim T-K et al (2013) Protective effects of ferulic acid in amyloid precursor protein plus presenilin-1 transgenic mouse model of Alzheimer disease. Biol Pharm Bull 36:140–143
Zhang L, Wang H, Wang T et al (2015) Ferulic acid ameliorates nerve injury induced by cerebral ischemia in rats. Exp Ther Med 9:972–976
Srinivasan M, Sudheer AR, Menon VP (2007) Ferulic acid: therapeutic potential through its antioxidant property. J Clin Biochem Nutr 40:92–100. doi:10.3164/jcbn.40.92
Cheng C-Y, Tang N-Y, Kao S-T, Hsieh C-L (2016) Ferulic acid administered at various time points protects against cerebral infarction by activating p38 MAPK/p90RSK/CREB/Bcl-2 anti-apoptotic signaling in the subacute phase of cerebral ischemia–reperfusion injury in rats. PLoS One 11:e0155748
Sung J-H, Gim S-A, Koh P-O (2014) Ferulic acid attenuates the cerebral ischemic injury-induced decrease in peroxiredoxin-2 and thioredoxin expression. Neurosci Lett 566:88–92. doi:10.1016/j.neulet.2014.02.040
Patenaude A, Murthy MRV, Mirault M-E (2005) Emerging roles of thioredoxin cycle enzymes in the central nervous system. Cell Mol Life Sci 62:1063–1080. doi:10.1007/s00018-005-4541-5
Gan Y, Ji X, Hu X et al (2012) Transgenic overexpression of peroxiredoxin-2 attenuates ischemic neuronal injury via suppression of a redox-sensitive pro-death signaling pathway. Antioxid Redox Signal 17:719–732. doi:10.1089/ars.2011.4298
Saitoh M, Nishitoh H, Fujii M et al (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17:2596–2606. doi:10.1093/emboj/17.9.2596
Ma YH, Su N, Chao XD et al (2012) Thioredoxin-1 attenuates post-ischemic neuronal apoptosis via reducing oxidative/nitrative stress. Neurochem Int 60:475–483. doi:10.1016/j.neuint.2012.01.029
Gim S-A, Sung J-H, Shah F-A et al (2013) Ferulic acid regulates the AKT/GSK-3β/CRMP-2 signaling pathway in a middle cerebral artery occlusion animal model. Lab Anim Res 29:63. doi:10.5625/lar.2013.29.2.63
Gao C, Holscher C, Liu Y, Li L (2012) GSK3: a key target for the development of novel treatments for type 2 diabetes mellitus and Alzheimer disease. Rev Neurosci 23:1–11
Yoshimura T, Kawano Y, Arimura N et al (2005) GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120:137–149. doi:10.1016/j.cell.2004.11.012
Gu Y, Hamajima N, Ihara Y (2000) Neurofibrillary tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, and Ser-522. Biochemistry 39:4267–4275
Petratos S, Li Q-X, George AJ et al (2008) The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 131:90–108. doi:10.1093/brain/awm260
Soutar MPM, Thornhill P, Cole AR, Sutherland C (2009) Increased CRMP2 phosphorylation is observed in Alzheimer’s disease; does this tell us anything about disease development? Curr Alzheimer Res 6:269–278
Verbeke KA, Boobis AR, Chiodini A et al (2015) Towards microbial fermentation metabolites as markers for health benefits of prebiotics. Nutr Res Rev 28:42–66. doi:10.1017/S0954422415000037
Tagliabue A, Elli M (2013) The role of gut microbiota in human obesity: recent findings and future perspectives. Nutr Metab Cardiovasc Dis 23:160–168. doi:10.1016/j.numecd.2012.09.002
Kasubuchi M, Hasegawa S, Hiramatsu T et al (2015) Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients 7:2839–2849. doi:10.3390/nu7042839
den Besten G, van Eunen K, Groen AK et al (2013) The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54:2325–2340. doi:10.1194/jlr.R036012
Gao Z, Yin J, Zhang J et al (2009) Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58:1509–1517. doi:10.2337/db08-1637
Fushimi T, Suruga K, Oshima Y et al (2006) Dietary acetic acid reduces serum cholesterol and triacylglycerols in rats fed a cholesterol-rich diet. Br J Nutr 95:916–924
Todesco T, Rao AV, Bosello O, Jenkins DJ (1991) Propionate lowers blood glucose and alters lipid metabolism in healthy subjects. Am J Clin Nutr 54:860–865
Saad MJA, Santos A, Prada PO (2016) Linking gut microbiota and inflammation to obesity and insulin resistance. Physiology (Bethesda) 31:283–293. doi:10.1152/physiol.00041.2015
Soldavini J, Kaunitz JD (2013) Pathobiology and potential therapeutic value of intestinal short-chain fatty acids in gut inflammation and obesity. Dig Dis Sci 58:2756–2766. doi:10.1007/s10620-013-2744-4
Brahe LK, Astrup A, Larsen LH (2013) Is butyrate the link between diet, intestinal microbiota and obesity-related metabolic diseases? Obes Rev 14:950–959. doi:10.1111/obr.12068
De Vadder F, Kovatcheva-Datchary P, Goncalves D et al (2014) Microbiota-generated metabolites promote metabolic benefits via gut–brain neural circuits. Cell 156:84–96. doi:10.1016/j.cell.2013.12.016
Delaere F, Duchampt A, Mounien L et al (2012) The role of sodium-coupled glucose co-transporter 3 in the satiety effect of portal glucose sensing. Mol Metab 2:47–53
Lacassagne O, Kessler JP (2000) Cellular and subcellular distribution of the amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor subunit GluR2 in the rat dorsal vagal complex. Neuroscience 99:557–563
Greene JG (2014) Causes and consequences of degeneration of the dorsal motor nucleus of the vagus nerve in Parkinson’s disease. Antioxid Redox Signal 21:649–667. doi:10.1089/ars.2014.5859
Lal S, Kirkup AJ, Brunsden AM et al (2001) Vagal afferent responses to fatty acids of different chain length in the rat. AJP: Gastrointest Liver Physiol 281:G907–G915
Sjogren MJC, Hellstrom PTO, Jonsson MAG et al (2002) Cognition-enhancing effect of vagus nerve stimulation in patients with Alzheimer’s disease: a pilot study. J Clin Psychiatry 63:972–980
Merrill CA, Jonsson MAG, Minthon L et al (2006) Vagus nerve stimulation in patients with Alzheimer’s disease: additional follow-up results of a pilot study through 1 year. J Clin Psychiatry 67:1171–1178
Vijay N, Morris ME (2014) Role of monocarboxylate transporters in drug delivery to the brain. Curr Pharm Des 20:1487–1498
Braniste V, Al-Asmakh M, Kowal C et al (2014) The gut microbiota influences blood–brain barrier permeability in mice. Sci Transl Med 6:263ra158. doi:10.1126/scitranslmed.3009759
Brown AJ, Goldsworthy SM, Barnes AA et al (2003) The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 278:11312–11319
Beharry AW, Sandesara PB, Roberts BM et al (2014) HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J Cell Sci 127:1441–1453. doi:10.1242/jcs.136390
Martins R, Lithgow GJ, Link W (2016) Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 15:196–207. doi:10.1111/acel.12427
Pino E, Amamoto R, Zheng L et al (2014) FOXO3 determines the accumulation of alpha-synuclein and controls the fate of dopaminergic neurons in the substantia nigra. Hum Mol Genet 23:1435–1452. doi:10.1093/hmg/ddt530
Xu P, Das M, Reilly J, Davis RJ (2011) JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 25:310–322. doi:10.1101/gad.1984311
Bahia PK, Pugh V, Hoyland K et al (2012) Neuroprotective effects of phenolic antioxidant tBHQ associate with inhibition of FoxO3a nuclear translocation and activity. J Neurochem 123:182–191. doi:10.1111/j.1471-4159.2012.07877.x
Edwards C, Canfield J, Copes N et al (2014) D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging (Albany NY) 6:621–644. doi:10.18632/aging.100683
Zhang M, Poplawski M, Yen K et al (2009) Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biol 7:e1000245. doi:10.1371/journal.pbio.1000245
Cohen E, Bieschke J, Perciavalle RM et al (2006) Opposing activities protect against age-onset proteotoxicity. Science 313:1604–1610. doi:10.1126/science.1124646
Koh H, Kim H, Kim MJ et al (2012) Silent information regulator 2 (Sir2) and Forkhead box O (FOXO) complement mitochondrial dysfunction and dopaminergic neuron loss in Drosophila PTEN-induced kinase 1 (PINK1) null mutant. J Biol Chem 287:12750–12758. doi:10.1074/jbc.M111.337907
Nankova BB, Agarwal R, MacFabe DF, La Gamma EF (2014) Enteric bacterial metabolites propionic and butyric acid modulate gene expression, including CREB-dependent catecholaminergic neurotransmission, in PC12 cells—possible relevance to autism spectrum disorders. PLoS One 9:e103740
Bourassa MW, Alim I, Bultman SJ, Ratan RR (2016) Butyrate, neuroepigenetics and the gut microbiome: can a high fiber diet improve brain health? Neurosci Lett 625:56–63. doi:10.1016/j.neulet.2016.02.009
Reigstad CS, Salmonson CE, Rainey JF et al (2015) Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J 29:1395–1403. doi:10.1096/fj.14-259598
Fukumoto S, Tatewaki M, Yamada T et al (2003) Short-chain fatty acids stimulate colonic transit via intraluminal 5-HT release in rats. Am J Physiol Regul Integr Comp Physiol 284:R1269–R1276
Gershon MD (2013) 5-Hydroxytryptamine (serotonin) in the gastrointestinal tract. Curr Opin Endocrinol Diabetes Obes 20:14–21. doi:10.1097/MED.0b013e32835bc703
DeCastro M, Nankova BB, Shah P et al (2005) Short chain fatty acids regulate tyrosine hydroxylase gene expression through a cAMP-dependent signaling pathway. Brain Res Mol Brain Res 142:28–38. doi:10.1016/j.molbrainres.2005.09.002
Govindarajan N, Agis-Balboa RC, Walter J et al (2011) Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J Alzheimers Dis 26:187–197. doi:10.3233/JAD-2011-110080
del Rio R, Noubade R, Saligrama N et al (2012) Histamine H4 receptor optimizes T regulatory cell frequency and facilitates anti-inflammatory responses within the central nervous system. J Immunol 188:541–547. doi:10.4049/jimmunol.1101498
Naddafi F, Mirshafiey A (2013) The neglected role of histamine in Alzheimer’s disease. Am J Alzheimers Dis Other Demen 28:327–336. doi:10.1177/1533317513488925
Dong H, Zhang W, Zeng X et al (2014) Histamine induces upregulated expression of histamine receptors and increases release of inflammatory mediators from microglia. Mol Neurobiol 49:1487–1500. doi:10.1007/s12035-014-8697-6
Schneider E, Rolli-Derkinderen M, Arock M, Dy M (2002) Trends in histamine research: new functions during immune responses and hematopoiesis. Trends Immunol 23:255–263
Morgan RK, McAllister B, Cross L et al (2007) Histamine 4 receptor activation induces recruitment of FoxP3+ T cells and inhibits allergic asthma in a murine model. J Immunol 178:8081–8089
Landete JM, de las Rivas B, Marcobal A, Munoz R (2008) Updated molecular knowledge about histamine biosynthesis by bacteria. Crit Rev Food Sci Nutr 48:697–714. doi:10.1080/10408390701639041
Thomas CM, Hong T, van Pijkeren JP et al (2012) Histamine derived from probiotic Lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLoS One 7:e31951
Alvarez XA, Franco A, Fernandez-Novoa L, Cacabelos R (1996) Blood levels of histamine, IL-1 beta, and TNF-alpha in patients with mild to moderate Alzheimer disease. Mol Chem Neuropathol 29:237–252. doi:10.1007/BF02815005
Stasiak A, Mussur M, Unzeta M et al (2011) The central histamine level in rat model of vascular dementia. J Physiol Pharmacol 62:549–558
Chen L, Xing T, Wang M et al (2011) Local infusion of ghrelin enhanced hippocampal synaptic plasticity and spatial memory through activation of phosphoinositide 3-kinase in the dentate gyrus of adult rats. Eur J Neurosci 33:266–275. doi:10.1111/j.1460-9568.2010.07491.x
Gahete MD, Cordoba-Chacon J, Kineman RD et al (2011) Role of ghrelin system in neuroprotection and cognitive functions: implications in Alzheimer’s disease. Peptides 32:2225–2228. doi:10.1016/j.peptides.2011.09.019
Steinert RE, Feinle-Bisset C, Asarian L et al (2017) Ghrelin, CCK, GLP-1, and PYY(3-36): secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol Rev 97:411–463. doi:10.1152/physrev.00031.2014
Guan XM, Yu H, Palyha OC et al (1997) Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res 48:23–29
Bauer PV, Hamr SC, Duca FA (2016) Regulation of energy balance by a gut–brain axis and involvement of the gut microbiota. Cell Mol Life Sci 73:737–755. doi:10.1007/s00018-015-2083-z
Fallucca F, Porrata C, Fallucca S, Pianesi M (2014) Influence of diet on gut microbiota, inflammation and type 2 diabetes mellitus. First experience with macrobiotic Ma-Pi 2 diet. Diabetes Metab Res Rev 30(Suppl 1):48–54. doi:10.1002/dmrr.2518
Queipo-Ortuno MI, Seoane LM, Murri M et al (2013) Gut microbiota composition in male rat models under different nutritional status and physical activity and its association with serum leptin and ghrelin levels. PLoS One 8:e65465
Dos Santos VV, Rodrigues ALS, De Lima TC et al (2013) Ghrelin as a neuroprotective and palliative agent in Alzheime’s and Parkinson’s disease. Curr Pharm Des 19:6773–6790
Liu Y, Chen L, Xu X et al (2009) Both ischemic preconditioning and ghrelin administration protect hippocampus from ischemia/reperfusion and upregulate uncoupling protein-2. BMC Physiol 9:17. doi:10.1186/1472-6793-9-17
Moon M, Choi JG, Nam DW et al (2011) Ghrelin ameliorates cognitive dysfunction and neurodegeneration in intrahippocampal amyloid-beta1-42 oligomer-injected mice. J Alzheimers Dis 23:147–159. doi:10.3233/JAD-2010-101263
Andrews ZB, Erion D, Beiler R et al (2009) Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J Neurosci 29:14057–14065. doi:10.1523/JNEUROSCI.3890-09.2009
Bayliss JA, Lemus M, Santos VV et al (2016) Acylated but not des-acyl ghrelin is neuroprotective in an MPTP mouse model of Parkinson’s disease. J Neurochem 137:460–471. doi:10.1111/jnc.13576
Schwarcz R, Bruno JP, Muchowski PJ, Wu H-Q (2012) Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13:465–477. doi:10.1038/nrn3257
Ruddick JP, Evans AK, Nutt DJ et al (2006) Tryptophan metabolism in the central nervous system: medical implications. Expert Rev Mol Med 8:1–27. doi:10.1017/S1462399406000068
Desbonnet L, Garrett L, Clarke G et al (2008) The probiotic Bifidobacteria infantis: an assessment of potential antidepressant properties in the rat. J Psychiatr Res 43:164–174. doi:10.1016/j.jpsychires.2008.03.009
Braidy N, Grant R, Adams S et al (2009) Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox Res 16:77–86. doi:10.1007/s12640-009-9051-z
Vamos E, Pardutz A, Klivenyi P et al (2009) The role of kynurenines in disorders of the central nervous system: possibilities for neuroprotection. J Neurol Sci 283:21–27. doi:10.1016/j.jnd.2009.02.326
Guillemin GJ, Williams KR, Smith DG et al (2003) Quinolinic acid in the pathogenesis of Alzheimer’s disease. Adv Exp Med Biol 527:167–176
Widner B, Leblhuber F, Walli J et al (2000) Tryptophan degradation and immune activation in Alzheimer’s disease. J Neural Transm 107:343–353
Clarke G, Grenham S, Scully P et al (2013) The microbiome–gut–brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry 18:666–673. doi:10.1038/mp.2012.77
Wikoff WR, Anfora AT, Liu J et al (2009) Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 106:3698–3703. doi:10.1073/pnas.0812874106
Freewan M, Rees MD, Plaza TSS et al (2013) Human indoleamine 2,3-dioxygenase is a catalyst of physiological heme peroxidase reactions: implications for the inhibition of dioxygenase activity by hydrogen peroxide. J Biol Chem 288:1548–1567. doi:10.1074/jbc.M112.410993
Tomas MSJ, Claudia Otero M, Ocana V, Elena Nader-Macias M (2004) Production of antimicrobial substances by lactic acid bacteria I: determination of hydrogen peroxide. Methods Mol Biol 268:337–346. doi:10.1385/1-59259-766-1:337
Rahman A, Ting K, Cullen KM et al (2009) The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS One 4:e6344. doi:10.1371/journal.pone.0006344
Guillemin GJ, Brew BJ, Noonan CE et al (2005) Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease hippocampus. Neuropathol Appl Neurobiol 31:395–404. doi:10.1111/j.1365-2990.2005.00655.x
Yamada A, Akimoto H, Kagawa S et al (2009) Proinflammatory cytokine interferon-gamma increases induction of indoleamine 2,3-dioxygenase in monocytic cells primed with amyloid beta peptide 1–42: implications for the pathogenesis of Alzheimer’s disease. J Neurochem 110:791–800. doi:10.1111/j.1471-4159.2009.06175.x
Giorgini F, Guidetti P, Nguyen Q et al (2005) A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet 37:526–531. doi:10.1038/ng1542
Forsythe P, Kunze WA, Bienenstock J (2012) On communication between gut microbes and the brain. Curr Opin Gastroenterol 28:557–562. doi:10.1097/MOG.0b013e3283572ffa
Lyte M (2014) Microbial endocrinology: host–microbiota neuroendocrine interactions influencing brain and behavior. Gut Microbes 5:381–389. doi:10.4161/gmic.28682
Barrett E, Ross RP, O’Toole PW et al (2012) gamma-Aminobutyric acid production by culturable bacteria from the human intestine. J Appl Microbiol 113:411–417. doi:10.1111/j.1365-2672.2012.05344.x
Sun J, Ling Z, Wang F et al (2016) Clostridium butyricum pretreatment attenuates cerebral ischemia/reperfusion injury in mice via anti-oxidation and anti-apoptosis. Neurosci Lett 613:30–35. doi:10.1016/j.neulet.2015.12.047
Ait-Belgnaoui A, Colom A, Braniste V et al (2014) Probiotic gut effect prevents the chronic psychological stress-induced brain activity abnormality in mice. Neurogastroenterol Motil 26:510–520. doi:10.1111/nmo.12295
Ezendam J, de Klerk A, Gremmer ER, van Loveren H (2008) Effects of Bifidobacterium animalis administered during lactation on allergic and autoimmune responses in rodents. Clin Exp Immunol 154:424–431. doi:10.1111/j.1365-2249.2008.03788.x
Ochoa-Reparaz J, Mielcarz DW, Wang Y et al (2010) A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol 3:487–495. doi:10.1038/mi.2010.29
Kobayashi T, Kato I, Nanno M et al (2010) Oral administration of probiotic bacteria, Lactobacillus casei and Bifidobacterium breve, does not exacerbate neurological symptoms in experimental autoimmune encephalomyelitis. Immunopharmacol Immunotoxicol 32:116–124. doi:10.3109/08923970903200716
Kobayashi T, Suzuki T, Kaji R et al (2012) Probiotic upregulation of peripheral IL-17 responses does not exacerbate neurological symptoms in experimental autoimmune encephalomyelitis mouse models. Immunopharmacol Immunotoxicol 34:423–433. doi:10.3109/08923973.2010.617755
Kwon H-K, Kim G-C, Kim Y et al (2013) Amelioration of experimental autoimmune encephalomyelitis by probiotic mixture is mediated by a shift in T helper cell immune response. Clin Immunol 146:217–227. doi:10.1016/j.clim.2013.01.001
Lavasani S, Dzhambazov B, Nouri M et al (2010) A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS One 5:e9009. doi:10.1371/journal.pone.0009009
Ohland CL, Kish L, Bell H et al (2013) Effects of Lactobacillus helveticus on murine behavior are dependent on diet and genotype and correlate with alterations in the gut microbiome. Psychoneuroendocrinology 38:1738–1747. doi:10.1016/j.psyneuen.2013.02.008
Davari S, Talaei SA, Alaei H, Salami M (2013) Probiotics treatment improves diabetes-induced impairment of synaptic activity and cognitive function: behavioral and electrophysiological proofs for microbiome–gut–brain axis. Neuroscience 240:287–296. doi:10.1016/j.neuroscience.2013.02.055
Gareau MG, Wine E, Rodrigues DM et al (2011) Bacterial infection causes stress-induced memory dysfunction in mice. Gut 60:307–317. doi:10.1136/gut.2009.202515
Rao AV, Bested AC, Beaulne TM et al (2009) A randomized, double-blind, placebo-controlled pilot study of a probiotic in emotional symptoms of chronic fatigue syndrome. Gut Pathog 1:6. doi:10.1186/1757-4749-1-6
Yan MH, Wang X, Zhu X (2013) Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med 62:90–101. doi:10.1016/j.freeradbiomed.2012.11.014
Distrutti E, O’Reilly J-A, McDonald C et al (2014) Modulation of intestinal microbiota by the probiotic VSL#3 resets brain gene expression and ameliorates the age-related deficit in LTP. PLoS One 9:e106503
Girard S-A, Bah TM, Kaloustian S et al (2009) Lactobacillus helveticus and Bifidobacterium longum taken in combination reduce the apoptosis propensity in the limbic system after myocardial infarction in a rat model. Br J Nutr 102:1420–1425. doi:10.1017/S0007114509990766
Liu J, Sun J, Wang F et al (2015) Neuroprotective effects of Clostridium butyricum against vascular dementia in mice via metabolic butyrate. Biomed Res Int 2015:412946. doi:10.1155/2015/412946
O’Sullivan E, Barrett E, Grenham S et al (2011) BDNF expression in the hippocampus of maternally separated rats: does Bifidobacterium breve 6330 alter BDNF levels? Benef Microbes 2:199–207. doi:10.3920/BM2011.0015
Bercik P, Park AJ, Sinclair D et al (2011) The anxiolytic effect of Bifidobacterium longum NCC3001 involves vagal pathways for gut–brain communication. Neurogastroenterol Motil 23:1132–1139. doi:10.1111/j.1365-2982.2011.01796.x
Le TK, Hosaka T, Le TT et al (2014) Oral administration of Bifidobacterium spp. improves insulin resistance, induces adiponectin, and prevents inflammatory adipokine expressions. Biomed Res Int 35:303–310
Bomhof MR, Saha DC, Reid DT et al (2014) Combined effects of oligofructose and Bifidobacterium animalis on gut microbiota and glycemia in obese rats. Obesity (Silver Spring) 22:763–771. doi:10.1002/oby.20632
Hoarau C, Martin L, Faugaret D et al (2008) Supernatant from bifidobacterium differentially modulates transduction signaling pathways for biological functions of human dendritic cells. PLoS One 3:e2753. doi:10.1371/journal.pone.0002753
Chang C-Y, Ke D-S, Chen J-Y (2009) Essential fatty acids and human brain. Acta Neurol Taiwan 18:231–241
Wall R, Marques TM, O’Sullivan O et al (2012) Contrasting effects of Bifidobacterium breve NCIMB 702258 and Bifidobacterium breve DPC 6330 on the composition of murine brain fatty acids and gut microbiota. Am J Clin Nutr 95:1278–1287. doi:10.3945/ajcn.111.026435
McCarthy J, O’Mahony L, O’Callaghan L et al (2003) Double blind, placebo controlled trial of two probiotic strains in interleukin 10 knockout mice and mechanistic link with cytokine balance. Gut 52:975–980
Rodes L, Khan A, Paul A et al (2013) Effect of probiotics Lactobacillus and Bifidobacterium on gut-derived lipopolysaccharides and inflammatory cytokines: an in vitro study using a human colonic microbiota model. J Microbiol Biotechnol 23:518–526
Sudo N, Chida Y, Aiba Y et al (2004) Postnatal microbial colonization programs the hypothalamic–pituitary–adrenal system for stress response in mice. J Physiol 558:263–275. doi:10.1113/jphysiol.2004.063388
Stone TW, Stoy N, Darlington LG (2013) An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci 34:136–143. doi:10.1016/j.tips.2012.09.006
Mao Y-K, Kasper DL, Wang B et al (2013) Bacteroides fragilis polysaccharide A is necessary and sufficient for acute activation of intestinal sensory neurons. Nat Commun 4:1465. doi:10.1038/ncomms2478
Kuley E, Ozogul F, Ozogul Y, Akyol I (2011) The function of lactic acid bacteria and brine solutions on biogenic amine formation by foodborne pathogens in trout fillets. Food Chem 129:1211–1216. doi:10.1016/j.foodchem.2011.05.113
Lin Q (2013) Submerged fermentation of Lactobacillus rhamnosus YS9 for gamma-aminobutyric acid (GABA) production. Braz J Microbiol 44:183–187
Stanaszek PM, Snell JF, O’Neill JJ (1977) Isolation, extraction, and measurement of acetylcholine from Lactobacillus plantarum. Appl Environ Microbiol 34:237–239
Sobko T, Huang L, Midtvedt T et al (2006) Generation of NO by probiotic bacteria in the gastrointestinal tract. Free Radic Biol Med 41:985–991. doi:10.1016/j.freeradbiomed.2006.06.020
Bajaj JS, Heuman DM, Hylemon PB et al (2014) Randomised clinical trial: Lactobacillus GG modulates gut microbiome, metabolome and endotoxemia in patients with cirrhosis. Aliment Pharmacol Ther 39:1113–1125. doi:10.1111/apt.12695
Fetissov SO, Hamze Sinno M, Coeffier M et al (2008) Autoantibodies against appetite-regulating peptide hormones and neuropeptides: putative modulation by gut microflora. Nutrition 24:348–359. doi:10.1016/j.nut.2007.12.006
Konstantinov SR, Smidt H, de Vos WM et al (2008) S layer protein A of Lactobacillus acidophilus NCFM regulates immature dendritic cell and T cell functions. Proc Natl Acad Sci USA 105:19474–19479. doi:10.1073/pnas.0810305105
Amdekar S, Singh V, Kumar A et al (2013) Lactobacillus casei and Lactobacillus acidophilus regulate inflammatory pathway and improve antioxidant status in collagen-induced arthritic rats. J Interferon Cytokine Res 33:1–8. doi:10.1089/jir.2012.0034
Bravo JA, Julio-Pieper M, Forsythe P et al (2012) Communication between gastrointestinal bacteria and the nervous system. Curr Opin Pharmacol 12:667–672. doi:10.1016/j.coph.2012.09.010
Bhathena J, Duchesneau CT (2012) Effect of orally administered microencapsulated FA-producing L. fermentum on markers of metabolic syndrome: an in vivo analysis. J Diabetes. doi:10.4172/2155-6156.S2-009
Tomaro-Duchesneau C, Saha S, Malhotra M et al (2012) Lactobacillus fermentum NCIMB 5221 has a greater ferulic acid production compared to other ferulic acid esterase producing Lactobacilli. Int J Probiotics Prebiotics 7(1):23–32
Grompone G, Martorell P, Llopis S et al (2012) Anti-inflammatory Lactobacillus rhamnosus CNCM I-3690 strain protects against oxidative stress and increases lifespan in Caenorhabditis elegans. PLoS One 7:e52493
Yan F, Cao H, Cover TL et al (2007) Soluble proteins produced by probiotic bacteria regulate intestinal epithelial cell survival and growth. Gastroenterology 132:562–575. doi:10.1053/j.gastro.2006.11.022
Sanchez B, Urdaci MC, Margolles A (2010) Extracellular proteins secreted by probiotic bacteria as mediators of effects that promote mucosa–bacteria interactions. Microbiology 156:3232–3242. doi:10.1099/mic.0.044057-0
Claes IJJ, Lebeer S, Shen C et al (2010) Impact of lipoteichoic acid modification on the performance of the probiotic Lactobacillus rhamnosus GG in experimental colitis. Clin Exp Immunol 162:306–314. doi:10.1111/j.1365-2249.2010.04228.x
Sagar S, Morgan ME, Chen S et al (2014) Bifidobacterium breve and Lactobacillus rhamnosus treatment is as effective as budesonide at reducing inflammation in a murine model for chronic asthma. Respir Res 15:46. doi:10.1186/1465-9921-15-46
Ushakova G, Fed’kiv O, Prykhod’ko O et al (2009) The effect of long-term lactobacilli (lactic acid bacteria) enteral treatment on the central nervous system of growing rats. J Nutr Biochem 20:677–684. doi:10.1016/j.jnutbio.2008.06.010
Pereira DIA, McCartney AL, Gibson GR (2003) An in vitro study of the probiotic potential of a bile-salt-hydrolyzing Lactobacillus fermentum strain, and determination of its cholesterol-lowering properties. Appl Environ Microbiol 69:4743–4752
Duary RK, Bhausaheb MA, Batish VK, Grover S (2012) Anti-inflammatory and immunomodulatory efficacy of indigenous probiotic Lactobacillus plantarum Lp91 in colitis mouse model. Mol Biol Rep 39:4765–4775. doi:10.1007/s11033-011-1269-1
Tsavkelova EA, Botvinko IV, Kudrin VS, Oleskin AV (2000) Detection of neurotransmitter amines in microorganisms with the use of high-performance liquid chromatography. Dokl Biochem 372:115–117
Chen Z, Guo L, Zhang Y et al (2014) Incorporation of therapeutically modified bacteria into gut microbiota inhibits obesity. J Clin Invest 124:3391–3406. doi:10.1172/JCI72517
Shishov VA, Kirovskaia TA, Kudrin VS, Oleskin AV (2009) Amine neuromediators, their precursors, and oxidation products in the culture of Escherichia coli K-12. Prikl Biokhim Mikrobiol 45:550–554
Pryde S, Duncan S, Hold G et al (2002) The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 217:133–139
Macfarlane S, Macfarlane GT (2003) Regulation of short-chain fatty acid production. Proc Nutr Soc 62:67–72
Author information
Authors and Affiliations
Contributions
This review was conceptualized and written by SW with supporting sections written and edited by NL and SYD. Advisement and manuscript suggestions were provided by SPS and SP.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest or competing financial interests.
Rights and permissions
About this article

Cite this article
Westfall, S., Lomis, N., Kahouli, I. et al. Microbiome, probiotics and neurodegenerative diseases: deciphering the gut brain axis. Cell. Mol. Life Sci. 74, 3769–3787 (2017). https://doi.org/10.1007/s00018-017-2550-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2550-9