
Abstract
At sites of inflammation and tumor growth, the local concentration of extracellular adenosine rapidly increases and plays a role in controlling the immune responses of nearby cells. Adenosine deaminases ADA1 and ADA2 (ADAs) decrease the level of adenosine by converting it to inosine, which serves as a negative feedback mechanism. Mutations in the genes encoding ADAs lead to impaired immune function, which suggests a crucial role for ADAs in immune system regulation. It is not clear why humans and other mammals possess two enzymes with adenosine deaminase activity. Here, we found that ADA2 binds to neutrophils, monocytes, NK cells and B cells that do not express CD26, a receptor for ADA1. Moreover, the analysis of CD4+ T-cell subset revealed that ADA2 specifically binds to regulatory T cells expressing CD39 and lacking the receptor for ADA1. Also, it was found that ADA1 binds to CD16− monocytes, while CD16+ monocytes preferably bind ADA2. A study of the blood samples from ADA2-deficient patients showed a dramatic reduction in the number of lymphocyte subsets and an increased concentration of TNF-α in plasma. Our results suggest the existence of a new mechanism, where the activation and survival of immune cells is regulated through the activities of ADA2 or ADA1 anchored to the cell surface.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Adenosine belongs to a class of small molecules that plays an important role in the regulation of various physiological processes including sleep control, body temperature maintenance and modulation of the immune system [1–3]. Although it is normally present at low levels in the body, adenosine can rapidly accumulate when damaged tissues release large amounts of ATP, which is subsequently degraded by ecto-enzymes CD39 and CD73 located on the cell surface [4]. Alternatively, adenosine can be transported across the cell membrane by nucleoside transporters along the equilibrium gradient [2]. Once it accumulates in the extracellular space, adenosine can bind and activate adenosine receptors, which are expressed by the majority of cells in the human body. The strength of the signal conducted by the adenosine receptors depends on their affinity for adenosine, the total number of receptors expressed on the cell surface, and the receptor type. These signals are transmitted by G proteins that are coupled to the four different adenosine receptors [1]. In general, activation of the A2A and A2B receptors in humans leads to an intracellular accumulation of cAMP, while adenosine binding to the A1 and A3 receptors decreases the intracellular concentration of cAMP [2]. Adenosine receptors are widely expressed by various cells, including cells of the immune, cardiovascular and nervous systems. Accordingly, several studies have found that these receptors contribute to different pathophysiological conditions, such as asthma and Parkinson’s disease. As a result, an array of adenosine receptor agonists and antagonists has been designed for use as potential drugs to tackle a variety of health problems [1, 5, 6].
Adenosine deaminases (ADAs) are primarily responsible for the regulation of adenosine levels, although their roles are not limited to this enzymatic function [7, 8]. Two different enzymes with identical adenosine deaminase activity have been found in humans, and are designated ADA1 and ADA2. Interest in ADA1 function was driven by the discovery that nearly 15 % of inherited immunodeficiencies are caused by the loss-of-function mutations in the ADA1 gene [9]. These disorders are collectively known as severe combined immunodeficiency (SCID), which is associated with a decrease in the number of circulating T and B cells due to the intracellular accumulation of toxic adenosine products that results in apoptosis [7]. Although the primary role of ADA1 is to regulate the intracellular adenosine concentration, it has been found that human ADA1 can bind dipeptidyl peptidase IV (CD26) and associate with A1, A2A and A2B adenosine receptors expressed on the surface of immune cells [7]. It was further proposed that ADA1 simultaneously binds to dendritic cells (DCs) and T cells via adenosine receptors and CD26, respectively, bridging the cells by forming an “immunological synapse” [7]. This could explain why ADA1-dependent stimulation of T-cell proliferation in the presence of DCs does not require ADA1 to be enzymatically active [10]. However, according to other reports, the enzymatic activity of ADA1 is required to counteract regulatory T-cell-mediated T-cell suppression [11–13]. According to the proposed scenario, ADA1 binds to CD26 on the surface of effector T cells and protects the effector T cells from the extracellular adenosine. It was suggested that in the absence of ADA1, adenosine reduces T-cell proliferation and cytokine release by activating the A2A adenosine receptor [3]. Studies examining the role of the second adenosine deaminase, ADA2, have been hampered due to a low abundance of this enzyme in human tissues, the absence of a similar enzyme in mice, and a general assumption that ADA2 may be an isoform of ADA1 [14]. Recently, we purified ADA2 as a by-product of commercially available immunoglobulin preparations and found that the gene coding for ADA2, CECR1, belongs to a new family of adenosine deaminase growth factors (ADGFs) [14]. Immune disorders such as systemic lupus erythematosus, tuberculosis and HIV infection are accompanied by a dramatic increase in ADA2 activity, and the level of ADA2 in pleural fluids is used as a marker for tuberculosis [15–18]. Furthermore, the concentration of ADA2 in serum correlates with the progression of tumor growth, which identifies ADA2 as a convenient biomarker for monitoring treatment in cancer patients [19]. Activated monocytes, macrophages and DCs at sites of inflammation and in the tumor microenvironment are thought to be the major source of ADA2 in plasma and pleural fluids [8]. In agreement with data from humans, hemocytes, the fly analogs of macrophages, are a source of extracellular ADGF-A in Drosophila [20], and ADGF-A is expressed by hemocytes during inflammatory responses [21]. Recently, the structure of ADA2 was determined, revealing striking differences between ADA2 and ADA1 both in the arrangement of their catalytic center and in their overall architectures [22]. ADA2 contains an open catalytic center, which explains why ADA2 has a 100-fold lower Km value for adenosine deamination compared with ADA1 [14, 23]. An N-terminal sequence that is absent in ADA1 was found to be required for both the dimerization of ADA2 and its interaction with proteoglycans. An additional chemokine-like domain was proposed to be a binding site for a putative ADA2 receptor. The fact that ADA2 is a secreted protein while ADA1 is predominantly an intracellular enzyme suggests that ADA2 released from antigen-presenting cells could be involved in cell–cell signaling. Although a specific ADA2 receptor and the target cells have not yet been identified, both proteoglycans and adenosine receptors are thought to interact with ADA2 on the cell surface [8].
Recently, patients with mutations in the CECR1 gene (DADA2 patients) have been identified [24, 25]. It was shown that ADA2 concentration in the plasma of these patients is reduced more than tenfold compared to that in healthy subjects. Patients with DADA2 display multiple health problems, including early onset systemic inflammation, recurrent ischemic strokes, and vasculitis/vasculopathy. Strikingly, the patients who are homozygous for a rare mutation, Gly47Arg, have symptoms of polyarteritis nodosa (PAN). Therefore, this missense mutation in ADA2 is the first identified cause of a monogenic vasculitis. The symptoms of the ADA2-deficient patients are distinct from patients with ADA1 deficiency, and they can be corrected by hematopoietic stem cell transplantation (HSCT) [26, 27]. Although the absence of either functional ADA1 or ADA2 leads to dysregulated immune function, the lack of one functional enzyme is not compensated for by the presence of the other enzyme, suggesting that ADA1 and ADA2 have distinct roles.
In this study, we show that different immune cells subsets bind selectively to ADA1 or ADA2 and propose a new mechanism for the regulation of immune cell function by adenosine deaminases. Dramatic changes in the composition of immune cells from DADA2 patients suggest a new role for ADA2 in the immune system homeostasis. Our findings help to explain clinical observations and elucidate the function of adenosine deaminases in the regulation of immune responses.
Results
ADA2 binds to myeloid cells and lymphocytes that do not express a receptor for ADA1
Previously, we have shown that ADA2 binds to heparin sulfate and suggested that the enzyme may interact with proteoglycans expressed on the cell surface of immune and cancer cells [8]. Also, ADA2 may associate with adenosine receptors and modulate their affinity to extracellular adenosine [8]. To define the cell types that primarily bind ADA2, we used recombinant ADA2. The binding of the enzyme to the blood immune cells was detected with CF633-labeled anti-ADA2 antibodies and analyzed by flow cytometry (Fig. 1). As it is shown in Fig. 1o, the highest fluorescein intensity for ADA2 binding to the cells was found on neutrophils. Monocytes, NK cells, and B cells were among the other cells binding the enzyme [8]. In contrast, both CD4+ and CD8+ T-cell subset and NKT cells did not bind any detectable amount of ADA2 and they express a receptor for ADA1, CD26 (Fig. 1j, l, n). Hence, human ADA1 and ADA2 bind to different types of immune cells, and only one ADA out of the two could be found on each cell type.

ADA2 binding to blood cells. The binding of ADA2 to neutrophils (a), monocytes (c), B cells (e) and NK cells (g), which was detected by CF633-labeled anti-ADA2 antibodies, is shown on the histograms (b, d, f, h, j, l, n). Red and green lines on the histograms correspond to the cells stained with anti-ADA2 antibodies in the presence or absence of ADA2, respectively. The expression of ADA1 receptor CD26 on CD4+ T cells (i), CD8+ T cells and NKT cells (m) is shown on dot plots j, l and n. o The graph showing the relative binding of ADA2 to different cell subsets from three donors. ADA2 is expressed in geometric mean fluorescein intensities (MFI) and the values are normalized to the MFI of neutrophils (100 %)
Further analysis of NK cells, monocytes, and CD4+ T cells revealed additional cell subsets expressing more ADA2 receptors compared to other cells of the same cell population. As shown in Fig. 2, NK cells expressing high level of CD56 (CD56high) bind much less ADA2 compared to other NK cells with lower levels of CD56 expression (Fig. 2c). Similarly, the monocyte subset expressing CD16 was found to have much higher capacity for ADA2 compared to the “classical” CD16-negative monocytes (Fig. 2f). Intriguingly, CD56+ NK cells, in contrast to CD56high NK cells, express high level of CD16, a low-affinity receptor for IgG, in analogy to CD16+ monocytes [28]. Previously, we showed that, CD4+ T cells have low levels of ADA2 binding (Fig. 1o). Further analysis of CD4+ T-cell subset revealed that the CD39+ CD25+ subset expresses a much higher level of ADA2 receptor compared to other CD4+ T cells (Fig. 2i). The level of ADA2 receptor expression on the cell subsets of three different healthy donors was found to be very similar (Fig. 2j, k). Interestingly, the analysis of ADA2 interaction with the immune cells from an ADA2-deficient (DADA2) patient showed a dramatic reduction in ADA2 binding compared to healthy donors (Fig. 2k). This result suggests that the expression of ADA2 receptor is downregulated in some DADA2 patients to compensate the absence of ADA2.

ADA2 binding to lymphocyte subsets. Dot plots show the binding of ADA2 (c, f, i) to NK (a, b), monocyte (d, e) and CD4+ T-cell subset (g, h) from a healthy donor, which was detected by CF633-labeled anti-ADA2 antibodies. ADA2 binding to cell subsets from three healthy donors is shown in the graph (j). The binding of ADA2 to the cell subsets is expressed in geometric mean fluorescein intensities (MFI), and the values obtained for the healthy donors and ADA2-deficient patients are compared in the table (k)
ADA2 binds to CD39+ CD25+ FOXP3+ subset of CD4+ regulatory T cells that do not express a receptor for ADA1
As it is shown in Fig. 2i, ADA2 binds to CD4+ T cells expressing cell surface markers that are typical for Treg cells. In the next experiment, we stained CD4+ T cells with antibodies against a transcription factor FoxP3 to determine specific Treg subsets interacting with ADA2. To stabilize ADA2 binding on the cells, we used enzymatically biotinylated ADA2, which was conjugated with a fluorescently labeled streptavidin in a molar ratio of 2:1 [8]. As shown in Fig. 3a and b, ADA2 binds to a Treg cell subset that expresses CD39 receptor. Moreover, FoxP3-negative CD4+ T cells that express CD39 demonstrated much lower level of ADA2 binding (Fig. 3c), suggesting that a receptor for ADA2 is specifically expressed on the CD39+ FoxP3+ subset of Treg cells. Contrarily, the level of CD26 expression on CD39+ FoxP3 was the lowest compared to other CD4+ T-cell subset. Further analysis of CD4+ cells expressing FoxP3 and CD26 also showed that both FoxP3+ and FoxP3− cells expressing a receptor for ADA1 have a low level of ADA2 binding (Fig. 3d, e). At the same time, ADA2 receptor and CD39 expression are much higher on the FoxP3+ subset that is negative for CD26. These data show that ADA2 specifically binds to Tregs expressing CD39 and lacking a receptor for ADA1, and, hence, that ADA2 can selectively regulate the activity of the FoxP3+ CD39+ CD4+ T-cell subset.

ADA2 binds to FoxP3+ CD39+ regulatory CD4+ T cells that express low levels of ADA1 receptor CD26. a A dot plot showing four T-cell subset determined by the level of CD39 and FoxP3 expression. b Mean fluorescent intensity (MFI) of ADA2–Streptavidin–FITC binding and the level of CD39 and FoxP3 expression in four CD4+ T cell subset shown in A. c A histogram showing the difference in ADA2 binding to CD39+ FoxP3+ and CD39+ FoxP3− subsets of CD4+ T cells. d A dot plot showing four T-cell subset determined by the level of CD26 and FoxP3 expression. e MFI of ADA2–Streptavidin–FITC binding and the level of CD26 and CD39 expression in four CD4+ T-cell subset shown in d
Human and mouse ADA1 preferentially bind to CD16− monocytes
We used recombinant mouse and human ADA1 expressed in human HEK-293T cells to analyze ADA1 binding to immune cells. The binding of biotinylated ADA1 to the cells was detected using streptavidin conjugated to a fluorochrome. As it was expected, human ADA1 binds to a fraction of lymphocytes expressing ADA1 receptor CD26 (Fig. 4c). The binding of biotinylated human ADA1 can be completely abolished in the presence of unmodified enzymes, proving that recombinant protein binds specifically to the cells. In accordance with previously reported results [29], it was found that monocytes could also be stained with ADA1. Analysis of monocyte subsets showed that ADA1 binding to monocytes inversely correlates with CD16 expression and that CD16− monocytes have a much higher capacity to bind ADA1. These results show that ADA1 and ADA2 bind different subsets of monocytes and, hence, that the enzymes have distinct roles in the regulation of the function of monocyte subsets. Interestingly, mouse ADA1 was also found to bind CD16− monocytes (not shown) and human monocyte-like THP1 cells (Fig. 4e). In contrast to human ADA1, mouse ADA1 did not bind the HuT78 T-cell line expressing CD26 receptor (Fig. 4e). This is in agreement with the experiments showing that mouse ADA1 does not interact with CD26 [30]. The binding of mouse and human ADA1 to a new receptor on THP1 cells was mutually exclusive (Fig. 4e), suggesting that the receptor is highly conserved among the two species.

ADA1 binding to monocyte subsets lymphocytes expressing CD26. Recombinant biotinylated hADA1 was bound to the cells following staining with Streptavidin PE. The binding of ADA1 to monocyte subsets (a, b) is expressed in geometric mean fluorescein intensities (MFI). c A histogram showing the staining of CD16− monocyte subset (region 3) with Streptavidin PE with ADA1 (blue line) and without ADA1 (red line). d, e Binding of biotinylated hADA1 to lymphocytes in the absence (d) and presence of competing wild-type hADA1. e Western blot showing the binding of biotinylated mouse ADA1 to THP1 cells in the absence (lane 1) or presence of unmodified human ADA1 (lane 2) and the binding of biotinylated mouse (lane 3) and human ADA1 (lane 4) to CD26+ HuT78 cells expressing CD26
Analysis of cell subsets in ADA2-deficient patients
It has been shown previously that extracellular adenosine binds to ADORs expressed on the cell surface of immune cells and that it negatively affects the cells’ activation [4]. Therefore, we analyzed the cell subsets in the DADA2 patients to find the cell subsets sensitive to adenosine and which might be affected in the absence of ADA2. The gating strategy and the result of the Flow cytometry analysis of frozen PBMCs from nine DADA2 patients are summarized in Table 1, and the most prominent difference between DADA2 patients and healthy individuals is illustrated in Fig. 5. Our data for healthy controls were similar to those of earlier published results on lymphocyte and monocyte subsets distribution in healthy controls of different ages [31]. Analysis of PBMCs from the DADA2 patients revealed several cell subsets that were either significantly reduced or almost absent. All nine patients had decreased levels of CD19+ CD25+ cells, memory T cells and NKT cells. A decrease in the number of NK cells and CD25+ CD4+ effector T cells was seen in the majority of patients. Some of the patients had either a remarkable decrease or a significant increase in CD16+ monocyte subset. The increase in CD16+ monocyte subset was observed in those patients with decreased percentages of monocytes in PBMCs. The overall percentage of B cells was found to be increased in five patients. While memory T cells and CD25+ B-cells were also reduced in pediatric patients controls (data not shown), the decrease in NK, NKT and CD16 subset of monocytes was a characteristic feature of DADA2 patients (Fig. 5). The patients’ parents who have a mutation in one copy of the ADA2 gene CECR1, and reduced levels of ADA2 in plasma, had a generally normal distribution of cell subsets, with the exception of CD16+ monocyte subsets (Table 2). Our data show that the absence of ADA2 affects major immune cell subsets, although the type of affected cells differs among DADA2 patients.

Analysis of cell subsets in DADA2 patients and healthy controls. a CD3− CD56+ NK and CD3+ CD56+ NKT cells, b CD14+ monocytes and c CD19+ B-cells are shown on the dot plots. The gating strategy for lymphocytes and monocytes is shown in Table 1
ADA2 regulates the level of TNF-α secreted by activated monocytes by decreasing the concentration of extracellular adenosine
It was reported that extracellular adenosine can bind to A2A adenosine receptors expressed on the cell surface of activated monocytes and downregulate cytokine secretion by these cells [32]. Here, we analyzed the effect of adding ADA2 to monocytes activated with LPS in the presence of increased concentrations of adenosine. As shown in Fig. 6a, the release of TNF-α by activated monocytes decreases with the increase of adenosine concentrations in the cell culture medium. Moreover, we found that TNF-α secretion is very sensitive to extracellular adenosine, and the concentration of TNF-α in the cell culture supernatants drops rapidly at very low adenosine concentrations close to those found in plasma (0.1–1 µM) [33], and is almost completely inhibited at 50-µM adenosine concentrations. The addition of ADA2 to the cell culture with 50 µM adenosine restores the level of TNF-α secretion (Fig. 6b). At the same time, the addition of a mutant ADA2 with a 100-fold reduced enzymatic activity (ADA2 H88G) had almost no effect on the level of cytokines secretion. This result demonstrates that ADA2 activates monocytes by depleting extracellular adenosine. Intriguingly, the analysis of TNF-α concentration in plasma of DADA2 patients and healthy donors showed that some of the DADA2 patients had increased levels of TNF-α in plasma (Fig. 6c). These results suggests that in the absence of ADA2, the immune system of DADA2 patients has to be adjusted to produce more TNF-α to compensate the immunosuppressive effect of the extracellular adenosine.

The level of TNF-α secretion by LPS-activated monocytes is controlled by extracellular adenosine and ADA2. a Changes in the TNF-α secretion level by LPS-activated monocytes in response to the increased adenosine concentration. b Addition of enzymatically active ADA2 but not ADA (H88G) mutant to adenosine-suppressed monocytes increases the level of TNF-α by LPS-activated monocytes in the presence of 50 µM adenosine. c Analysis of ADA2 and TNF-α concentration in plasma of healthy donors and DADA2 patients and their relatives
Discussion
It has been shown that extracellular adenosine regulates the activity of lymphocytes. Accordingly, it was reported that adenosine, bound to A2A and A2B adenosine receptors, can suppress the activation of the effector T cells, NK cells, NKT cells, neutrophils, and monocytes [34, 35]. Adenosine deaminases are natural antagonists of adenosine receptors. However, the role of adenosine deaminases in the regulation of extracellular adenosine concentration, and their effect on the immune cell function, have not been thoroughly studied. Initially, human ADA1 was thought to be mainly an intracellular enzyme, which plays a key role in adenosine catabolism inside the cells [7]. Later, hADA1 was detected on the cell surface of neurons, erythrocytes, platelets, epithelial cells, and immune cells [29, 36, 37] and found in a complex with CD26 in human plasma [38]. Although the mechanism of ADA1 delivery to the cell surface remains unclear [39], it was suggested that ecto-ADA1 can act extraenzymatically as a growth factor, and can even serve as an intercellular adaptor [10]. In contrast to ADA1, ADA2 is secreted by myeloid cells, and its enzymatic activity is, thus, linked to its extracellular function [8]. Recent findings demonstrated that ADA2 deficiency results in dysregulation of immune, neural, and cardiovascular systems [24, 25]. DADA2 patients display a variable clinical phenotype, including systemic inflammation, aplastic anemia, and vasculopathy/vasculitis, symptoms that are not compensated for by ADA1. In this study, we showed that this could be explained by differential binding of ADA1 and ADA2 to the immune cells. This suggests a new mechanism, where ADAs bound to different subsets of the immune cells regulate the function of the corresponding immune cells by depleting extracellular adenosine.
While ADA1 binds to T cells and NKT cells expressing the CD26 receptor, we found that ADA2 binds to neutrophils, monocytes, B cells, NK cells, and CD39+ Treg cells that do not express CD26 (Figs. 1, 7). Furthermore, a detailed analysis of monocyte and NK cell subsets revealed that CD16+ cell subsets of monocytes and NK cells preferably bind ADA2 (Figs. 2, 7). Therefore, the absence of ADA2 would result in higher sensitivity of the ADA2-binding cells to changes in extracellular adenosine. Our hypothesis is further supported by the analysis of immune cell subsets from DADA2 patients that revealed striking abnormalities and inter-patient variability in the frequency of cell subsets compared to healthy individuals (Fig. 5; Table 1). This difference could be a result of the compensatory mechanisms developed in ADA2-deficient cells in response to epigenetic and environmental factors [40]. However, it is also known that ADA2 is required for the differentiation and homeostasis of immune cells. B cells from DADA2 patients show a defect in terminal maturation, and they are more prone to apoptosis [25]. A more recent study reported that the absolute number of NK cells, T cells, and B cells was reduced in DADA2 patients, and that some patients had neutropenia—the major subset of cells that bind ADA2 [40]. While a decrease in memory T cells and CD25 B cells could be seen in children with other autoimmune diseases (not shown), the low frequency of NK, NKT, and CD16+ monocytes was associated with DADA2 (Fig. 5). The lack of NK cells could be a risk factor for cancer development in adults with ADA2 deficiency [40]. The absence of the CD25 subset of B cells may also explain the low levels of immunoglobulin in DADA2 patients [41].

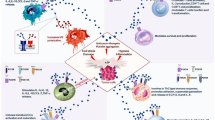
A scheme showing the binding of human ADA1 and ADA2 to immune cell subsets and their possible role in the regulation of immune cell activity. It is shown that ADA2 is secreted by monocytes and it may bind proteoglycans, adenosine, or other unknown receptors expressed on the surface of CD16+ monocytes, CD16+ NK cells, CD16+ neutrophils, CD39+ Treg cells and B cells. In contrast, extracellular ADA1 could be found on the effector T cells expressing ADA1 receptor CD26. ADA1 may also bind CD16− monocytes and epithelial/stromal cells via adenosine or other unknown receptors and participate in bridging of T cells and monocytes. Some of the cells express ecto-enzymes CD39 and CD73 hydrolyzing ATP into AMP and AMP into adenosine, respectively. Adenosine can bind to A2A receptors, expressed on the same or other cells and regulate the cell function. ADA2 and ADA1 bound to the cells can also regulate the cells’ function either by decreasing the concentration of extracellular adenosine or by activating their corresponding receptors
It has been suggested that CD39-expressing regulatory T cells could modulate the T-cell effector function through the production of extracellular adenosine [42]. Mouse Treg cells are activated by the binding of extracellular adenosine to the A2A receptor [42]. Therefore, if the latter is also true for the human Tregs, ADA2 binding to CD39+ Treg cells might inhibit Tregs activation by extracellular adenosine (Fig. 7). Hence, ADA2 secreted by myeloid cells can bind to Tregs and block their immunosuppressive functions [43]. The decrease in the cell number of DADA2 patients could be explained by their sensitivity to extracellular adenosine, as well as to the altered activity of Treg cells. Interestingly, immune cells expressing CD39, such as neutrophils, monocytes, B cells, and CD39+ Treg cells, bind ADA2. This suggests that the CD39+ cells, assisted by CD73-expressing cells [44], produce immunosuppressive extracellular adenosine, and ADA2 bound to their cell surface may control the cells’ activation in the adenosine environment (Fig. 7).
We identified a receptor for ADA1 expressed on CD16-monocytes (Figs. 4, 7). Both mouse and human ADA1 were found to bind this receptor in an exclusive manner, indicating that the receptor is conserved among the species. It has been previously suggested that A1 adenosine receptor expressed on DCs may associate with ADA1 bound to CD26 receptor on T cells [7]. Therefore, it is possible to suggest that CD16− monocytes can also express adenosine receptor that binds ADA1, allowing a contact formation between monocytes and CD26+ T cells (Fig. 7). Strikingly, CD16− monocytes express much less intracellular ADA1 compared to the CD16+ subset [45]. This might be required to keep the receptor on the CD16− subset free from endogenous ADA1, enabling it to form a contact with ADA1 bound to CD26 on T cells. Our results show that monocyte subsets could be defined by the level of ADA1 and ADA2 receptors’ expression. ADAs, in turn, could control the differentiation and activation of monocytes. It was suggested that ADA2 might be involved in the macrophage polarization, with monocytes from DADA2 patients differentiated into M1 type, but not M2 [25]. This may also contribute to the excessive TNF-α production by M1-polarized macrophages (Fig. 6c) [46, 47].
Although the level of adenosine in human plasma is extremely low, the adenosine concentration in inflamed tissues can reach substantial concentrations to affect the function of immune cells [2]. As it is shown in Fig. 6, even a small increase in adenosine concentration can lead to a substantial decrease in TNF-α secretion by activated monocytes. However, the addition of ADA2 decreases adenosine concentration and restores the level of TNF-α. A tenfold increase in the ADA2 concentration in pleural fluids of patients with tuberculosis (Porcel, 2009) suggests that ADA2 is actively secreted by activated myeloid cells in the sites of inflammation. While healthy individuals can successfully restore cytokine production levels by decreasing the level of adenosine with ADA2 bound to the activated monocytes (Fig. 6) or neutrophils, DADA2 patients must develop different compensatory mechanisms to survive during bacterial infections or other pathologies related to the immune system function. For instance, the lack of regulation of monocytes activation in the absence of ADA2 may result in the overproduction of pro-inflammatory cytokines, such as TNF-α, to counteract the immunosuppressive adenosine signaling, resulting in a decrease in the production of these cytokines (Fig. 6). Although this could be beneficial in combating pathogens during bacterial and viral infections, it may have negative side effects, such as damage to blood vessels and strokes, due to the excessive production of pro-inflammatory cytokines [48]. As was shown previously, ADA2 modulates the affinity of adenosine receptors (ADORs) to their agonists [8], suggesting that the enzyme associates with ADORs. Therefore, the absence of ADA2 might also affect the function and expression level of ADORs (Fig. 2k), resulting in additional negative drawbacks for DADA2 patients.
Our results show that human adenosine deaminases bind to distinct lymphocyte subsets, expressing specific receptors for ADA1 and ADA2. Adenosine deaminases bound to the cell surface of these cells can regulate their activity by decreasing the concentration of extracellular adenosine. However, they may also modulate the function of their receptors or participate in the formation of immunological synapses independently of their enzymatic activity. Therefore, extracellular deaminases and their inhibitors or neutralizing antibodies could become promising drugs in the management of cancers and inflammation.
Methods
ADAs cloning and modification
To stably express human ADA2 and ADA1 in cells transfected with a replication-defective lentiviral vector, the open reading frames (ORFs) of the genes were PCR-amplified with the following primers: (F) 5′-ATCTCGAGCCACCATGTTGGTGGATGGCCCATCTG-3′ and (R) 5′-TAGGATCCTCACTTTGTAGCCACATCTGC-3′ (hADA2), (F) 5′-AGCTCGAGACCGGTCCACCATGGCCCAGACGCCCGCCT-3′ and (R) 5′-ATGGATCCGCTAGCTCAGAGGTTCTGCCCTGCAG-3′ (hADA1), (F) 5′-AGCTCGAGACCGGTCCACCATGGCCCAGACACCCGCAT-3′ and (R) 5′-TCAGATCTATTGGTATTCTCTGTAGAGC-3′ (mADA1). The PCR products were then subcloned into the pCR2.1-TOPO plasmid, excised by restriction digest with XhoI and BamHI, and ligated into a XhoI/BamHI-digested self-inactivating (SIN) transfer plasmid (pHR-cPPT-hB7-SIN) [49]. To express hADA2 with a His-tag and biotinylation site, the wild-type hADA2 gene in lentiviral vector was digested with Xho I and Sac I, and a His–biotin-tagged gene in insect cell expression plasmid [22] was digested with Sac I and Sal I. The 5′ end fragment of hADA2 with native secretion signal, and 3′ end fragment of the modified hADA2 with the HIS-tag and biotinylation site, were separated through agarose gel electrophoresis, purified, and ligated. Subsequently, new hADA2 with its native secretion signal and a His-tag and biotinylation site was amplified with primers (F) 5′-ATCTCGAGCCACCATGTTGGTGGATGGCCCATCTG-3′ and (R) 5′-GCAGATCTGCTAGCGTCGACTTAGTGATGATGATG-3′ using the ligated hADA2 as template. The PCR products was then subcloned into the pCR2.1-TOPO plasmid, named as pCR2.1-hADA2-His-bio, excised by restriction digest with Xho I and Bgl II, and ligated into an XhoI/BamHI-digested self-inactivating (SIN) transfer plasmid (pHR-cPPT-hB7-SIN). The resultant plasmid was named as pHR-cPPT-hADA2-His-bio, with the hADA2 gene sequenced and confirmed. To introduce the mutation of H88G, a set of primers containing the mutation of interest were synthesized. Subsequently, two PCR products were amplified using the primers (F) 5′-ATCTCGAGCCACCATGTTGGTGGATGGCCCATCTG-3′ and the reverse primer containing the mutation 5′-GTCACGATGCCAATGTCACCGAGGTGCAAGGCAGCC-3′, as well as the forward primer containing the desired mutation 5′-GGCTGCCTTGCACCTCGGTGACATTGGCATCGTGA-3′ and the primer (R) 5′-GCTCTCCACTGAGCTCATACACCGGCAGCAG-3′. A third round of fusing PCR was done using both of the PCR products as template, and primers (F) 5′-ATCTCGAGCCACCATGTTGGTGGATGGCCCATCTG-3′ and (R) 5′-GCTCTCCACTGAGCTCATACACCGGCAGCAG-3′. To subclone the fusion PCR product containing H88G mutation into the hADA2 gene, the plasmid pCR2.1-hADA2-His-bio was first digested with Hind III and Spe I to remove the Sac I restriction site from the plasmid backbone, self-ligated, and named as pCR2.1-hADA2-His-bio-HS. The fusion PCR product was then digested with Xho I and Sac I, and ligated with pCR2.1-hADA2-His-bio-HS that was digested with the same enzymes. Subsequently, the mutant hADA2 gene was released through Xho I/BamH I digestion and inserted into pHR-cPPT-hADA2-His-bio that was digested with the same enzymes, with the hADA2 gene sequenced and confirmed.
ADAs expression and purification
HEK-293T cells were transfected with the lentiviral transfer plasmid expressing an ADA gene or mutant, pCMV-VSV-G envelope and pCMVΔR8.2 packaging plasmids using an optimized calcium phosphate transfection method [50]. Subsequently, lentiviral vectors were concentrated by ultracentrifugation from the conditioned medium of the transfected HEK-293T cells, and were then used to infect new 293T cells. Finally, recombinant ADA2 with or without a 6× His-tag and biotinylation site was purified from conditioned medium of the lentivirus-transduced 293T cells, as described [14, 22]. For some experiments, ADA2 was enzymatically biotinylated and then conjugated with streptavidin labeled with fluorescein isothiocyanate (FITC) from eBioscience, as described previously [8]. The recombinant human and mouse ADA1 was purified from the 293T cell lysates [23]. The 293T cells expressing hADA1 were lysed through three freeze/thaw cycles using liquid nitrogen and a 42 °C water bath. The cell lysate was re-suspended in 50 ml of ice-cold 50 mM Tris–HCl buffer (pH 6.8) containing 50 mM NaCl, 10 μM Zn(AcO)2, and 0.02 % NaN3 (buffer A). The supernatant was separated from the cell debris by centrifugation at 4000g for 20 min, filtered and applied to a DEAE-Sepharose (GE Healthcare) equilibrated with buffer A. The flow through containing ADA1 was collected, and the pH was adjusted to 8.4 with Tris base. The enzyme was applied to a DEAE-Sepharose column equilibrated with buffer B (50 mM Tris–HCl pH 8.5, 10 μM Zn(AcO)2, and 0.02 % NaN3). ADA1 bound to the column was eluted using 0–500-mM NaCl gradient. The fractions containing ADA activity were pooled, concentrated using 10-kDa centrifuge ultraconcentrators (Millipore), and further purified on a Superdex 200 column (GE Healthcare) equilibrated with phosphate-buffered saline (PBS) containing 10 μM Zn(AcO)2 and 0.02 % NaN3. Purified ADA1 was chemically biotinylated using a kit from G Biosciences. Endotoxin removal resin (Pierce) was used to eliminate endotoxin in recombinant ADAs for the functional assays. The endotoxin level in the preparations was monitored using the LAL endotoxin assay kit (GenScript).
Analysis of blood cells and human plasma from healthy donors and DADA2 patients
All the patients were evaluated at the NIH Clinical Center. The patients enrolled in the study had been approved by institute review board, and the patients or their parents provided written informed consent. All the authors vouch for the accuracy and completeness of the data and analyses reported and for the fidelity of the study to the protocol. Blood cells were obtained from fresh blood after the red blood cells lysis with RBC buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.3). PBMCs were purified from the buffy coats of healthy donors (Red Cross, Helsinki, Finland) or the fresh blood of DADA2 patients via a Ficoll density gradient and stored frozen at −80 °C before the analysis. Blood cells or PBMCs were stained with various cell surface markers from BD Pharmingen. The flow cytometry data were analyzed using Flowing Software (http://www.flowingsoftware.com/). Intracellular staining with anti-FoxP3 antibodies was performed with a FoxP3 staining kit (eBioscience). To stain the cells with unmodified ADA2 and anti-ADA2 antibodies, the cells were first incubated with 30 µM ADA2 in 40 µl of FACS buffer (2 % FCS in PBS) for 10 min at 4 °C, then washed with FACS buffer and stained with 3 µg/ml anti-ADA2 rabbit polyclonal antibodies. Anti-ADA2 polyclonal antibodies were affinity-purified on protein G Sepharose (GE Healthcare) from the plasma of rabbits immunized with recombinant ADA2 [22] following an additional step of affinity purification on ADA2-streptavidin column. The anti-ADA2 antibodies were either labeled with fluorescein isothiocyanate (FITC) or CF633 dye (Sigma). The labeling kits were purchased from G-Biosciences. In all experiments, except FoxP3 staining of T cells, we used unmodified ADA2 and anti-ADA2 antibodies, as indicated in the text. However, for the experiment with FoxP3 staining, which involves multiple washes, we had to create a complex containing two ADA2 molecules linked via streptavidin to stabilize the binding of ADA2 to the cells [8]. ADA2 concentration in the plasma of healthy donors and DADA2 patients was detected using a previously described assay [25] and TNF-α concentration was measured using an ELISA kit (Bio Legends).
Monocytes isolation and culture
Human CD14+ monocytes were purified from buffy coats of healthy donors (Red Cross, Helsinki, Finland) by first isolating PBMCs with a Ficoll density gradient and, subsequently, selecting for CD14+ monocytes using anti-CD14-conjugated magnetic microbeads (Miltenyi), as described previously (Zavialov et al. [8]). The recovered cells were 95–99 % CD14+, as determined by flow cytometry using the FITC-conjugated anti-human CD14 mAb (BD Biosciences). RPMI 1640, supplemented with 1 % nonessential amino acids, 1 % sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 0.1 % 2-mercaptoethanol and 10 % FBS, was used as a complete medium in the cell cultures of monocytes. Monocytes were cultured in suspension in 5-ml polypropylene tubes (Falcon) at 0.5 × 106 cells/ml in 0.5 ml of the culture medium. Monocytes were activated with 10 ng/ml LPS in the presence of adenosine, 15 µg/ml ADA2 or ADA2 (H88G) as indicated. After 20 h, culture in the tubes at 37 °C 5 % CO2, the cells were separated by centrifugation (300g, 5 min), and the supernatants were collected to measure TNF-α concentration by ELISA (Bio Legends).
Western blot analysis of ADA1 binding to THP1 and HuT78 cells
The cells were grown in a complete RPMI medium. Each of the four experiments used ten million THP1 or HuT78 cells. The cells were stained with 100 µg/ml of biotinylated mADA1 or hADA1 in 60 µl of FACS buffer (PBS, 2 % FCS, 0.02 % NaN3) for 10 min at 4 °C. In the competition experiment, THP1 cells were pre-incubated with 100 µg/ml hADA1 in 400 µl of FACS buffer for 10 min. Then the cells were centrifuged for 5 min at 300g to remove the supernatant with unbound hADA1 and then stained with mADA1, as described above. After the staining with biotinylated ADAs, the cells were washed three times with cold 1 ml of FACS buffer and lysed on ice with 50 µl of 20 mM Tris HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 %Triton X-100, 0.1 % SDS, 1 mM PMSF. The cell lysates were sonicated in the Bioruptor bath sonicator (Diagenode) and 25 µl was applied on 4–20 % Mini-PROTEAN RGX Gel (Bio-Rad). The proteins from the gel were transferred to a nitrocellulose membrane, and biotinylated proteins were visualized using avidin-HRP (eBioscience) and chemiluminescent Western blot detection reagent (Thermo Fisher).
References
Hasko G, Linden J, Cronstein B, Pacher P (2008) Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 7(9):759–770. doi:10.1038/nrd2638
Fredholm BB (2007) Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 14(7):1315–1323. doi:10.1038/sj.cdd.4402132
Linden J, Cekic C (2013) Regulation of lymphocyte function by adenosine. Arterioscler Thromb Vasc Biol 32(9):2097–2103. doi:10.1161/ATVBAHA.111.226837
Eltzschig HK, Sitkovsky MV, Robson SC (2012) Purinergic signaling during inflammation. N Engl J Med 367(24):2322–2333. doi:10.1056/NEJMra1205750
Burnstock G (2008) Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov 7(7):575–590. doi:10.1038/nrd2605
Jacobson KA, Gao ZG (2006) Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5(3):247–264. doi:10.1038/nrd1983
Franco R, Pacheco R, Gatell JM, Gallart T, Lluis C (2007) Enzymatic and extraenzymatic role of adenosine deaminase 1 in T-cell-dendritic cell contacts and in alterations of the immune function. Crit Rev Immunol 27(6):495–509
Zavialov AV, Gracia E, Glaichenhaus N, Franco R, Lauvau G (2010) Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J Leukoc Biol 88(2):279–290. doi:10.1189/jlb.1109764
Buckley RH (2004) Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol 22:625–655
Pacheco R, Martinez-Navio JM, Lejeune M, Climent N, Oliva H, Gatell JM, Gallart T, Mallol J, Lluis C, Franco R (2005) CD26, adenosine deaminase, and adenosine receptors mediate costimulatory signals in the immunological synapse. Proc Natl Acad Sci USA 102(27):9583–9588. doi:10.1073/pnas.0501050102
Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, Lang S, Jackson EK, Gorelik E, Whiteside TL (2010) Generation and accumulation of immunosuppressive adenosine by human CD4+ CD25 high FOXP3+ regulatory T cells. J Biol Chem 285(10):7176–7186. doi:10.1074/jbc.M109.047423
Mandapathil M, Szczepanski M, Harasymczuk M, Ren J, Cheng D, Jackson EK, Gorelik E, Johnson J, Lang S, Whiteside TL (2012) CD26 expression and adenosine deaminase activity in regulatory T cells (Treg) and CD4(+) T effector cells in patients with head and neck squamous cell carcinoma. Oncoimmunology 1(5):659–669. doi:10.4161/onci.20387
Naval-Macabuhay I, Casanova V, Navarro G, Garcia F, Leon A, Miralles L, Rovira C, Martinez-Navio JM, Gallart T, Mallol J, Gatell JM, Lluis C, Franco R, McCormick PJ, Climent N (2016) Adenosine deaminase regulates Treg expression in autologous T cell-dendritic cell cocultures from patients infected with HIV-1. J Leukoc Biol 99(2):349–359. doi:10.1189/jlb.3A1214-580RR
Zavialov AV, Engstrom A (2005) Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity. Biochem J 391(Pt 1):51–57
Chittiprol S, Satishchandra P, Bhimasenarao RS, Rangaswamy GR, Sureshbabu SV, Subbakrishna DK, Satish KS, Desai A, Ravi V, Shetty KT (2007) Plasma adenosine deaminase activity among HIV1 Clade C seropositives: relation to CD4 T cell population and antiretroviral therapy. Clin Chim Int J Clin Chem 377(1–2):133–137. doi:10.1016/j.cca.2006.09.006
Porcel JM (2009) Tuberculous pleural effusion. Lung 187(5):263–270. doi:10.1007/s00408-009-9165-3
Saghiri R, Ghashghai N, Movaseghi S, Poursharifi P, Jalilfar S, Bidhendi MA, Ghazizadeh L, Ebrahimi-Rad M (2012) Serum adenosine deaminase activity in patients with systemic lupus erythematosus: a study based on ADA1 and ADA2 isoenzymes pattern. Rheumatol Int 32(6):1633–1638. doi:10.1007/s00296-011-1836-8
Zemlin AE, Burgess LJ, Carstens ME (2009) The diagnostic utility of adenosine deaminase isoenzymes in tuberculous pleural effusions. Int J Tuberc Lung Dis 13(2):214–220
Roberts EL, Roberts OT (2012) Plasma adenosine deaminase isoform 2 in cancer patients undergoing chemotherapy. Br J Biomed Sci 69(1):11–13
Dolezal T, Dolezelova E, Zurovec M, Bryant PJ (2005) A role for adenosine deaminase in drosophila larval development. PLoS Biol 3(7):e201
Novakova M, Dolezal T (2011) Expression of drosophila adenosine deaminase in immune cells during inflammatory response. PLoS One 6(3):e17741. doi:10.1371/journal.pone.0017741
Zavialov AV, Yu X, Spillmann D, Lauvau G, Zavialov AV (2010) Structural basis for the growth factor activity of human adenosine deaminase ADA2. J Biol Chem 285(16):12367–12377. doi:10.1074/jbc.M109.083527
Liu C, Skaldin M, Wu C, Lu Y, Zavialov AV (2016) Application of ADA1 as a new marker enzyme in sandwich ELISA to study the effect of adenosine on activated monocytes. Sci Rep 6:31370. doi:10.1038/srep31370
Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, Zlotogorski A, Berkun Y, Press JJ, Mukamel M, Voth I, Hashkes PJ, Harel L, Hoffer V, Ling E, Yalcinkaya F, Kasapcopur O, Lee MK, Klevit RE, Renbaum P, Weinberg-Shukron A, Sener EF, Schormair B, Zeligson S, Marek-Yagel D, Strom TM, Shohat M, Singer A, Rubinow A, Pras E, Winkelmann J, Tekin M, Anikster Y, King MC, Levy-Lahad E (2014) Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med 370(10):921–931. doi:10.1056/NEJMoa1307362
Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Stone DL, Chae JJ, Rosenzweig SD, Bishop K, Barron KS, Kuehn HS, Hoffmann P, Negro A, Tsai WL, Cowen EW, Pei W, Milner JD, Silvin C, Heller T, Chin DT, Patronas NJ, Barber JS, Lee CC, Wood GM, Ling A, Kelly SJ, Kleiner DE, Mullikin JC, Ganson NJ, Kong HH, Hambleton S, Candotti F, Quezado MM, Calvo KR, Alao H, Barham BK, Jones A, Meschia JF, Worrall BB, Kasner SE, Rich SS, Goldbach-Mansky R, Abinun M, Chalom E, Gotte AC, Punaro M, Pascual V, Verbsky JW, Torgerson TR, Singer NG, Gershon TR, Ozen S, Karadag O, Fleisher TA, Remmers EF, Burgess SM, Moir SL, Gadina M, Sood R, Hershfield MS, Boehm M, Kastner DL, Aksentijevich I (2014) Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 370(10):911–920. doi:10.1056/NEJMoa1307361
Van Eyck L, Jr., Hershfield MS, Pombal D, Kelly SJ, Ganson NJ, Moens L, Frans G, Schaballie H, De Hertogh G, Dooley J, Bossuyt X, Wouters C, Liston A, Meyts I (2015) Hematopoietic stem cell transplantation rescues the immunologic phenotype and prevents vasculopathy in patients with adenosine deaminase 2 deficiency. J Allergy Clin Immunol 135(1):283–287 e285. doi:10.1016/j.jaci.2014.10.010
van Montfrans J, Zavialov A, Zhou Q (2014) Mutant ADA2 in vasculopathies. N Engl J Med 371(5):478. doi:10.1056/NEJMc1405506#SA1
Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, Zimmer J (2016) Human CD56bright NK cells: an update. J Immunol 196(7):2923–2931. doi:10.4049/jimmunol.1502570
Aran JM, Colomer D, Matutes E, Vives-Corrons JL, Franco R (1991) Presence of adenosine deaminase on the surface of mononuclear blood cells: immunochemical localization using light and electron microscopy. J Histochem Cytochem 39(8):1001–1008
Dong RP, Tachibana K, Hegen M, Munakata Y, Cho D, Schlossman SF, Morimoto C (1997) Determination of adenosine deaminase binding domain on CD26 and its immunoregulatory effect on T cell activation. J Immunol 159(12):6070–6076
Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, Wara DW, Douglas SD, Luzuriaga K, McFarland EJ, Yogev R, Rathore MH, Levy W, Graham BL, Spector SA, Pediatric ACTG (2003) Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol 112(5):973–980. doi:10.1016/j.jaci.2003.07.003
Zhang JG, Hepburn L, Cruz G, Borman RA, Clark KL (2005) The role of adenosine A2A and A2B receptors in the regulation of TNF-alpha production by human monocytes. Biochem Pharmacol 69(6):883–889. doi:10.1016/j.bcp.2004.12.008
Moser GH, Schrader J, Deussen A (1989) Turnover of adenosine in plasma of human and dog blood. Am J Physiol 256(4 Pt 1):C799–806
Cekic C, Linden J (2016) Purinergic regulation of the immune system. Nat Rev Immunol 16(3):177–192. doi:10.1038/nri.2016.4
Barletta KE, Ley K, Mehrad B (2012) Regulation of neutrophil function by adenosine. Arterioscler Thromb Vasc Biol 32(4):856–864. doi:10.1161/ATVBAHA.111.226845
Franco R, Aran JM, Colomer D, Matutes E, Vives-Corrons JL (1990) Association of adenosine deaminase with erythrocyte and platelet plasma membrane: an immunological study using light and electron microscopy. J Histochem Cytochem 38(5):653–658
Gines S, Marino M, Mallol J, Canela EI, Morimoto C, Callebaut C, Hovanessian A, Casado V, Lluis C, Franco R (2002) Regulation of epithelial and lymphocyte cell adhesion by adenosine deaminase–CD26 interaction. Biochem J 361(Pt 2):203–209
Ratech H, Hirschhorn R (1981) Serum adenosine deaminase in normals and in a patient with adenosine deaminase deficient-severe combined immunodeficiency. Clin Chim Acta Int J Clin Chem 115(3):341–347
Cordero OJ, Salgado FJ, Fernandez-Alonso CM, Herrera C, Lluis C, Franco R, Nogueira M (2001) Cytokines regulate membrane adenosine deaminase on human activated lymphocytes. J Leukoc Biol 70(6):920–930
Van Montfrans JM, Hartman EA, Braun KP, Hennekam EA, Hak EA, Nederkoorn PJ, Westendorp WF, Bredius RG, Kollen WJ, Scholvinck EH, Legger GE, Meyts I, Liston A, Lichtenbelt KD, Giltay JC, Van Haaften G, De Vries Simons GM, Leavis H, Sanders CJ, Bierings MB, Nierkens S, Van Gijn ME (2016) Phenotypic variability in patients with ADA2 deficiency due to identical homozygous R169Q mutations. Rheumatology. doi:10.1093/rheumatology/kev439
Schepp J, Bulashevska A, Mannhardt-Laakmann W, Cao H, Yang F, Seidl M, Kelly S, Hershfield M, Grimbacher B (2016) Deficiency of adenosine deaminase 2 causes antibody deficiency. J Clin Immunol 36(3):179–186. doi:10.1007/s10875-016-0245-x
Ohta A, Sitkovsky M (2014) Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol 5:304. doi:10.3389/fimmu.2014.00304
Gasteiger G, Rudensky AY (2014) Interactions between innate and adaptive lymphocytes. Nat Rev Immunol 14(9):631–639. doi:10.1038/nri3726
Schuler PJ, Saze Z, Hong CS, Muller L, Gillespie DG, Cheng D, Harasymczuk M, Mandapathil M, Lang S, Jackson EK, Whiteside TL (2014) Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin Exp Immunol 177(2):531–543. doi:10.1111/cei.12354
Ancuta P, Liu KY, Misra V, Wacleche VS, Gosselin A, Zhou X, Gabuzda D (2009) Transcriptional profiling reveals developmental relationship and distinct biological functions of CD16+ and CD16− monocyte subsets. BMC Genom 10:403. doi:10.1186/1471-2164-10-403
Martinon F, Aksentijevich I (2015) New players driving inflammation in monogenic autoinflammatory diseases. Nat Rev Rheumatol 11(1):11–20. doi:10.1038/nrrheum.2014.158
Giannelou A, Zhou Q, Kastner DL (2014) When less is more: primary immunodeficiency with an autoinflammatory kick. Curr Opin Allergy Clin Immunol 14(6):491–500. doi:10.1097/ACI.0000000000000117
Belot A, Wassmer E, Twilt M, Lega JC, Zeef LA, Oojageer A, Kasher PR, Mathieu AL, Malcus C, Demaret J, Fabien N, Collardeau-Frachon S, Mechtouff L, Derex L, Walzer T, Rice GI, Durieu I, Crow YJ (2014) Mutations in CECR1 associated with a neutrophil signature in peripheral blood. Pediatric Rheumatol Online J 12:44. doi:10.1186/1546-0096-12-44
Wu C, Lu Y (2010) High-titre retroviral vector system for efficient gene delivery into human and mouse cells of haematopoietic and lymphocytic lineages. J Gen Virol 91(Pt 8):1909–1918. doi:10.1099/vir.0.020255-0
Wu C, Lu Y (2007) Inclusion of high molecular weight dextran in calcium phosphate-mediated transfection significantly improves gene transfer efficiency. Cell Mol Biol 53(4):67–74
Acknowledgments
This study was supported by a Grant 256053 from the Finnish Academy to A.Z.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Additional information
Y. Kaljas, C. Liu, M. Skaldin and C. Wu equal contribution.
Rights and permissions
About this article

Cite this article
Kaljas, Y., Liu, C., Skaldin, M. et al. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell. Mol. Life Sci. 74, 555–570 (2017). https://doi.org/10.1007/s00018-016-2357-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2357-0